
Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Preclinical Proof of Concept and Mechanism of Action for Using HSPC for Cystinosis
3. Ex Vivo Gene Modified Cell Therapy: A Safer Approach Than Allogeneic Transplantation
4. Preclinical Studies for the Autologous Gene-Modified Hematopoietic Stem Cell Approach for Cystinosis
5. Investigational New Drug-Enabling Studies
5.1. Pharmacology/Toxicology Studies
5.2. Manufacturing Development
5.3. Clinical Design
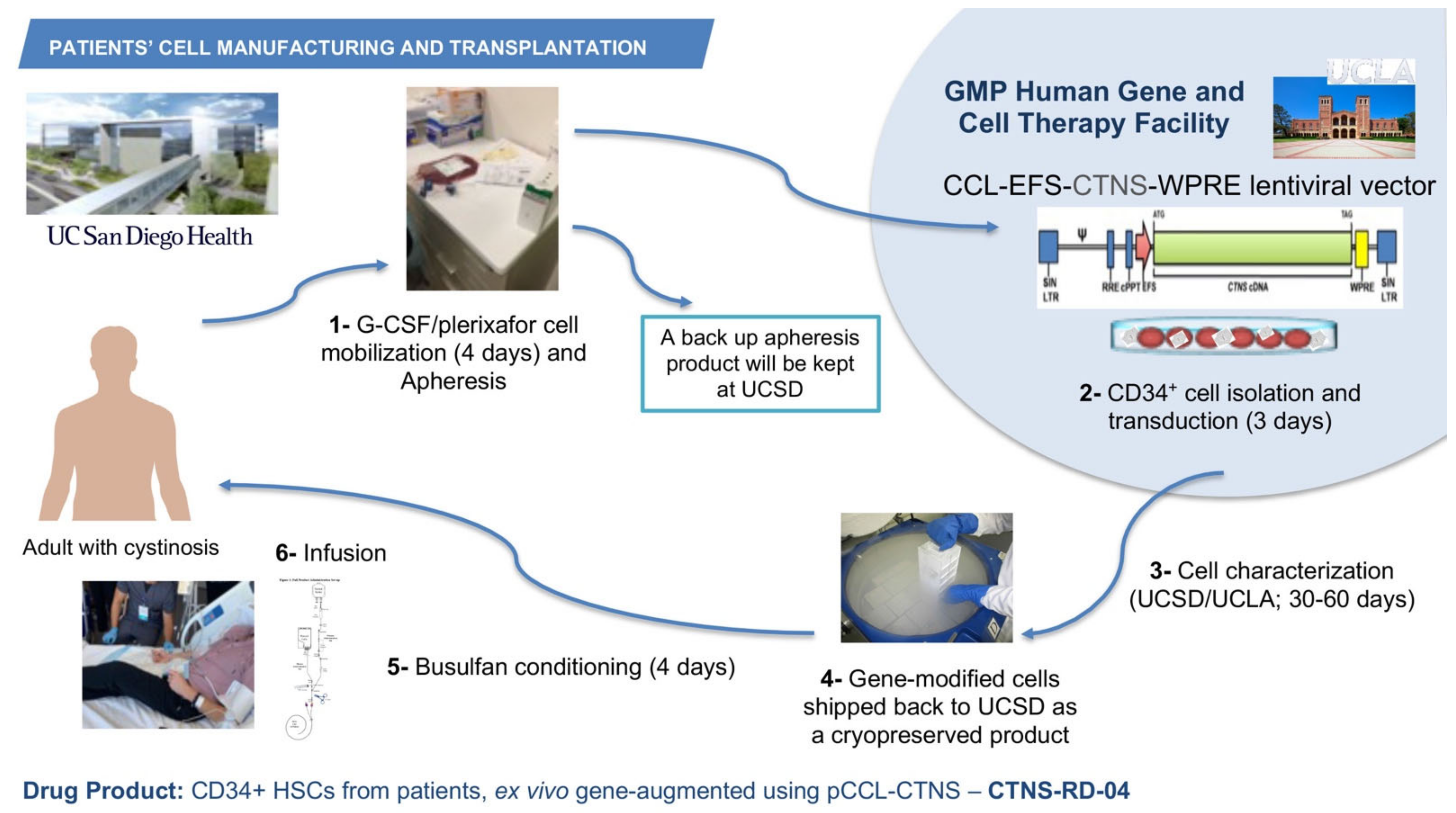
6. Autologous Gene-Modified HSPC Clinical Trial for Cystinosis
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Levy, M.; Feingold, J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000, 58, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Haffner, D.; Weinfurth, A.; Manz, F.; Schmidt, H.; Bremer, H.J.; Mehls, O.; Scharer, K. Long-term outcome of paediatric patients with hereditary tubular disorders. Nephron 1999, 83, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Gahl, W. Nephropathic cystinosis: Late complications of a multisystemic disease. Pediatr. Nephrol. 2008, 23, 863–878. [Google Scholar] [CrossRef]
- Gahl, W.A.; Kuehl, E.M.; Iwata, F.; Lindblad, A.; Kaiser-Kupfer, M.I. Corneal crystals in nephropathic cystinosis: Natural history and treatment with cysteamine eyedrops. Mol. Genet. Metab. 2000, 71, 100–120. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Wong, V.; Seegmiller, J.E. The early diagnosis of cystinosis. J. Pediatr. 1969, 74, 114–116. [Google Scholar] [CrossRef]
- Dufier, J.L.; Dhermy, P.; Gubler, M.C.; Gagnadoux, M.F.; Broyer, M. Ocular changes in long-term evolution of infantile cystinosis. Ophthalmic Paediatr. Genet. 1987, 8, 131–137. [Google Scholar] [CrossRef]
- Tsilou, E.; Zhou, M.; Gahl, W.; Sieving, P.C.; Chan, C.C. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: Report of a case and review of the literature. Surv. Ophthalmol. 2007, 52, 97–105. [Google Scholar] [CrossRef]
- Ueda, M.; O’Brien, K.; Rosing, D.R.; Ling, A.; Kleta, R.; McAreavey, D.; Bernardini, I.; Gahl, W.A. Coronary artery and other vascular calcifications in patients with cystinosis after kidney transplantation. Clin. J. Am. Soc. Nephrol. 2006, 1, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Keser, A.G.; Topaloglu, R.; Bilginer, Y.; Besbas, N. Long-term endocrinologic complications of cystinosis. Minerva Pediatr. 2014, 66, 123–130. [Google Scholar]
- Klusmann, M.; Van′t Hoff, W.; Monsell, F.; Offiah, A.C. Progressive destructive bone changes in patients with cystinosis. Skeletal Radiol. 2013, 43, 387–391. [Google Scholar] [CrossRef]
- Bacchetta, J.; Greco, M.; Bertholet-Thomas, A.; Nobili, F.; Zustin, J.; Cochat, P.; Emma, F.; Boivin, G. Skeletal implications and management of cystinosis: Three case reports and literature review. Bonekey Rep. 2016, 5, 828. [Google Scholar] [CrossRef]
- Ballantyne, A.O.; Trauner, D.A. Neurobehavioral consequences of a genetic metabolic disorder: Visual processing deficits in infantile nephropathic cystinosis. Neuropsychiatry Neuropsychol. Behav. Neurol. 2000, 13, 254–263. [Google Scholar]
- Scarvie, K.M.; Ballantyne, A.O.; Trauner, D.A. Visuomotor performance in children with infantile nephropathic cystinosis. Percep. Mot. Skills 1996, 82, 67–75. [Google Scholar] [CrossRef]
- Trauner, D.A.; Chase, C.; Scheller, J.; Katz, B.; Schneider, J.A. Neurologic and cognitive deficits in children with cystinosis. J. Pediatr. 1988, 112, 912–914. [Google Scholar] [CrossRef]
- Trauner, D.A.; Spilkin, A.M.; Williams, J.; Babchuck, L. Specific cognitive deficits in young children with cystinosis: Evidence for an early effect of the cystinosin gene on neural function. J. Pediatr. 2007, 151, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Trauner, D.A.; Williams, J.; Ballantyne, A.O.; Spilkin, A.M.; Crowhurst, J.; Hesselink, J. Neurological impairment in nephropathic cystinosis: Motor coordination deficits. Pediatr. Nephrol. 2010, 25, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Viltz, L.; Trauner, D.A. Effect of age at treatment on cognitive performance in patients with cystinosis. J. Pediatr. 2013, 163, 489–492. [Google Scholar] [CrossRef]
- Anikster, Y.; Lacbawan, F.; Brantly, M.; Gochuico, B.L.; Avila, N.A.; Travis, W.; Gahl, W.A. Pulmonary dysfunction in adults with nephropathic cystinosis. Chest 2001, 119, 394–401. [Google Scholar] [CrossRef]
- Sonies, B.C.; Almajid, P.; Kleta, R.; Bernardini, I.; Gahl, W.A. Swallowing dysfunction in 101 patients with nephropathic cystinosis: Benefit of long-term cysteamine therapy. Medicine 2005, 84, 137–146. [Google Scholar] [CrossRef]
- Berger, J.R.; Dillon, D.A.; Young, B.A.; Goldstein, S.J.; Nelson, P. Cystinosis of the brain and spinal cord with associated vasculopathy. J. Neurol. Sci. 2009, 184, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Trauner, D.A.; Fahmy, R.F.; Mishler, D.A. Oral motor dysfunction and feeding difficulties in nephropathic cystinosis. Pediatr. Neurol. 2001, 24, 365–368. [Google Scholar] [CrossRef]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Kalatzis, V.; Cherqui, S.; Antignac, C.; Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001, 20, 5940–5949. [Google Scholar] [CrossRef]
- Cherqui, S.; Kalatzis, V.; Trugnan, G.; Antignac, C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J. Biol. Chem. 2001, 276, 13314–13321. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef]
- Johnson, J.L.; Napolitano, G.; Monfregola, J.; Rocca, C.J.; Cherqui, S.; Catz, S.D. Upregulation of the Rab27a-Dependent Trafficking and Secretory Mechanisms Improves Lysosomal Transport, Alleviates Endoplasmic Reticulum Stress, and Reduces Lysosome Overload in Cystinosis. Mol. Cell. Biol. 2013, 33, 2950–2962. [Google Scholar] [CrossRef]
- Napolitano, G.; Johnson, J.L.; He, J.; Rocca, C.J.; Monfregola, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2015, 7, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Pejovic, V.; Kerisit, K.G.; Junius, S.; Thoene, J.G. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cdelta. J. Am. Soc. Nephrol. 2006, 17, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Sansanwal, P.; Kambham, N.; Sarwal, M.M. Caspase-4 may play a role in loss of proximal tubules and renal injury in nephropathic cystinosis. Pediatr. Nephrol. 2010, 25, 105–109. [Google Scholar] [CrossRef]
- Dohil, R.; Fidler, M.; Gangoiti, J.A.; Kaskel, F.; Schneider, J.A.; Barshop, B.A. Twice-daily cysteamine bitartrate therapy for children with cystinosis. J. Pediatr. 2010, 156, 71–75.e3. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A. Approval of cysteamine for patients with cystinosis. Pediatr. Nephrol. 1995, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Brodin-Sartorius, A.; Tete, M.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef]
- Emma, F.; Hoff, W.V.; Hohenfellner, K.; Topaloglu, R.; Greco, M.; Ariceta, G.; Bettini, C.; Bockenhauer, D.; Veys, K.; Pape, L.; et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int. 2021, 100, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Balog, J.Z.; Kleta, R. Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy. Ann. Intern. Med. 2007, 147, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S. Cysteamine therapy: A treatment for cystinosis, not a cure. Kidney Int. 2012, 81, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Bougneres, P.; Schmidt, M.; Kalle, C.V.; Fischer, A.; Cavazzana-Calvo, M.; Aubourg, P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012, 507, 187–198. [Google Scholar] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Gentner, B.; Bernardo, M.E.M.E.; Tucci, F.; Zonari, E.; Fumagalli, F.; Pontesilli, S.; Acquati, S.; Silvani, P.; Ciceri, F.; Rovelli, A.; et al. Extensive Metabolic Correction of Hurler Disease by Hematopoietic Stem Cell-Based Gene Therapy: Preliminary Results from a Phase I/II Trial. Blood 2019, 134, 607. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Sevin, C.; Hamard, G.; Kalatzis, V.; Sich, M.; Pequignot, M.O.; Gogat, K.; Abitbol, M.; Broyer, M.; Gubler, M.C.; et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol. Cell. Biol. 2002, 22, 7622–7632. [Google Scholar] [CrossRef] [PubMed]
- Nevo, N.; Chol, M.; Bailleux, A.; Kalatzis, V.; Morisset, L.; Devuyst, O.; Gubler, M.C.; Antignac, C. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol. Dial. Transpl. 2010, 25, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Chevronnay, H.P.G.; Janssens, V.; van der Smissen, P.; N′Kuli, F.; Nevo, N.; Guiot, Y.; Levtchenko, E.; Marbaix, E.; Pierreux, C.E.; Cherqui, S.; et al. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J. Am. Soc. Nephrol. 2014, 25, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Serratrice, N.; Hippert, C.; Payet, O.; Arndt, C.; Cazevieille, C.; Maurice, T.; Hamel, C.; Malecaze, F.; Antignac, C.; et al. The ocular anomalies in a cystinosis animal model mimic disease pathogenesis. Pediatr. Res. 2007, 62, 156–162. [Google Scholar] [CrossRef]
- Simpson, J.; Nien, C.J.; Flynn, K.; Jester, B.; Cherqui, S.; Jester, J. Quantitative in vivo and ex vivo confocal microscopy analysis of corneal cystine crystals in the Ctns knockout mouse. Mol. Vis. 2011, 17, 2212–2220. [Google Scholar]
- Challen, G.A.; Boles, N.; Lin, K.K.; Goodell, M.A. Mouse hematopoietic stem cell identification and analysis. Cytometry A 2009, 75, 14–24. [Google Scholar] [CrossRef]
- Syres, K.; Harrison, F.; Tadlock, M.; Jester, J.V.; Simpson, J.; Roy, S.; Salomon, D.R.; Cherqui, S. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 2009, 114, 2542–2552. [Google Scholar] [CrossRef]
- Yeagy, B.A.; Harrison, F.; Gubler, M.C.; Koziol, J.A.; Salomon, D.R.; Cherqui, S. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011, 79, 1198–1206. [Google Scholar] [CrossRef]
- Rocca, C.J.; Kreymerman, A.; Ur, S.N.; Frizzi, K.E.; Naphade, S.; Lau, A.; Tran, T.; Calcutt, N.A.; Goldberg, J.L.; Cherqui, S. Treatment of Inherited Eye Defects by Systemic Hematopoietic Stem Cell Transplantation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7214–7223. [Google Scholar] [CrossRef]
- Chevronnay, H.P.G.; Janssens, V.; van der Smissen, P.; Liao, X.H.; Abid, Y.; Nevo, N.; Antignac, C.; Refetoff, S.; Cherqui, S.; Pierreux, C.E.; et al. A mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: Decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing. Endocrinology 2015, 156, 2349–2364. [Google Scholar] [CrossRef][Green Version]
- Chevronnay, H.P.G.; Jansen, V.; van der Smissen, P.; Rocca, C.J.; Liao, X.H.; Refetoff, S.; Pierreux, C.E.; Cherqui, S.; Courtoy, P. Hematopoietic stem cell transplantation can normalize thyroid function in a cystinosis mouse model. Endocrinology 2016, 157, 1363–1371. [Google Scholar] [CrossRef]
- Biffi, A.; de Palma, M.; Quattrini, A.; del Carro, U.; Amadio, S.; Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.; et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Investig. 2004, 113, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, C.; Villani, G.R.; di Napoli, D.; Reyero, E.G.; Lombardo, A.; Naldini, L.; di Natale, P. Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum. Gene Ther. 2005, 16, 81–90. [Google Scholar] [CrossRef]
- Massaro, G.; Geard, A.F.; Liu, W.; Coombe-Tennant, O.; Waddington, S.N.; Baruteau, J.; Gissen, P.; Rahim, A.A. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules 2021, 11, 611. [Google Scholar] [CrossRef]
- Naphade, S.; Sharma, J.; Chevronnay, H.P.G.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef]
- Goodman, S.; Naphade, S.; Khan, M.; Sharma, J.; Cherqui, S. Macrophage polarization impacts tunneling nanotube formation and intercellular organelle trafficking. Sci. Rep. 2019, 9, 14529. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L. Acute graft-versus-host disease: Differing risk with differing graft sources and conditioning intensity. Best Pract. Res. Clin. Haematol. 2008, 21, 177–192. [Google Scholar] [CrossRef]
- Pallera, A.M.; Schwartzberg, L.S. Managing the toxicity of hematopoietic stem cell transplant. J. Support Oncol. 2004, 2, 223–237; discussion 237–228, 241, 246–227. [Google Scholar]
- Cutler, C.; Li, S.; Ho, V.T.; Koreth, J.; Alyea, E.; Soiffer, R.J.; Antin, J.H. Extended follow-up of methotrexate-free immunosuppression using sirolimus and tacrolimus in related and unrelated donor peripheral blood stem cell transplantation. Blood 2007, 109, 3108–3114. [Google Scholar] [CrossRef]
- Geyer, M.B.; Jacobson, J.S.; Freedman, J.; George, D.; Moore, V.; van de Ven, C.; Satwani, P.; Bhatia, M.; Garvin, J.H.; Bradley, M.B.; et al. A comparison of immune reconstitution and graft-versus-host disease following myeloablative conditioning versus reduced toxicity conditioning and umbilical cord blood transplantation in paediatric recipients. Br. J. Haematol. 2011, 155, 218–234. [Google Scholar] [CrossRef]
- Schleuning, M.; Judith, D.; Jedlickova, Z.; Stubig, T.; Heshmat, M.; Baurmann, H.; Schwerdtfeger, R. Calcineurin inhibitor-free GVHD prophylaxis with sirolimus, mycophenolate mofetil and ATG in Allo-SCT for leukemia patients with high relapse risk: An observational cohort study. Bone Marrow. Transpl. 2009, 43, 717–723. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Elmonem, M.A.; Veys, K.; Arcolino, F.O.; van Dyck, M.; Benedetti, M.C.; Diomedi-Camassei, F.; de Hertogh, G.; van den Heuvel, L.P.; Renard, M.; Levtchenko, E. Allogeneic HSCT transfers wild-type cystinosin to nonhematological epithelial cells in cystinosis: First human report. Am. J. Transpl. 2018, 18, 2823–2828. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Berry, C.C.; Malani, N.; Leboulch, P.; Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M.; Bushman, F.D. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood 2010, 115, 4356–4366. [Google Scholar] [CrossRef]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 2021, 29, 489–504. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Stan, R.; Krishnan, A.; Li, H.; Rossi, J.J.; Zaia, J.A. Development of hematopoietic stem cell based gene therapy for HIV-1 infection: Considerations for proof of concept studies and translation to standard medical practice. Viruses 2013, 5, 2898–2919. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Papanikolaou, E.; Georgomanoli, M.; Anagnou, N.P. Towards more successful gene therapy clinical trials for beta-thalassemia. Curr. Mol. Med. 2013, 13, 1314–1330. [Google Scholar] [CrossRef]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; de Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Zhang, L.; Thrasher, A.J.; Gaspar, H.B. Current progress on gene therapy for primary immunodeficiencies. Gene Ther. 2013, 20, 963–969. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Booth, C.; Shaw, K.L.; Xu-Bayford, J.; Garabedian, E.; Trevisan, V.; Carbonaro-Sarracino, D.A.; Soni, K.; Terrazas, D.; Snell, K.; et al. Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 2021, 384, 2002–2013. [Google Scholar] [CrossRef]
- Demaison, C.; Parsley, K.; Brouns, G.; Scherr, M.; Battmer, K.; Kinnon, C.; Grez, M.; Thrasher, A.J. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 2002, 13, 803–813. [Google Scholar] [CrossRef]
- Zanta-Boussif, M.A.; Charrier, S.; Brice-Ouzet, A.; Martin, S.; Opolon, P.; Thrasher, A.J.; Hope, T.J.; Galy, A. Validation of a mutated PRE sequence allowing high and sustained transgene expression while abrogating WHV-X protein synthesis: Application to the gene therapy of WAS. Gene Ther. 2009, 16, 605–619. [Google Scholar] [CrossRef]
- Kingsman, S.M.; Mitrophanous, K.; Olsen, J.C. Potential oncogene activity of the woodchuck hepatitis post-transcriptional regulatory element (WPRE). Gene Ther. 2005, 12, 3–4. [Google Scholar] [CrossRef]
- Wakabayashi-Ito, N.; Nagata, S. Characterization of the regulatory elements in the promoter of the human elongation factor-1 alpha gene. J. Biol. Chem. 1994, 269, 29831–29837. [Google Scholar] [CrossRef]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological Promoters Reduce the Genotoxic Risk of Integrating Gene Vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.; Yeagy, B.A.; Rocca, C.J.; Kohn, D.B.; Salomon, D.R.; Cherqui, S. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol. Ther. 2013, 21, 433–444. [Google Scholar] [CrossRef]
- Ronen, K.; Negre, O.; Roth, S.; Colomb, C.; Malani, N.; Denaro, M.; Brady, T.; Fusil, F.; Gillet-Legrand, B.; Hehir, K.; et al. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat beta-thalassemia. Mol. Ther. 2011, 19, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, P.I.; Higashimoto, T.; Urbinati, F.; Modlich, U.; Nestheide, S.; Xia, P.; Fox, C.; Corsinotti, A.; Baum, C.; Malik, P. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol. Ther. 2009, 17, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Bengali, M.; Goodman, S.; Sun, X.; Dohil, M.A.; Dohil, R.; Newbury, R.; Lobry, T.; Hernandez, L.; Antignac, C.; Jain, S.; et al. Non-invasive intradermal imaging of cystine crystals in cystinosis. PLoS ONE 2021, 16, e0247846. [Google Scholar] [CrossRef] [PubMed]
- Yannaki, E.; Karponi, G.; Zervou, F.; Constantinou, V.; Bouinta, A.; Tachynopoulou, V.; Kotta, K.; Jonlin, E.; Papayannopoulou, T.; Anagnostopoulos, A.; et al. Hematopoietic stem cell mobilization for gene therapy: Superior mobilization by the combination of granulocyte-colony stimulating factor plus plerixafor in patients with beta-thalassemia major. Hum. Gene Ther. 2013, 24, 852–860. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherqui, S. Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside. Cells 2021, 10, 3273. https://doi.org/10.3390/cells10123273
Cherqui S. Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside. Cells. 2021; 10(12):3273. https://doi.org/10.3390/cells10123273
Chicago/Turabian StyleCherqui, Stephanie. 2021. "Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside" Cells 10, no. 12: 3273. https://doi.org/10.3390/cells10123273
APA StyleCherqui, S. (2021). Hematopoietic Stem Cell Gene Therapy for Cystinosis: From Bench-to-Bedside. Cells, 10(12), 3273. https://doi.org/10.3390/cells10123273