
Identifying Plectin Isoform Functions through Animal Models


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
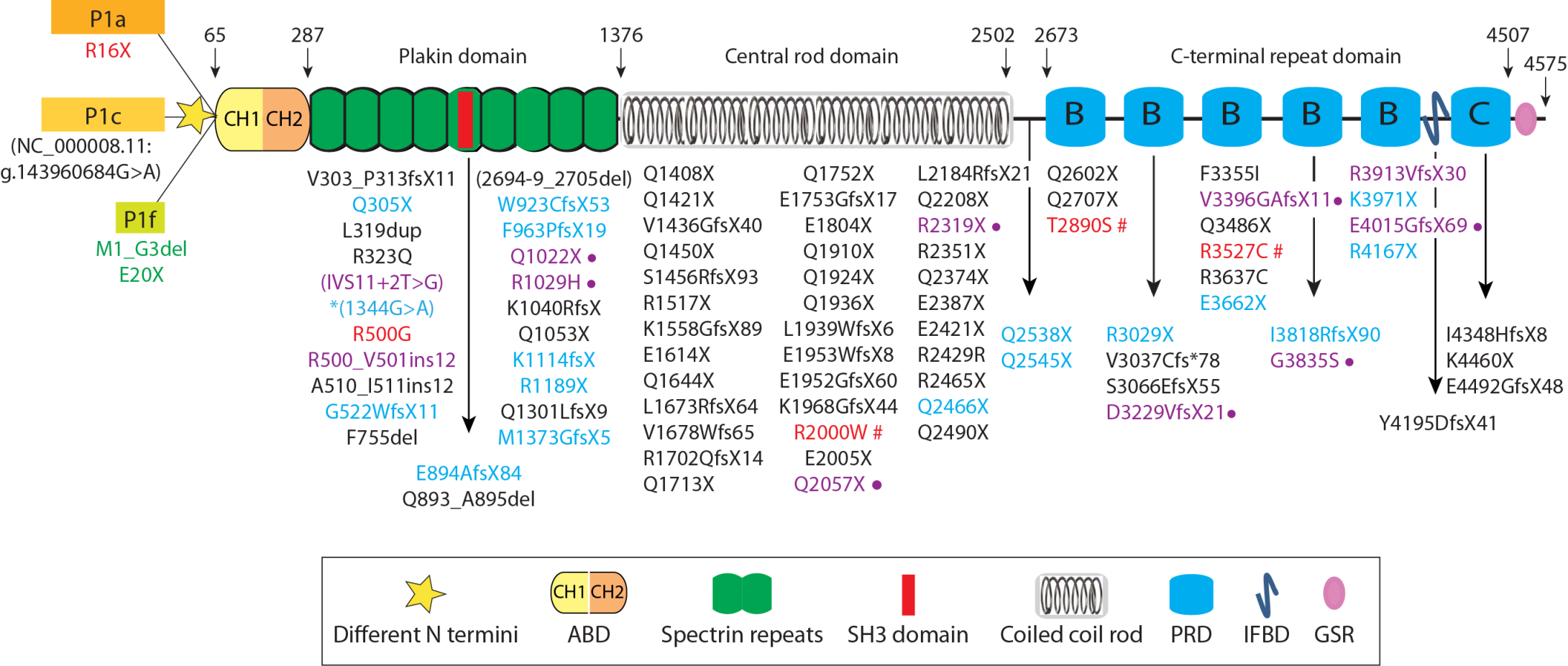
2. Plectin-Null Mice
3. Conditional, Tissue-Restricted Plectin KOs
3.1. Plectin Ablation in Stratified Epithelia
3.2. Plectin Ablation in Single Epithelia
3.3. Plectin Ablation in Endothelial Cells
3.4. Plectin Ablation in Schwann Cells
3.5. Plectin Ablation in Skeletal and Heart Muscle
4. Isoform-Specific Plectin KOs
4.1. Plectin Isoform 1 (P1)-Deficient Mice: P1-KO
4.2. Plectin Isoform 1b (P1b)-Deficient Mice: P1b-KO
4.3. Plectin Isoform 1d (P1d)-Deficient Mice: P1d-KO
4.4. Plectin Isoform 1c (P1c)-Deficient Mice: P1c-KO
4.5. Human Knockouts for Isoforms P1a and P1f
5. Plectin Knock-In Mouse Models
5.1. EBS-Ogna Mice
5.2. Rodless Plectin Mice
6. Zebrafish and C. elegans
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castañón, M.J.; Walko, G.; Winter, L.; Wiche, G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem. Cell Biol. 2013, 140, 33–53. [Google Scholar] [CrossRef]
- Wiche, G.; Castañón, M.J. Intermediate Filament Linker Proteins: Plectin and BPAG1. In Encyclopedia of Biological Chemistry, 3rd ed.; Jez, J., Ed.; Elsevier: New York, NY, USA, 2021; pp. 200–219. [Google Scholar]
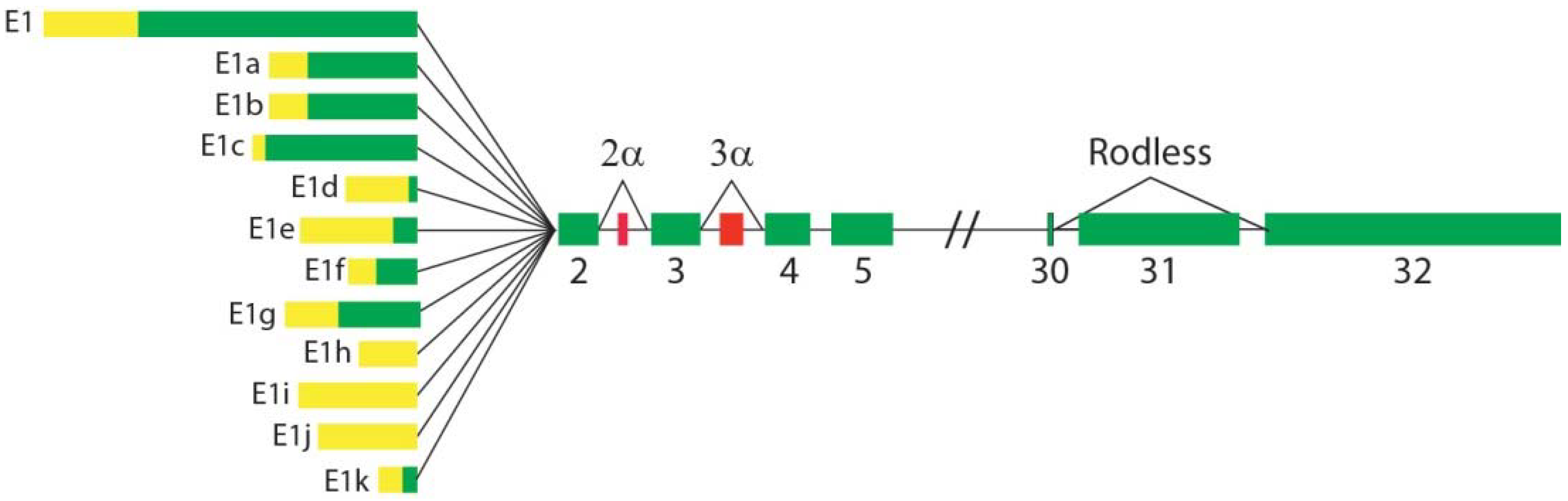
- Elliott, C.E.; Becker, B.; Oehler, S.; Castañón, M.J.; Hauptmann, R.; Wiche, G. Plectin transcript diversity: Identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics 1997, 42, 115–125. [Google Scholar] [CrossRef]
- Fuchs, P.; Zörer, M.; Rezniczek, G.A.; Spazierer, D.; Oehler, S.; Castañón, M.J.; Hauptmann, R.; Wiche, G. Unusual 5′ transcript complexity of plectin isoforms: Novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 1999, 8, 2461–2472. [Google Scholar] [CrossRef]
- Liu, C.G.; Maercker, C.; Castañón, M.J.; Hauptmann, R.; Wiche, G. Human plectin: Organization of the gene, sequence analysis, and chromosome localization (8q24). Proc. Natl. Acad. Sci. USA 1996, 93, 4278–4283. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Abrahamsberg, C.; Fuchs, P.; Spazierer, D.; Wiche, G. Plectin 5’-transcript diversity: Short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 2003, 12, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Denk, H. Occurrence and immunolocalization of plectin in tissues. J. Cell Biol. 1983, 97, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Aberer, W. Identification of plectin in different human cell types and immunolocalization at epithelial basal cell surface membranes. Exp. Cell Res. 1984, 155, 43–49. [Google Scholar] [CrossRef]
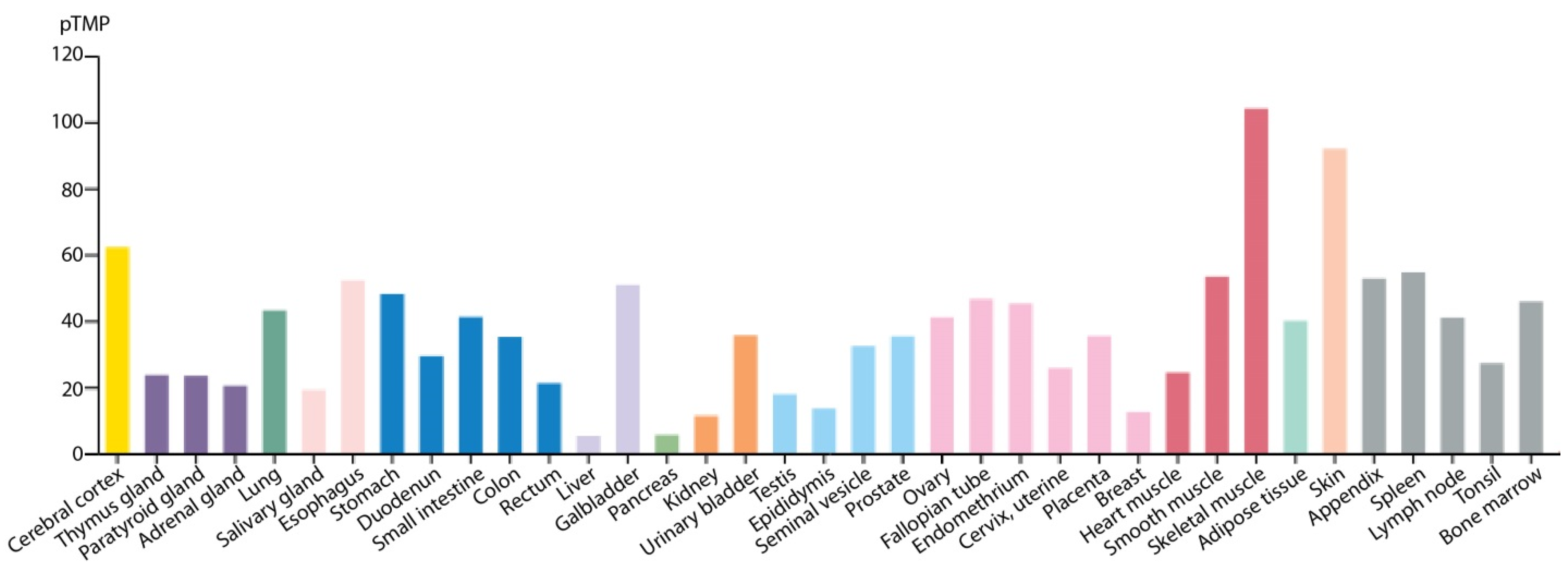
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Wiche, G. The many faces of plectin and plectinopathies: Pathology and mechanisms. Acta NeuroPathol. 2013, 125, 77–93. [Google Scholar] [CrossRef]
- Capecchi, M.R. Altering the genome by homologous recombination. Science 1989, 244, 1288–1292. [Google Scholar] [CrossRef]
- Gu, H.; Marth, J.D.; Orban, P.C.; Mossmann, H.; Rajewsky, K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science 1994, 265, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Andrä, K.; Lassmann, H.; Bittner, R.; Shorny, S.; Fässler, R.; Propst, F.; Wiche, G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997, 11, 3143–3156. [Google Scholar] [CrossRef] [PubMed]
- Kucherlapati, R.S.; Spencer, J.; Moore, P.D. Homologous recombination catalyzed by human cell extracts. Mol. Cell. Biol. 1985, 5, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Gache, Y.; Chavanas, S.; Lacour, J.P.; Wiche, G.; Owaribe, K.; Meneguzzi, G.; Ortonne, J.P. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J. Clin. Investig. 1996, 97, 2289–2298. [Google Scholar] [CrossRef]
- Smith, F.J.; Eady, R.A.; Leigh, I.M.; McMillan, J.R.; Rugg, E.L.; Kelsell, D.P.; Bryant, S.P.; Spurr, N.K.; Geddes, J.F.; Kirtschig, G.; et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat. Genet. 1996, 13, 450–457. [Google Scholar] [CrossRef]
- Andrä, K.; Kornacker, I.; Jörgl, A.; Zörer, M.; Spazierer, D.; Fuchs, P.; Fischer, I.; Wiche, G. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (−/−) keratinocytes. J. Investig. Dermatol. 2003, 120, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Andrä, K.; Nikolic, B.; Stöcher, M.; Drenckhahn, D.; Wiche, G. Not just scaffolding: Plectin regulates actin dynamics in cultured cells. Genes Dev. 1998, 12, 3442–3451. [Google Scholar] [CrossRef]
- Ackerl, R.; Walko, G.; Fuchs, P.; Fischer, I.; Schmuth, M.; Wiche, G. Conditional targeting of plectin in prenatal and adult mouse stratified epithelia causes keratinocyte fragility and lesional epidermal barrier defects. J. Cell Sci. 2007, 120, 2435–2443. [Google Scholar] [CrossRef]
- Tarutani, M.; Itami, S.; Okabe, M.; Ikawa, M.; Tezuka, T.; Yoshikawa, K.; Kinoshita, T.; Takeda, J. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc. Natl. Acad. Sci. USA 1997, 94, 7400–7405. [Google Scholar] [CrossRef]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef]
- Indra, A.K.; Warot, X.; Brocard, J.; Bornert, J.M.; Xiao, J.H.; Chambon, P.; Metzger, D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: Comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999, 27, 4324–4327. [Google Scholar] [CrossRef] [PubMed]
- Jirouskova, M.; Nepomucka, K.; Oyman-Eyrilmez, G.; Kalendova, A.; Havelkova, H.; Sarnova, L.; Chalupsky, K.; Schuster, B.; Benada, O.; Miksatkova, P.; et al. Plectin controls biliary tree architecture and stability in cholestasis. J. Hepatol. 2018, 68, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Krausova, A.; Buresova, P.; Sarnova, L.; Oyman-Eyrilmez, G.; Skarda, J.; Wohl, P.; Bajer, L.; Sticova, E.; Bartonova, L.; Pacha, J.; et al. Plectin ensures intestinal epithelial integrity and protects colon against colitis. Mucosal Immunol. 2021, 14, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.; Wögenstein, K.L.; Österreicher, C.H.; Guldiken, N.; Chen, Y.; Doler, C.; Wiche, G.; Boor, P.; Haybaeck, J.; Strnad, P.; et al. Epiplakin attenuates experimental mouse liver injury by chaperoning keratin reorganization. J. Hepatol. 2015, 62, 1357–1366. [Google Scholar] [CrossRef]
- Wu, S.H.; Hsu, J.S.; Chen, H.L.; Chien, M.M.; Wu, J.F.; Ni, Y.H.; Liou, B.Y.; Ho, M.C.; Jeng, Y.M.; Chang, M.H.; et al. Plectin Mutations in Progressive Familial Intrahepatic Cholestasis. Hepatology 2019, 70, 2221–2224. [Google Scholar] [CrossRef]
- Fontao, L.; Stutzmann, J.; Gendry, P.; Launay, J.F. Regulation of the type II hemidesmosomal plaque assembly in intestinal epithelial cells. Exp. Cell Res. 1999, 250, 298–312. [Google Scholar] [CrossRef]
- el Marjou, F.; Janssen, K.P.; Chang, B.H.; Li, M.; Hindie, V.; Chan, L.; Louvard, D.; Chambon, P.; Metzger, D.; Robine, S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 2004, 39, 186–193. [Google Scholar] [CrossRef]
- De Arcangelis, A.; Hamade, H.; Alpy, F.; Normand, S.; Bruyere, E.; Lefebvre, O.; Mechine-Neuville, A.; Siebert, S.; Pfister, V.; Lepage, P.; et al. Hemidesmosome integrity protects the colon against colitis and colorectal cancer. Gut 2017, 66, 1748–1760. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Rus, S.; Wolfram, M.; Brunner, D.; Goldmann, W.H.; Bonakdar, N.; Fischer, I.; Reipert, S.; Zuzuarregui, A.; Walko, G.; et al. Plectin reinforces vascular integrity by mediating crosstalk between the vimentin and the actin networks. J. Cell Sci. 2015, 128, 4138–4150. [Google Scholar] [CrossRef]
- Alva, J.A.; Zovein, A.C.; Monvoisin, A.; Murphy, T.; Salazar, A.; Harvey, N.L.; Carmeliet, P.; Iruela-Arispe, M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006, 235, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the vimentin network under control: Cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jörgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014, 28, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Rouan, F.; Kofler, B.; Rezniczek, G.A.; Kornacker, I.; Muss, W.; Hametner, R.; Klausegger, A.; Huber, A.; Pohla-Gubo, G.; et al. A Compound Heterozygous One Amino-Acid Insertion/Nonsense Mutation in the Plectin Gene Causes Epidermolysis Bullosa Simplex with Plectin Deficiency. Am. J. Pathol. 2001, 158, 617–625. [Google Scholar] [CrossRef]
- Kiritsi, D.; Pigors, M.; Tantcheva-Poor, I.; Wessel, C.; Arin, M.J.; Kohlhase, J.; Bruckner-Tuderman, L.; Has, C. Epidermolysis bullosa simplex ogna revisited. J. Investig. Dermatol. 2013, 133, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulou, Z.; Liu, L.; Ozoemena, L.; Branco, C.C.; Senra, R.; Reis-Rego, A.; Mota-Vieira, L. A novel PLEC nonsense homozygous mutation (c.7159G > T; p.Glu2387*) causes epidermolysis bullosa simplex with muscular dystrophy and diffuse alopecia: A case report. BMC Dermatol. 2018, 18, 1. [Google Scholar] [CrossRef]
- Khan, F.F.; Khan, N.; Rehman, S.; Ejaz, A.; Ali, U.; Erfan, M.; Ahmed, Z.M.; Naeem, M. Identification and Computational Analysis of Novel Pathogenic Variants in Pakistani Families with Diverse Epidermolysis Bullosa Phenotypes. Biomolecules 2021, 11, 620. [Google Scholar] [CrossRef]
- Fuchs, P.; Zörer, M.; Reipert, S.; Rezniczek, G.A.; Propst, F.; Walko, G.; Fischer, I.; Bauer, J.; Leschnik, M.W.; Luscher, B.; et al. Targeted inactivation of a developmentally regulated neural plectin isoform (plectin 1c) in mice leads to reduced motor nerve conduction velocity. J. Biol. Chem. 2009, 284, 26502–26509. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Konieczny, P.; Nikolic, B.; Reipert, S.; Schneller, D.; Abrahamsberg, C.; Davies, K.E.; Winder, S.J.; Wiche, G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J. Cell Biol. 2007, 176, 965–977. [Google Scholar] [CrossRef]
- Cai, H.; Erdman, R.A.; Zweier, L.; Chen, J.; Shaw, J.H.T.; Baylor, K.A.; Stecker, M.M.; Carey, D.J.; Chan, Y.M. The sarcoglycan complex in Schwann cells and its role in myelin stability. Exp. Neurol. 2007, 205, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Wögenstein, K.L.; Winter, L.; Fischer, I.; Feltri, M.L.; Wiche, G. Stabilization of the dystroglycan complex in Cajal bands of myelinating Schwann cells through plectin-mediated anchorage to vimentin filaments. Glia 2013, 61, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Feltri, M.L.; D’Antonio, M.; Previtali, S.; Fasolini, M.; Messing, A.; Wrabetz, L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann. N. Y. Acad. Sci. 1999, 883, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Previtali, S.C.; Nodari, A.; Taveggia, C.; Pardini, C.; Dina, G.; Villa, A.; Wrabetz, L.; Quattrini, A.; Feltri, M.L. Expression of laminin receptors in schwann cell differentiation: Evidence for distinct roles. J. Neurosci. 2003, 23, 5520–5530. [Google Scholar] [CrossRef]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Hijikata, T.; Murakami, T.; Imamura, M.; Fujimaki, N.; Ishikawa, H. Plectin is a linker of intermediate filaments to Z-discs in skeletal muscle fibers. J. Cell Sci. 1999, 112 Pt 6, 867–876. [Google Scholar] [CrossRef]
- Reipert, S.; Steinböck, F.; Fischer, I.; Bittner, R.E.; Zeöld, A.; Wiche, G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp. Cell Res. 1999, 252, 479–491. [Google Scholar] [CrossRef]
- Schröder, R.; Warlo, I.; Herrmann, H.; van der Ven, P.F.; Klasen, C.; Blümcke, I.; Mundegar, R.R.; Fürst, D.O.; Goebel, H.H.; Magin, T.M. Immunogold EM reveals a close association of plectin and the desmin cytoskeleton in human skeletal muscle. Eur. J. Cell Biol. 1999, 78, 288–295. [Google Scholar] [CrossRef]
- Bruning, J.C.; Michael, M.D.; Winnay, J.N.; Hayashi, T.; Horsch, D.; Accili, D.; Goodyear, L.J.; Kahn, C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 1998, 2, 559–569. [Google Scholar] [CrossRef]
- Keller, C.; Hansen, M.S.; Coffin, C.M.; Capecchi, M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004, 18, 2608–2613. [Google Scholar] [CrossRef]
- Konieczny, P.; Fuchs, P.; Reipert, S.; Kunz, W.S.; Zeöld, A.; Fischer, I.; Paulin, D.; Schröder, R.; Wiche, G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 2008, 181, 667–681. [Google Scholar] [CrossRef]
- Winter, L.; Staszewska, I.; Mihailovska, E.; Fischer, I.; Goldmann, W.H.; Schröder, R.; Wiche, G. Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J. Clin. Investig. 2014, 124, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Celik, C.; Uysal, H.; Heper, A.O.; Karaoglan, B. Epidermolysis bullosa simplex associated with muscular dystrophy and cardiac involvement. J. Clin. Neuromuscul. Dis. 2005, 6, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Bolling, M.C.; Pas, H.H.; de Visser, M.; Aronica, E.; Pfendner, E.G.; van den Berg, M.P.; Diercks, G.F.; Suurmeijer, A.J.; Jonkman, M.F. PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J. Investig. Dermatol. 2010, 130, 1178–1181. [Google Scholar] [CrossRef]
- Winter, L.; Turk, M.; Harter, P.N.; Mittelbronn, M.; Kornblum, C.; Norwood, F.; Jungbluth, H.; Thiel, C.T.; Schlotzer-Schrehardt, U.; Schröder, R. Downstream effects of plectin mutations in epidermolysis bullosa simplex with muscular dystrophy. Acta Neuropathol. Commun. 2016, 4, 44. [Google Scholar] [CrossRef]
- Villa, C.R.; Ryan, T.D.; Collins, J.J.; Taylor, M.D.; Lucky, A.W.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy associated with epidermolysis bullosa simplex with muscular dystrophy and PLEC1 mutation. Neuromuscul. Disord. 2015, 25, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Helgadottir, A.; Gretarsdottir, S.; Benonisdottir, S.; Magnusdottir, A.; Davidsson, O.B.; Rajamani, S.; Roden, D.M.; et al. A Missense Variant in PLEC Increases Risk of Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 2157–2168. [Google Scholar] [CrossRef]
- Selcen, D.; Juel, V.C.; Hobson-Webb, L.D.; Smith, E.C.; Stickler, D.E.; Bite, A.V.; Ohno, K.; Engel, A.G. Myasthenic syndrome caused by plectinopathy. Neurology 2011, 76, 327–336. [Google Scholar] [CrossRef]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Mihailovska, E.; Raith, M.; Valencia, R.G.; Fischer, I.; Al Banchaabouchi, M.; Herbst, R.; Wiche, G. Neuromuscular synapse integrity requires linkage of acetylcholine receptors to postsynaptic intermediate filament networks via rapsyn-plectin 1f complexes. Mol. Biol. Cell 2014, 25, 4130–4149. [Google Scholar] [CrossRef]
- Agbulut, O.; Li, Z.; Perie, S.; Ludosky, M.A.; Paulin, D.; Cartaud, J.; Butler-Browne, G. Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell Motil. Cytoskelet. 2001, 49, 51–66. [Google Scholar] [CrossRef]
- Eiber, N.; Frob, F.; Schowalter, M.; Thiel, C.; Clemen, C.S.; Schröder, R.; Hashemolhosseini, S. Lack of Desmin in Mice Causes Structural and Functional Disorders of Neuromuscular Junctions. Front. Mol. Neurosci. 2020, 13, 567084. [Google Scholar] [CrossRef]
- Ferry, A.; Messeant, J.; Parlakian, A.; Lemaitre, M.; Roy, P.; Delacroix, C.; Lilienbaum, A.; Hovhannisyan, Y.; Furling, D.; Klein, A.; et al. Desmin prevents muscle wasting, exaggerated weakness and fragility, and fatigue in dystrophic mdx mouse. J. Physiol. 2020, 598, 3667–3689. [Google Scholar] [CrossRef]
- Schröder, R.; Mundegar, R.R.; Treusch, M.; Schlegel, U.; Blümcke, I.; Owaribe, K.; Magin, T.M. Altered distribution of plectin/HD1 in dystrophinopathies. Eur. J. Cell Biol. 1997, 74, 165–171. [Google Scholar] [CrossRef]
- Raith, M.; Valencia, R.G.; Fischer, I.; Orthofer, M.; Penninger, J.M.; Spuler, S.; Rezniczek, G.A.; Wiche, G. Linking cytoarchitecture to metabolism: Sarcolemma-associated plectin affects glucose uptake by destabilizing microtubule networks in mdx myofibers. Skelet Muscle 2013, 3, 14. [Google Scholar] [CrossRef]
- Even, P.C.; Decrouy, A.; Chinet, A. Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochem. J. 1994, 304 Pt 2, 649–654. [Google Scholar] [CrossRef]
- Zhang, T.; Haws, P.; Wu, Q. Multiple variable first exons: A mechanism for cell- and tissue-specific gene regulation. Genome Res. 2004, 14, 79–89. [Google Scholar] [CrossRef][Green Version]
- Rezniczek, G.A.; Winter, L.; Walko, G.; Wiche, G. Functional and Genetic Analysis of Plectin in Skin and Muscle. Methods Enzymol. 2016, 569, 235–259. [Google Scholar] [CrossRef] [PubMed]
- House, C.M.; Frew, I.J.; Huang, H.L.; Wiche, G.; Traficante, N.; Nice, E.; Catimel, B.; Bowtell, D.D. A binding motif for Siah ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2003, 100, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Vannier, C.; Pesty, A.; San-Roman, M.J.; Schmidt, A.A. The Bin/amphiphysin/Rvs (BAR) domain protein endophilin B2 interacts with plectin and controls perinuclear cytoskeletal architecture. J. Biol. Chem. 2013, 288, 27619–27637. [Google Scholar] [CrossRef] [PubMed]
- Hijikata, T.; Nakamura, A.; Isokawa, K.; Imamura, M.; Yuasa, K.; Ishikawa, R.; Kohama, K.; Takeda, S.; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through beta-synemin, alpha-dystrobrevin and actin. J. Cell Sci. 2008, 121, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Wilhelmsen, K.; Kuikman, I.; Janssen, H.; Hodzic, D.; Sonnenberg, A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 2007, 120, 3384–3394. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Kreft, M.; Secades, P.; Janssen, H.; Sonnenberg, A. Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol. Biol. Cell 2013, 24, 2454–2466. [Google Scholar] [CrossRef] [PubMed]
- Staszewska, I.; Fischer, I.; Wiche, G. Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum. Mol. Genet. 2015, 24, 7373–7389. [Google Scholar] [CrossRef]
- Abrahamsberg, C.; Fuchs, P.; Osmanagic-Myers, S.; Fischer, I.; Propst, F.; Elbe-Bürger, A.; Wiche, G. Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2005, 102, 18449–18454. [Google Scholar] [CrossRef]
- Rapaport, D. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 2003, 4, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 2008, 181, 903–911. [Google Scholar] [CrossRef]
- Winter, L.; Kuznetsov, A.V.; Grimm, M.; Zeold, A.; Fischer, I.; Wiche, G. Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum. Mol. Genet. 2015, 24, 4530–4544. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells 2020, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Mado, K.; Chekulayev, V.; Shevchuk, I.; Puurand, M.; Tepp, K.; Kaambre, T. On the role of tubulin, plectin, desmin, and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C657–C667. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Kunz, W.S.; Rouan, F.; Pfendner, E.; Tolksdorf, K.; Kappes-Horn, K.; Altenschmidt-Mehring, M.; Knoblich, R.; van der Ven, P.F.; Reimann, J.; et al. Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J. Neuropathol. Exp. Neurol. 2002, 61, 520–530. [Google Scholar] [CrossRef]
- Schröder, R.; Pacholsky, D.; Reimann, J.; Matten, J.; Wiche, G.; Fürst, D.O.; van der Ven, P.F. Primary longitudinal adhesion structures: Plectin-containing precursors of costameres in differentiating human skeletal muscle cells. Histochem Cell Biol. 2002, 118, 301–310. [Google Scholar] [CrossRef]
- Schröder, R.; Fürst, D.O.; Klasen, C.; Reimann, J.; Herrmann, H.; van der Ven, P.F.M. Association of plectin with Z-discs is a prerequisite for the formation of the intermyofibrillar desmin cytoskeleton. Lab. Investig. 2000, 80, 455–464. [Google Scholar] [CrossRef][Green Version]
- Li, Z.; Colucci-Guyon, E.; Pincon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef]
- Carlsson, L.; Li, Z.; Paulin, D.; Thornell, L.E. Nestin is expressed during development and in myotendinous and neuromuscular junctions in wild type and desmin knock-out mice. Exp. Cell Res. 1999, 251, 213–223. [Google Scholar] [CrossRef]
- Bellin, R.M.; Huiatt, T.W.; Critchley, D.R.; Robson, R.M. Synemin may function to directly link muscle cell intermediate filaments to both myofibrillar Z-lines and costameres. J. Biol. Chem. 2001, 276, 32330–32337. [Google Scholar] [CrossRef]
- Newey, S.E.; Howman, E.V.; Ponting, C.P.; Benson, M.A.; Nawrotzki, R.; Loh, N.Y.; Davies, K.E.; Blake, D.J. Syncoilin, a novel member of the intermediate filament superfamily that interacts with alpha-dystrobrevin in skeletal muscle. J. Biol. Chem. 2001, 276, 6645–6655. [Google Scholar] [CrossRef] [PubMed]
- Errante, L.D.; Wiche, G.; Shaw, G. Distribution of plectin, an intermediate filament-associated protein, in the adult rat central nervous system. J. Neurosci. Res. 1994, 37, 515–528. [Google Scholar] [CrossRef]
- Lie, A.A.; Schröder, R.; Blumcke, I.; Magin, T.M.; Wiestler, O.D.; Elger, C.E. Plectin in the human central nervous system: Predominant expression at pia/glia and endothelia/glia interfaces. Acta Neuropathol. 1998, 96, 215–221. [Google Scholar] [CrossRef]
- Walko, G.; Vukasinovic, N.; Gross, K.; Fischer, I.; Sibitz, S.; Fuchs, P.; Reipert, S.; Jungwirth, U.; Berger, W.; Salzer, U.; et al. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011, 7, e1002396. [Google Scholar] [CrossRef]
- Koszka, C.; Leichtfried, F.E.; Wiche, G. Identification and spatial arrangement of high molecular weight proteins (Mr 300 000-330 000) co-assembling with microtubules from a cultured cell line (rat glioma C6). Eur. J. Cell Biol. 1985, 38, 149–156. [Google Scholar]
- Herrmann, H.; Wiche, G. Plectin and IFAP-300K are homologous proteins binding to microtubule- associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J. Biol. Chem. 1987, 262, 1320–1325. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Verkhovsky, A.B.; Borisy, G.G. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996, 135, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Valencia, R.G.; Mihailovska, E.; Winter, L.; Bauer, K.; Fischer, I.; Walko, G.; Jorgacevski, J.; Potokar, M.; Zorec, R.; Wiche, G. Plectin dysfunction in neurons leads to tau accumulation on microtubules affecting neuritogenesis, organelle trafficking, pain sensitivity and memory. Neuropathol. Appl. Neurobiol. 2021, 47, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Lesniewicz, K.; Lüscher-Firzlaff, J.; Poreba, E.; Fuchs, P.; Walsemann, G.; Wiche, G.; Lüscher, B. Overlap of the gene encoding the novel poly(ADP-ribose) polymerase Parp10 with the plectin 1 gene and common use of exon sequences. Genomics 2005, 86, 38–46. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Rice, S.J.; Tselepi, M.; Sorial, A.K.; Aubourg, G.; Shepherd, C.; Almarza, D.; Skelton, A.J.; Pangou, I.; Deehan, D.; Reynard, L.N.; et al. Prioritization of PLEC and GRINA as Osteoarthritis Risk Genes Through the Identification and Characterization of Novel Methylation Quantitative Trait Loci. Arthritis Rheumatol. 2019, 71, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Sorial, A.K.; Hofer, I.M.J.; Tselepi, M.; Cheung, K.; Parker, E.; Deehan, D.J.; Rice, S.J.; Loughlin, J. Multi-tissue epigenetic analysis of the osteoarthritis susceptibility locus mapping to the plectin gene PLEC. Osteoarthr. Cartil. 2020, 28, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Gostynska, K.B.; Nijenhuis, M.; Lemmink, H.; Pas, H.H.; Pasmooij, A.M.; Lang, K.K.; Castañón, M.J.; Wiche, G.; Jonkman, M.F. Mutation in exon 1a of PLEC, leading to disruption of plectin isoform 1a, causes autosomal-recessive skin-only epidermolysis bullosa simplex. Hum. Mol. Genet. 2015, 24, 3155–3162. [Google Scholar] [CrossRef]
- Gundesli, H.; Talim, B.; Korkusuz, P.; Balci-Hayta, B.; Cirak, S.; Akarsu, N.A.; Topaloglu, H.; Dincer, P. Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am. J. Hum. Genet. 2010, 87, 834–841. [Google Scholar] [CrossRef]
- Mroczek, M.; Durmus, H.; Topf, A.; Parman, Y.; Straub, V. Four Individuals with a Homozygous Mutation in Exon 1f of the PLEC Gene and Associated Myasthenic Features. Genes 2020, 11, 716. [Google Scholar] [CrossRef]
- Deev, R.V.; Bardakov, S.N.; Mavlikeev, M.O.; Yakovlev, I.A.; Umakhanova, Z.R.; Akhmedova, P.G.; Magomedova, R.M.; Chekmaryeva, I.A.; Dalgatov, G.D.; Isaev, A.A. Glu20Ter Variant in PLEC 1f Isoform Causes Limb-Girdle Muscle Dystrophy with Lung Injury. Front. Neurol. 2017, 8, 367. [Google Scholar] [CrossRef]
- Eisenberg, J.L.; Beaumont, K.G.; Takawira, D.; Hopkinson, S.B.; Mrksich, M.; Budinger, G.R.; Jones, J.C. Plectin-containing, centrally localized focal adhesions exert traction forces in primary lung epithelial cells. J. Cell Sci. 2013, 126, 3746–3755. [Google Scholar] [CrossRef]
- Koss-Harnes, D.; Hoyheim, B.; Anton-Lamprecht, I.; Gjesti, A.; Jorgensen, R.S.; Jahnsen, F.L.; Olaisen, B.; Wiche, G.; Gedde-Dahl, T., Jr. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: Two identical de novo mutations. J. Investig. Dermatol. 2002, 118, 87–93. [Google Scholar] [CrossRef]
- Olaisen, B.; Gedde-Dahl, T., Jr. GPT—epidermolysis bullosa simplex (EBS Ogna) linkage in man. Hum. Hered. 1973, 23, 189–196. [Google Scholar] [CrossRef]
- Bolling, M.C.; Jongbloed, J.D.H.; Boven, L.G.; Diercks, G.F.H.; Smith, F.J.D.; Irwin McLean, W.H.; Jonkman, M.F. Plectin mutations underlie epidermolysis bullosa simplex in 8% of patients. J. Investig. Dermatol. 2014, 134, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Armel, T.Z.; Leinwand, L.A. Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 6291–6296. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bosl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef]
- Wiche, G.; Becker, B.; Luber, K.; Weitzer, G.; Castañón, M.J.; Hauptmann, R.; Stratowa, C.; Stewart, M. Cloning and sequencing of rat plectin indicates a 466-kD polypeptide chain with a three-domain structure based on a central alpha-helical coiled coil. J. Cell Biol. 1991, 114, 83–99. [Google Scholar] [CrossRef]
- Fuchs, P.; Spazierer, D.; Wiche, G. Plectin rodless isoform expression and its detection in mouse brain. Cell. Mol. Neurobiol. 2005, 25, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; van Wilpe, S.; Kuikman, I.; Litjens, S.H.; Sonnenberg, A. Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol. Biol. Cell 2004, 15, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Natsuga, K.; Nishie, W.; Akiyama, M.; Nakamura, H.; Shinkuma, S.; McMillan, J.R.; Nagasaki, A.; Has, C.; Ouchi, T.; Ishiko, A.; et al. Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex. Hum. Mutat. 2010, 31, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Klausegger, A.; Waddell, L.B.; Grasern, N.; Lloyd, L.; Tran, K.; North, K.N.; Bauer, J.W.; McKelvie, P.; Chow, C.W.; et al. Epidermolysis bullosa with late-onset muscular dystrophy and plectin deficiency. Muscle Nerve 2011, 44, 135–141. [Google Scholar] [CrossRef]
- Charlesworth, A.; Gagnoux-Palacios, L.; Bonduelle, M.; Ortonne, J.P.; De Raeve, L.; Meneguzzi, G. Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J. Investig. Dermatol. 2003, 121, 1344–1348. [Google Scholar] [CrossRef][Green Version]
- Sawamura, D.; Goto, M.; Sakai, K.; Nakamura, H.; McMillan, J.R.; Akiyama, M.; Shirado, O.; Oyama, N.; Satoh, M.; Kaneko, F.; et al. Possible involvement of exon 31 alternative splicing in phenotype and severity of epidermolysis bullosa caused by mutations in PLEC1. J. Investig. Dermatol. 2007, 127, 1537–1540. [Google Scholar] [CrossRef]
- Ketema, M.; Secades, P.; Kreft, M.; Nahidiazar, L.; Janssen, H.; Jalink, K.; de Pereda, J.M.; Sonnenberg, A. The rod domain is not essential for the function of plectin in maintaining tissue integrity. Mol. Biol. Cell 2015, 26, 2402–2417. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Gregor, M.; Walko, G.; Burgstaller, G.; Reipert, S.; Wiche, G. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 2006, 174, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Kettleborough, R.N.; Busch-Nentwich, E.M.; Harvey, S.A.; Dooley, C.M.; de Bruijn, E.; van Eeden, F.; Sealy, I.; White, R.J.; Herd, C.; Nijman, I.J.; et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 2013, 496, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Thisse, B.; Heyer, V.; Lux, A.; Alunni, V.; Degrave, A.; Seiliez, I.; Kirchner, J.; Parkhill, J.P.; Thisse, C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004, 77, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Frank, M.; Thisse, C.I.; Thisse, B.V.; Uitto, J. Zebrafish: A model system to study heritable skin diseases. J. Investig. Dermatol. 2011, 131, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Bührdel, J.B.; Hirth, S.; Kessler, M.; Westphal, S.; Forster, M.; Manta, L.; Wiche, G.; Schoser, B.; Schessl, J.; Schröder, R.; et al. In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2015, 461, 217–223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosher, J.M.; Hahn, B.S.; Legouis, R.; Sookhareea, S.; Weimer, R.M.; Gansmuller, A.; Chisholm, A.D.; Rose, A.M.; Bessereau, J.L.; Labouesse, M. The Caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J. Cell Biol. 2003, 161, 757–768. [Google Scholar] [CrossRef]
- Francis, R.; Waterston, R.H. Muscle cell attachment in Caenorhabditis elegans. J. Cell Biol. 1991, 114, 465–479. [Google Scholar] [CrossRef]
- Hresko, M.C.; Schriefer, L.A.; Shrimankar, P.; Waterston, R.H. Myotactin, a novel hypodermal protein involved in muscle-cell adhesion in Caenorhabditis elegans. J. Cell Biol. 1999, 146, 659–672. [Google Scholar] [CrossRef]
- Zhang, H.; Labouesse, M. The making of hemidesmosome structures in vivo. Dev. Dyn. 2010, 239, 1465–1476. [Google Scholar] [CrossRef]
- Zhang, H.; Landmann, F.; Zahreddine, H.; Rodriguez, D.; Koch, M.; Labouesse, M. A tension-induced mechanotransduction pathway promotes epithelial morphogenesis. Nature 2011, 471, 99–103. [Google Scholar] [CrossRef]
- Suman, S.K.; Daday, C.; Ferraro, T.; Vuong-Brender, T.; Tak, S.; Quintin, S.; Robin, F.; Grater, F.; Labouesse, M. The plakin domain of C. elegans VAB-10/plectin acts as a hub in a mechanotransduction pathway to promote morphogenesis. Development 2019, 146, dev183780. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; Buey, R.M.; Sonnenberg, A.; de Pereda, J.M. The structure of the plakin domain of plectin reveals a non-canonical SH3 domain interacting with its fourth spectrin repeat. J. Biol. Chem. 2011, 286, 12429–12438. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; Manso, J.A.; Buey, R.M.; Carballido, A.M.; Carabias, A.; Sonnenberg, A.; de Pereda, J.M. The Structure of the Plakin Domain of Plectin Reveals an Extended Rod-like Shape. J. Biol. Chem. 2016, 291, 18643–18662. [Google Scholar] [CrossRef] [PubMed]
- Daday, C.; Kolsek, K.; Gräter, F. The mechano-sensing role of the unique SH3 insertion in plakin domains revealed by Molecular Dynamics simulations. Sci. Rep. 2017, 7, 11669. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castañón, M.J.; Wiche, G. Identifying Plectin Isoform Functions through Animal Models. Cells 2021, 10, 2453. https://doi.org/10.3390/cells10092453
Castañón MJ, Wiche G. Identifying Plectin Isoform Functions through Animal Models. Cells. 2021; 10(9):2453. https://doi.org/10.3390/cells10092453
Chicago/Turabian StyleCastañón, Maria J., and Gerhard Wiche. 2021. "Identifying Plectin Isoform Functions through Animal Models" Cells 10, no. 9: 2453. https://doi.org/10.3390/cells10092453
APA StyleCastañón, M. J., & Wiche, G. (2021). Identifying Plectin Isoform Functions through Animal Models. Cells, 10(9), 2453. https://doi.org/10.3390/cells10092453