
Alpha-Synuclein and Lipids: The Elephant in the Room?



Abstract
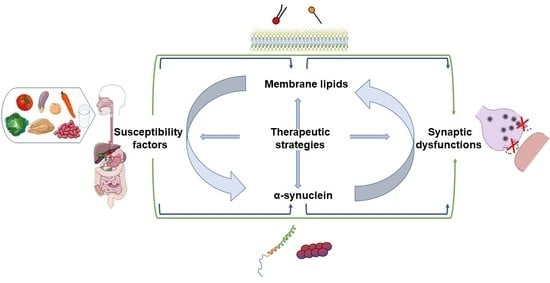
1. Introduction
2. α-Synuclein and Its Relationship with Lipid Membranes
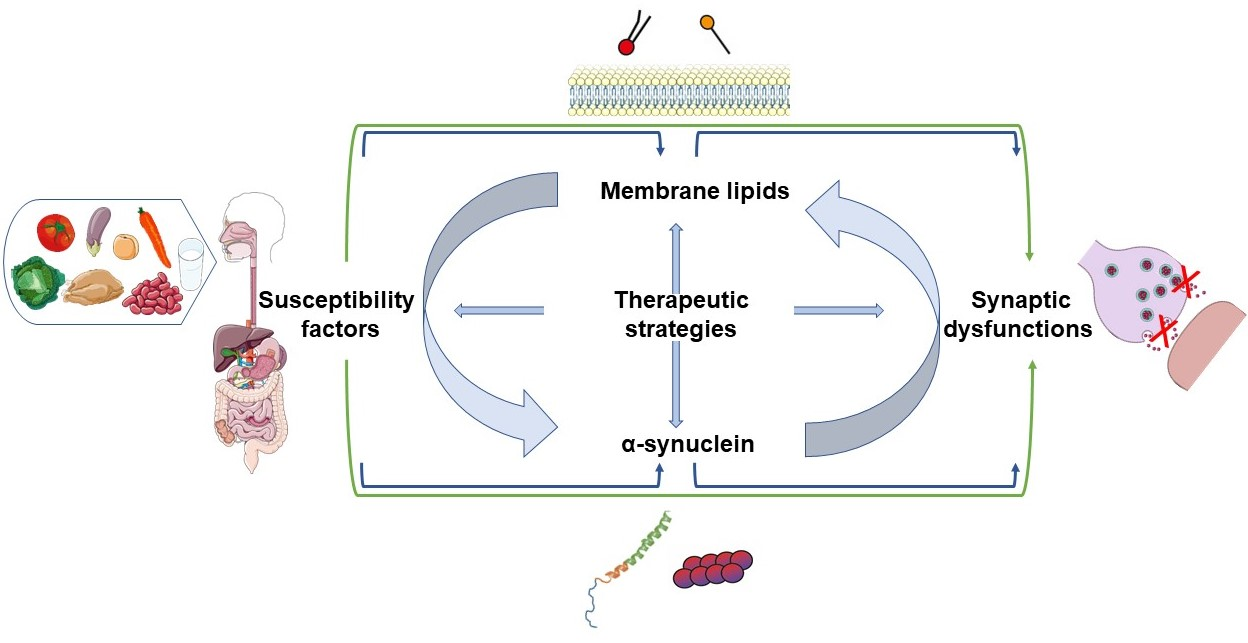
2.1. α-Synuclein Structure and Interaction with Lipids
2.2. α-Synuclein and Lipid Bilayers
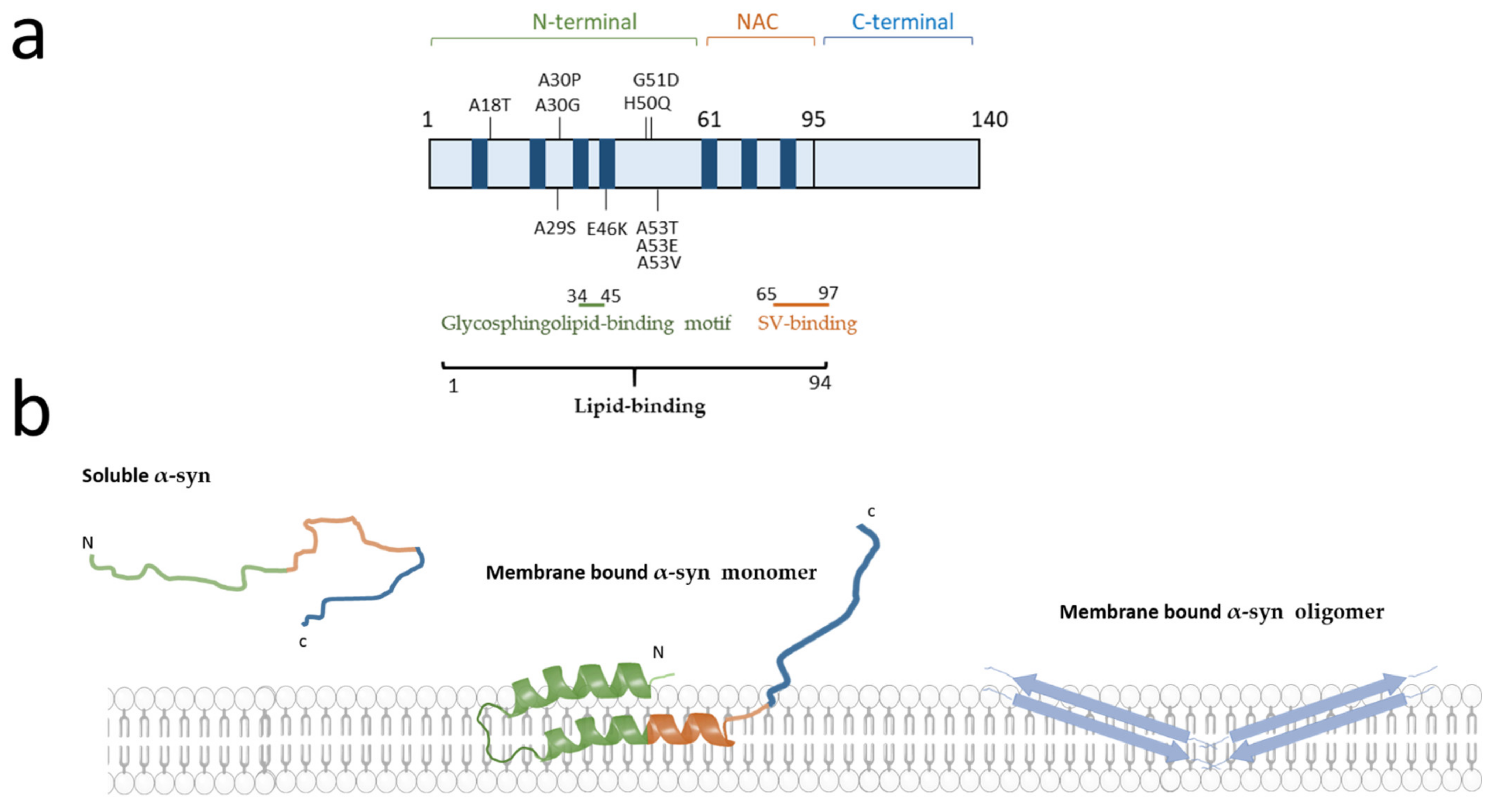
2.2.1. Presynaptic Membrane Composition and α-Synuclein Binding Affinity
2.2.2. Lipid Rafts and α-Synuclein Interaction
2.3. α-Synuclein and Synaptic Vesicles
2.3.1. α-Synuclein and Membrane Curvature
2.3.2. α-Synuclein Affinity According to Vesicle Composition
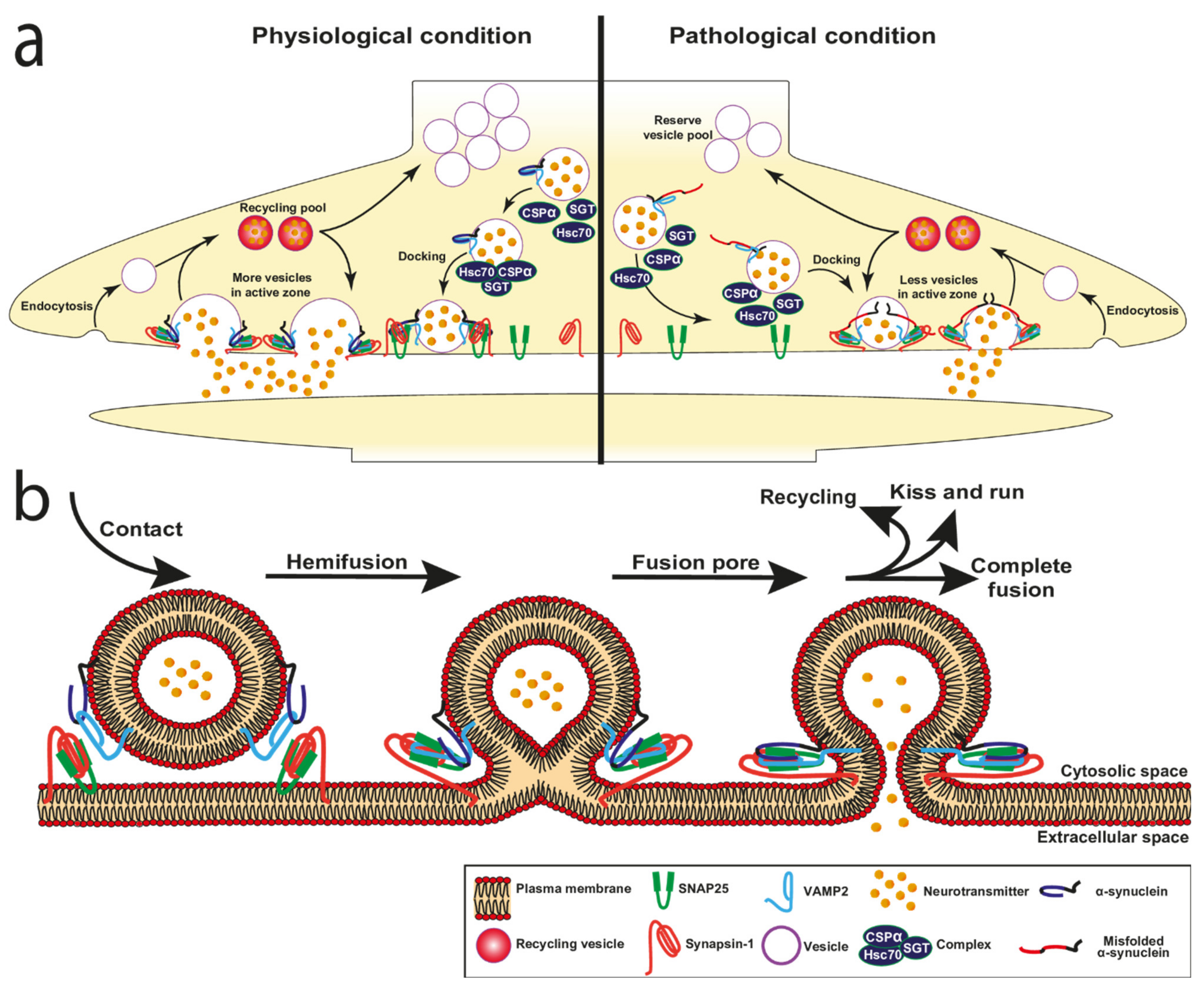
3. α-Synuclein Function in Exocytosis
3.1. α-Synuclein and Vesicle Docking
3.2. α-Synuclein and Fusion Pore
3.3. α-Synuclein and the Cooperation with SNARE Proteins in Exocytosis
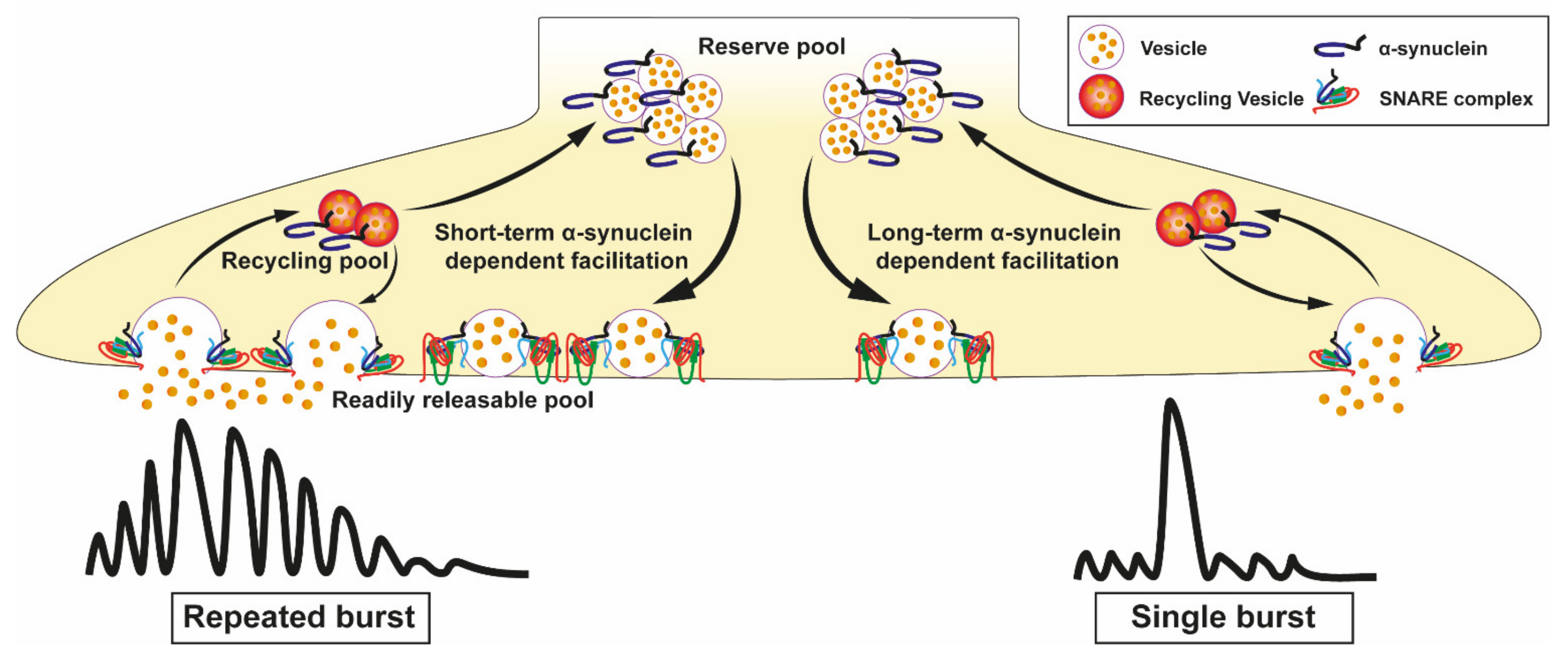
3.4. Loss and Overexpression of α-Synuclein in Neurotransmitter Release
3.5. Vesicle Recycling
3.6. Aberrant α-Synuclein in PD-Lipid Binding and Synaptic Function
3.6.1. Oligomerisation of Pathogenic α-Synuclein and Lipid Binding
3.6.2. Fine Regulation of α-Synuclein on Synaptic Activity
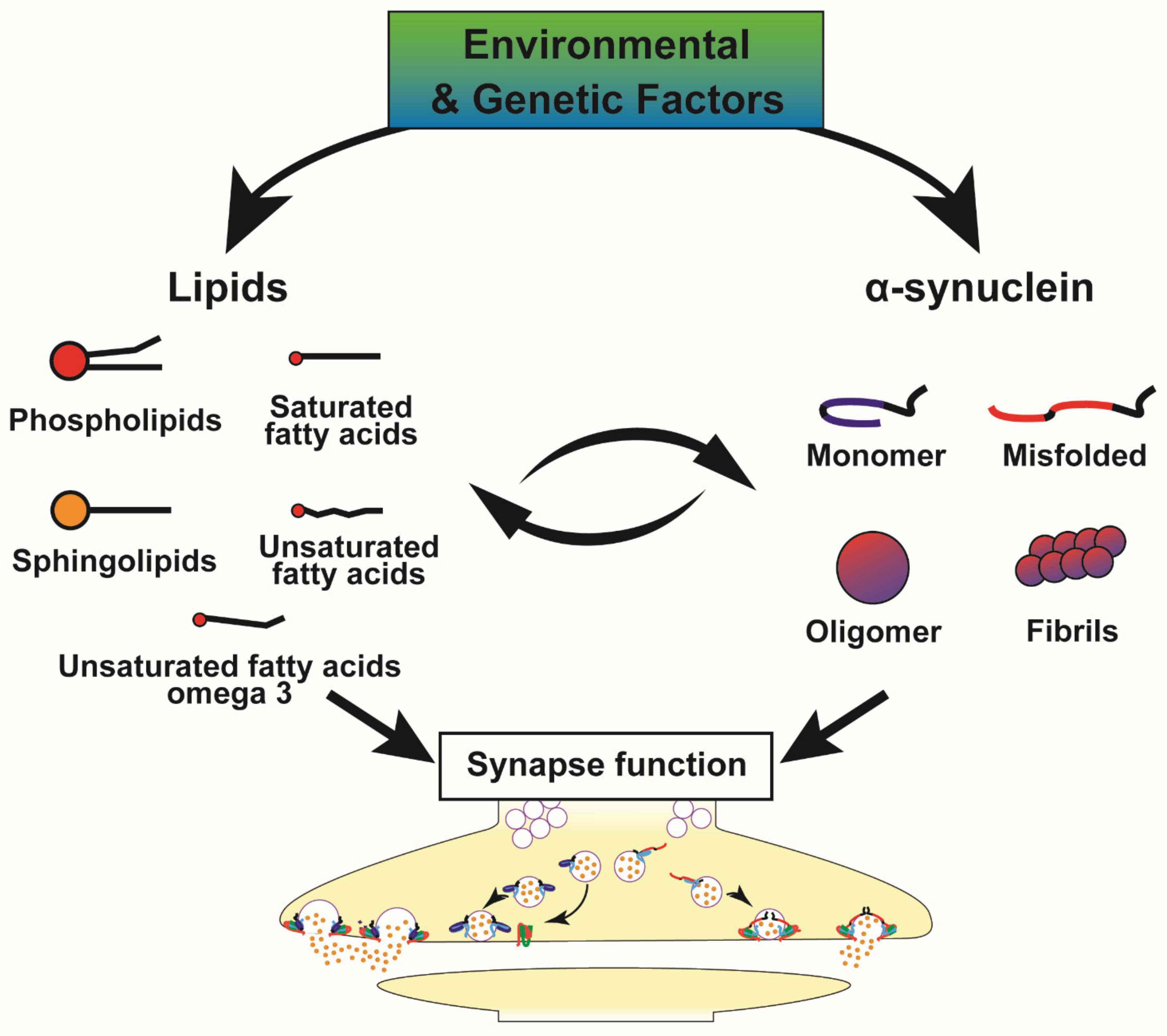
4. Metabolic Alterations and Genetic Susceptibility Factors in PD, Implications for the α-Syn-Lipid Interplay
5. Future Directions
5.1. Towards Further Fundamental Advances
5.2. Towards Target Identification and Pharmacological Strategies
5.2.1. Targeting Membrane Lipids or α-Synuclein Membrane Affinity
5.2.2. Environmental Factors and Potential Therapeutic Strategies
5.2.3. Targeting Synaptic Proteins
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kieburtz, K.; Wunderle, K.B. Parkinson’s disease: Evidence for environmental risk factors. Mov. Disord. 2013, 28, 8–13. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef]
- Campion, D.; Martin, C.; Heilig, R.; Charbonnier, F.; Moreau, V.; Flaman, J.M.; Petit, J.L.; Hannequin, D.; Brice, A.; Frebourg†, T. The NACP/synuclein gene: Chromosomal assignment and screening for alterations in Alzheimer disease. Genomics 1995, 26, 254–257. [Google Scholar] [CrossRef]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Divane, A.; Goedert, M. Assignment of Human α-Synuclein (SNCA) and β-Synuclein (SNCB) Genes to Chromosomes 4q21 and 5q35. Genomics 1995, 27, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Hoffman-Zacharska, D.; Koziorowski, D.; Ross, O.A.; Milewski, M.; Poznanski, J.A.; Jurek, M.; Wszolek, Z.K.; Soto-Ortolaza, A.; Awek, J.A.S.; Janik, P.; et al. Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 1057–1060. [Google Scholar] [CrossRef]
- Liu, H.; Koros, C.; Strohäker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibáñez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Brás, J.; Gibbons, E.; Guerreiro, R. Genetics of synucleins in neurodegenerative diseases. Acta Neuropathol. 2021, 141, 471–490. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Dürr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet (Lond. Engl.) 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Marchese, D.; Botta-Orfila, T.; Cirillo, D.; Rodriguez, J.A.; Livi, C.M.; Fernández-Santiago, R.; Ezquerra, M.; Martí, M.J.; Bechara, E.; Tartaglia, G.G. Discovering the 3′ UTR-mediated regulation of alpha-synuclein. Nucleic Acids Res. 2017, 45, 12888–12903. [Google Scholar] [CrossRef]
- Tseng, E.; Rowell, W.J.; Glenn, O.-C.; Hon, T.; Barrera, J.; Kujawa, S.; Chiba-Falek, O. The Landscape of SNCA Transcripts Across Synucleinopathies: New Insights From Long Reads Sequencing Analysis. Front. Genet. 2019, 10, 584. [Google Scholar] [CrossRef]
- Langmyhr, M.; Henriksen, S.P.; Cappelletti, C.; van de Berg, W.D.J.; Pihlstrøm, L.; Toft, M. Allele-specific expression of Parkinson’s disease susceptibility genes in human brain. Sci. Rep. 2021, 11, 504. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, e12188. [Google Scholar] [CrossRef]
- Pankratz, N.; Dumitriu, A.; Hetrick, K.N.; Sun, M.; Latourelle, J.C.; Wilk, J.B.; Halter, C.; Doheny, K.F.; Gusella, J.F.; Nichols, W.C.; et al. Copy Number Variation in Familial Parkinson Disease. PLoS ONE 2011, 6, e20988. [Google Scholar] [CrossRef]
- Perez-Rodriguez, D.; Kalyva, M.; Leija-Salazar, M.; Lashley, T.; Tarabichi, M.; Chelban, V.; Gentleman, S.; Schottlaender, L.; Franklin, H.; Vasmatzis, G.; et al. Investigation of somatic CNVs in brains of synucleinopathy cases using targeted SNCA analysis and single cell sequencing. Acta Neuropathol. Commun. 2019, 7, 219. [Google Scholar] [CrossRef]
- Mokretar, K.; Pease, D.; Taanman, J.-W.; Soenmez, A.; Ejaz, A.; Lashley, T.; Ling, H.; Gentleman, S.; Houlden, H.; Holton, J.L.; et al. Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 2018, 141, 2419–2431. [Google Scholar] [CrossRef]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Züchner, S.; Konidari, I.; Wang, G.; et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef]
- Lesage, S.; Houot, M.; Mangone, G.; Tesson, C.; Bertrand, H.; Forlani, S.; Anheim, M.; Brefel-Courbon, C.; Broussolle, E.; Thobois, S.; et al. Genetic and Phenotypic Basis of Autosomal Dominant Parkinson’s Disease in a Large Multi-Center Cohort. Front. Neurol. 2020, 11, 682. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.; Brundin, P. Prion-like propagation of pathology in Parkinson disease. Handb. Clin. Neurol. 2018, 153, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- Massey, A.R.; Monogue, B.; Chen, Y.; Lesteberg, K.; Johnson, M.E.; Bergkvist, L.; Steiner, J.A.; Ma, J.; Mahalingam, R.; Kleinschmidt-Demasters, B.K.; et al. Alpha-synuclein supports interferon stimulated gene expression in neurons. bioRxiv 2020. [Google Scholar] [CrossRef]
- Burré, J. The synaptic function of α-synuclein. J. Parkinson’s Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Bonini, N.M.; Giasson, B.I. Snaring the Function of α-Synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Man, W.K.; Tahirbegi, B.; Vrettas, M.D.; Preet, S.; Ying, L.; Vendruscolo, M.; De Simone, A.; Fusco, G. The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition. Nat. Commun. 2021, 12, 927. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.; Scheller, R. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. alpha-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Bussell, R.; Eliezer, D. A Structural and Functional Role for 11-mer Repeats in α-Synuclein and Other Exchangeable Lipid Binding Proteins. J. Mol. Biol. 2003, 329, 763–778. [Google Scholar] [CrossRef]
- Musteikytė, G.; Jayaram, A.K.; Xu, C.K.; Vendruscolo, M.; Krainer, G.; Knowles, T.P.J. Interactions of α-synuclein oligomers with lipid membranes. Biochim. Biophys. Acta–Biomembr. 2021, 1863, 183536. [Google Scholar] [CrossRef] [PubMed]
- Adão, R.; Cruz, P.F.; Vaz, D.C.; Fonseca, F.; Pedersen, J.N.; Ferreira-da-Silva, F.; Brito, R.M.M.; Ramos, C.H.I.; Otzen, D.; Keller, S.; et al. DIBMA nanodiscs keep α-synuclein folded. Biochim. Biophys. Acta–Biomembr. 2020, 1862, 183314. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Fuller, N.; Rand, R.P.; St, George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Raben, D.M.; Barber, C.N. Phosphatidic acid and neurotransmission. Adv. Biol. Regul. 2017, 63, 15–21. [Google Scholar] [CrossRef]
- Fantini, J.; Carlus, D.; Yahi, N. The fusogenic tilted peptide (67–78) of α-synuclein is a cholesterol binding domain. Biochim. Biophys. Acta–Biomembr. 2011, 1808, 2343–2351. [Google Scholar] [CrossRef]
- Mahfoud, R.; Garmy, N.; Maresca, M.; Yahi, N.; Puigserver, A.; Fantini, J. Identification of a Common Sphingolipid-binding Domain in Alzheimer, Prion, and HIV-1 Proteins. J. Biol. Chem. 2002, 277, 11292–11296. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-Terminus of the Intrinsically Disordered Protein α-Synuclein Triggers Membrane Binding and Helix Folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease–lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef]
- Bisaglia, M.; Tessari, I.; Pinato, L.; Bellanda, M.; Giraudo, S.; Fasano, M.; Bergantino, E.; Bubacco, L.; Mammi, S. A Topological Model of the Interaction between α-Synuclein and Sodium Dodecyl Sulfate Micelles. Biochemistry 2005, 44, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R.; Browne, G. Conformational Properties of a-Synuclein in its Free and Lipid-associated States. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Watson, H. Biological membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Carpenter, T.S.; Bhatia, H.; Bremer, P.-T.; Marrink, S.J.; Lightstone, F.C. Computational Lipidomics of the Neuronal Plasma Membrane. Biophys. J. 2017, 113, 2271–2280. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 175909141878188. [Google Scholar] [CrossRef]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef]
- Man, W.K.; De Simone, A.; Barritt, J.D.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. A Role of Cholesterol in Modulating the Binding of α-Synuclein to Synaptic-Like Vesicles. Front. Neurosci. 2020, 14, 18. [Google Scholar] [CrossRef]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.H.; Noble, T.; et al. Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014.e8. [Google Scholar] [CrossRef]
- Xicoy, H.; Brouwers, J.F.; Wieringa, B.; Martens, G.J.M. Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease. Cells 2020, 9, 1966. [Google Scholar] [CrossRef]
- Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 2021, 9, 491. [Google Scholar] [CrossRef]
- Lv, Z.; Hashemi, M.; Banerjee, S.; Zagorski, K.; Rochet, J.-C.; Lyubchenko, Y.L. Assembly of α-synuclein aggregates on phospholipid bilayers. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 802–812. [Google Scholar] [CrossRef]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 2009, 132, 3285–3297. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Liou, L.-C.; Ren, Q.; Zhang, Z.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Phosphatidylethanolamine deficiency disrupts α-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, E3976–E3985. [Google Scholar] [CrossRef]
- Lou, X.; Kim, J.; Hawk, B.J.; Shin, Y.-K. α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 2017, 474, 2039–2049. [Google Scholar] [CrossRef]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef]
- Kim, W.S.; Halliday, G.M. Changes in sphingomyelin level affect alpha-synuclein and ABCA5 expression. J. Parkinson’s Dis. 2012, 2, 41–46. [Google Scholar] [CrossRef]
- Mesa-Herrera, F.; Taoro-González, L.; Valdés-Baizabal, V.-B.; Diaz, M.; Marín, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 Specifically Interacts with α-Synuclein and Inhibits Fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-Synuclein Aggregation by Exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef]
- Lee, Y.J.; Wang, S.; Slone, S.R.; Yacoubian, T.A.; Witt, S.N. Defects in Very Long Chain Fatty Acid Synthesis Enhance Alpha-Synuclein Toxicity in a Yeast Model of Parkinson’s Disease. PLoS ONE 2011, 6, e15946. [Google Scholar] [CrossRef][Green Version]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Jensen, P.H.; Marsh, D. α-Synuclein Association with Phosphatidylglycerol Probed by Lipid Spin Labels. Biochemistry 2003, 42, 12919–12926. [Google Scholar] [CrossRef] [PubMed]
- Schommer, J.; Marwarha, G.; Nagamoto-Combs, K.; Ghribi, O. Palmitic Acid-Enriched Diet Increases α-Synuclein and Tyrosine Hydroxylase Expression Levels in the Mouse Brain. Front. Neurosci. 2018, 12, 552. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of α-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R.; et al. Inhibiting Stearoyl-CoA Desaturase Ameliorates α-Synuclein Cytotoxicity. Cell Rep. 2018, 25, 2742–2754.e31. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.; Fabelo, N.; Martín, V.; Garcia-Esparcia, P.; Ferrer, I.; Quinto-Alemany, D.; Díaz, M. Anomalies occurring in lipid profiles and protein distribution in frontal cortex lipid rafts in dementia with Lewy bodies disclose neurochemical traits partially shared by Alzheimer’s and Parkinson’s diseases. Neurobiol. Aging 2017, 49, 52–59. [Google Scholar] [CrossRef]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. α-Synuclein Neuropathology is Controlled by Nuclear Hormone Receptors and Enhanced by Docosahexaenoic Acid in A Mouse Model for Parkinson’s Disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Pyszko, J.; Strosznajder, J.B. Sphingosine Kinase 1 and Sphingosine-1-Phosphate in Oxidative Stress Evoked by 1-Methyl-4-Phenylpyridinium (MPP+) in Human Dopaminergic Neuronal Cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mallett, V.; Vanderperre, B.; Tavassoly, O.; Dauvilliers, Y.; Wu, R.Y.J.; Ruskey, J.A.; Leblond, C.S.; Ambalavanan, A.; Laurent, S.B.; et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov. Disord. 2019, 34, 526–535. [Google Scholar] [CrossRef]
- Pyszko, J.A.; Strosznajder, J.B. Original article The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol. 2014, 52, 260–269. [Google Scholar] [CrossRef]
- Bae, E.-J.; Lee, H.-J.; Jang, Y.-H.; Michael, S.; Masliah, E.; Min, D.S.; Lee, S.-J. Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Differ. 2014, 21, 1132–1141. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Peñuelas, N.; Vila, M.; Laguna, A. Autophagic- and Lysosomal-Related Biomarkers for Parkinson’s Disease: Lights and Shadows. Cells 2019, 8, 1317. [Google Scholar] [CrossRef]
- Belarbi, K.; Cuvelier, E.; Bonte, M.-A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.-C. Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 59. [Google Scholar] [CrossRef]
- Van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D.J. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef]
- Brekk, O.R.; Korecka, J.A.; Crapart, C.C.; Huebecker, M.; MacBain, Z.K.; Rosenthal, S.A.; Sena-Esteves, M.; Priestman, D.A.; Platt, F.M.; Isacson, O.; et al. Upregulating β-hexosaminidase activity in rodents prevents α-synuclein lipid associations and protects dopaminergic neurons from α-synuclein-mediated neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 127. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Bringing order to chaos. J. Lipid Res. 2003, 44, 655–667. [Google Scholar] [CrossRef]
- Zabrocki, P.; Bastiaens, I.; Delay, C.; Bammens, T.; Ghillebert, R.; Pellens, K.; De Virgilio, C.; Van Leuven, F.; Winderickx, J. Phosphorylation, lipid raft interaction and traffic of α-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta 2008, 1783, 1767–1780. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.I.; Anthony, M.D.; Edwards, R.H. Lipid Rafts Mediate the Synaptic Localization of -Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef]
- Perissinotto, F.; Stani, C.; De Cecco, E.; Vaccari, L.; Rondelli, V.; Posocco, P.; Parisse, P.; Scaini, D.; Legname, G.; Casalis, L. Iron-mediated interaction of alpha synuclein with lipid raft model membranes. Nanoscale 2020, 12, 7631–7640. [Google Scholar] [CrossRef]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.P.; Haque, F.; Rochet, J.-C.; Hovis, J.S. α-Synuclein-Induced Tubule Formation in Lipid Bilayers. J. Phys. Chem. B 2011, 115, 5886–5893. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Westphal, C.H.; Chandra, S.S. Monomeric Synucleins Generate Membrane Curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef]
- Shen, H.; Pirruccello, M.; De Camilli, P. SnapShot: Membrane Curvature Sensors and Generators. Cell 2012, 150, 1300–1300.e2. [Google Scholar] [CrossRef]
- Wang, H.-L.; Lu, C.-S.; Yeh, T.-H.; Shen, Y.-M.; Weng, Y.-H.; Huang, Y.-Z.; Chen, R.-S.; Liu, Y.-C.; Cheng, Y.-C.; Chang, H.-C.; et al. Combined Assessment of Serum Alpha-Synuclein and Rab35 is a Better Biomarker for Parkinson’s Disease. J. Clin. Neurol. 2019, 15, 488–495. [Google Scholar] [CrossRef]
- Schechter, M.; Atias, M.; Abd Elhadi, S.; Davidi, D.; Gitler, D.; Sharon, R. α-Synuclein facilitates endocytosis by elevating the steady-state levels of phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2020, 295, 18076–18090. [Google Scholar] [CrossRef]
- Tosatto, L.; Andrighetti, A.O.; Plotegher, N.; Antonini, V.; Tessari, I.; Ricci, L.; Bubacco, L.; Dalla Serra, M. Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta 2012, 1818, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.R.; Rhoades, E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-terminal Acetylation of α-Synuclein Changes the Affinity for Lipid Membranes but not the Structural Properties of the Bound State. Sci. Rep. 2020, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-Terminal Acetylation of α-Synuclein on Its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.I.; Jiang, Z.; Strub, M.-P.; Lee, J.C. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated α-synuclein. J. Biol. Chem. 2018, 293, 11195–11205. [Google Scholar] [CrossRef] [PubMed]
- Samuel, F.; Flavin, W.P.; Iqbal, S.; Pacelli, C.; Sri Renganathan, S.D.; Trudeau, L.-E.; Campbell, E.M.; Fraser, P.E.; Tandon, A. Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem. 2016, 291, 4374–4385. [Google Scholar] [CrossRef]
- Dikiy, I.; Fauvet, B.; Jovičić, A.; Mahul-Mellier, A.-L.; Desobry, C.; El-Turk, F.; Gitler, A.D.; Lashuel, H.A.; Eliezer, D. Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by PD Mutations. ACS Chem. Biol. 2016, 11, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Lindau, M.; Alvarez de Toledo, G. The fusion pore. Biochim. Biophys. Acta 2003, 1641, 167–173. [Google Scholar] [CrossRef]
- Pevsner, J.; Hsu, S.-C.; Braun, J.E.A.; Calakos, N.; Ting, A.E.; Bennett, M.K.; Scheller, R.H. Specificity and regulation of a synaptic vesicle docking complex. Neuron 1994, 13, 353–361. [Google Scholar] [CrossRef]
- Bost, A.; Shaib, A.H.; Schwarz, Y.; Niemeyer, B.A.; Becherer, U. Large dense-core vesicle exocytosis from mouse dorsal root ganglion neurons is regulated by neuropeptide Y. Neuroscience 2017, 346, 1–13. [Google Scholar] [CrossRef]
- Staal, R.G.W.; Mosharov, E.V.; Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat. Neurosci. 2004, 7, 341–346. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef]
- Wolozin, B. Cholesterol and the biology of Alzheimer’s disease. Neuron 2004, 41, 7–10. [Google Scholar] [CrossRef]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide—Insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Doh, M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AbetaP (1-40) and (1-42) peptides. FASEB J. 2002, 16, 1526–1536. [Google Scholar] [CrossRef]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ Oligomers Are Rapidly Sequestered from Brain ISF In Vivo and Bind GM1 Ganglioside on Cellular Membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T. α-Synuclein, Especially the Parkinson’s Disease-associated Mutants, Forms Pore-like Annular and Tubular Protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Runwal, G.; Edwards, R.H. The Membrane Interactions of Synuclein: Physiology and Pathology. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Bohannon, K.P.; Bittner, M.A.; Axelrod, D.; Holz, R.W. Identification of β-synuclein on secretory granules in chromaffin cells and the effects of α- and β-synuclein on post-fusion BDNF discharge and fusion pore expansion. Neurosci. Lett. 2019, 699, 134–139. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Dingjan, I.; Linders, P.T.A.; Verboogen, D.R.J.; Revelo, N.H.; Ter Beest, M.; van den Bogaart, G. Endosomal and Phagosomal SNAREs. Physiol. Rev. 2018, 98, 1465–1492. [Google Scholar] [CrossRef]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of α-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Choi, M.-G.; Kim, M.J.; Kim, D.-G.; Yu, R.; Jang, Y.-N.; Oh, W.-J. Sequestration of synaptic proteins by alpha-synuclein aggregates leading to neurotoxicity is inhibited by small peptide. PLoS ONE 2018, 13, e0195339. [Google Scholar] [CrossRef]
- Sharma, M.; Burré, J.; Südhof, T.C. CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef]
- Hoffmann, C.; Sansevrino, R.; Morabito, G.; Logan, C.; Vabulas, R.M.; Ulusoy, A.; Ganzella, M.; Milovanovic, D. Synapsin Condensates Recruit alpha-Synuclein. J. Mol. Biol. 2021, 433, 166961. [Google Scholar] [CrossRef]
- Brose, N. For Better or for Worse: Complexins Regulate SNARE Function and Vesicle Fusion. Traffic 2008, 9, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lai, Y.; Li, X.; Wang, M.; Leitz, J.; Hu, Y.; Zhang, Y.; Choi, U.B.; Cipriano, D.; Pfuetzner, R.A.; et al. C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proc. Natl. Acad. Sci. USA 2016, 113, E7590–E7599. [Google Scholar] [CrossRef]
- Liu, J.; Bu, B.; Crowe, M.; Li, D.; Diao, J.; Ji, B. Membrane packing defects in synaptic vesicles recruit complexin and synuclein. Phys. Chem. Chem. Phys. 2021, 23, 2117–2125. [Google Scholar] [CrossRef]
- Chandra, S.; Fornai, F.; Kwon, H.-B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for α- and β-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-Synuclein Cooperates with CSPα in Preventing Neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Yasuda, T.; Fukaya, M.; Yamamori, S.; Itakura, M.; Nihira, T.; Hayakawa, H.; Kawanami, A.; Kataoka, M.; Nagai, M.; et al. Accumulation of α-Synuclein Triggered by Presynaptic Dysfunction. J. Neurosci. 2012, 32, 17186–17196. [Google Scholar] [CrossRef]
- Faustini, G.; Longhena, F.; Varanita, T.; Bubacco, L.; Pizzi, M.; Missale, C.; Benfenati, F.; Björklund, A.; Spano, P.; Bellucci, A. Synapsin III deficiency hampers α-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta Neuropathol. 2018, 136, 621–639. [Google Scholar] [CrossRef]
- Longhena, F.; Faustini, G.; Varanita, T.; Zaltieri, M.; Porrini, V.; Tessari, I.; Poliani, P.L.; Missale, C.; Borroni, B.; Padovani, A.; et al. Synapsin III is a key component of α-synuclein fibrils in Lewy bodies of PD brains. Brain Pathol. 2018, 28, 875–888. [Google Scholar] [CrossRef]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic Vesicle Depletion Correlates with Attenuated Synaptic Responses to Prolonged Repetitive Stimulation in Mice Lacking α-Synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Buchman, V.L.; Cragg, S.J.; Wade-Martins, R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci. 2008, 27, 947–957. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef]
- Fountaine, T.M.; Venda, L.L.; Warrick, N.; Christian, H.C.; Brundin, P.; Channon, K.M.; Wade-Martins, R. The effect of α-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur. J. Neurosci. 2008, 28, 2459–2473. [Google Scholar] [CrossRef]
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of Vesicular Monoamine Transporter-2 Activity in α-Synuclein Stably Transfected SH-SY5Y Cells. Cell. Mol. Neurobiol. 2008, 28, 35–47. [Google Scholar] [CrossRef]
- Bu, M.; Farrer, M.J.; Khoshbouei, H. Dynamic control of the dopamine transporter in neurotransmission and homeostasis. NPJ Park. Dis. 2021, 7, 22. [Google Scholar] [CrossRef]
- Wersinger, C.; Prou, D.; Vernier, P.; Sidhu, A. Modulation of dopamine transporter function by α-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J. 2003, 17, 2151–2153. [Google Scholar] [CrossRef]
- Lee, F.J.S.; Liu, F.; Pristupa, Z.B.; Niznik, H.B. Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001, 15, 916–926. [Google Scholar] [CrossRef]
- Gaugler, M.N.; Genc, O.; Bobela, W.; Mohanna, S.; Ardah, M.T.; El-Agnaf, O.M.; Cantoni, M.; Bensadoun, J.-C.; Schneggenburger, R.; Knott, G.W.; et al. Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012, 123, 653–669. [Google Scholar] [CrossRef]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Bjorklund, A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, M.; Cataldi, S.; Choi, S.J.; Edwards, R.H.; Mosharov, E.V.; Sulzer, D. A dual role for α-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32701–32710. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Selkoe, D.; Dettmer, U. Vesicle trafficking and lipid metabolism in synucleinopathy. Acta Neuropathol. 2021, 141, 491–510. [Google Scholar] [CrossRef]
- Yavich, L.; Oksman, M.; Tanila, H.; Kerokoski, P.; Hiltunen, M.; van Groen, T.; Puoliväli, J.; Männistö, P.T.; García-Horsman, A.; MacDonald, E.; et al. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human α-synuclein. Neurobiol. Dis. 2005, 20, 303–313. [Google Scholar] [CrossRef]
- Bellani, S.; Sousa, V.L.; Ronzitti, G.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. The regulation of synaptic function by α-synuclein. Commun. Integr. Biol. 2010, 3, 106–109. [Google Scholar] [CrossRef]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. α-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Pavia-Collado, R.; Miquel-Rio, L.; Coppola-Segovia, V.; Ferrés-Coy, A.; Ruiz-Bronchal, E.; Galofré, M.; Paz, V.; Campa, L.; Revilla, R.; et al. Anti-α-synuclein ASO delivered to monoamine neurons prevents α-synuclein accumulation in a Parkinson’s disease-like mouse model and in monkeys. EBioMedicine 2020, 59, 102944. [Google Scholar] [CrossRef]
- Hill, E.; Gowers, R.; Richardson, M.J.E.; Wall, M.J. α-Synuclein Aggregates Increase the Conductance of Substantia Nigra Dopamine Neurons, an Effect Partly Reversed by the KATP Channel Inhibitor Glibenclamide. Eneuro 2021, 8, ENEURO.0330-20.2020. [Google Scholar] [CrossRef]
- Soll, L.G.; Eisen, J.N.; Vargas, K.J.; Medeiros, A.T.; Hammar, K.M.; Morgan, J.R. α-Synuclein-112 Impairs Synaptic Vesicle Recycling Consistent With Its Enhanced Membrane Binding Properties. Front. Cell Dev. Biol. 2020, 8, 405. [Google Scholar] [CrossRef]
- Busch, D.J.; Oliphint, P.A.; Walsh, R.B.; Banks, S.M.L.; Woods, W.S.; George, J.M.; Morgan, J.R. Acute increase of α-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol. Biol. Cell 2014, 25, 3926–3941. [Google Scholar] [CrossRef]
- Darios, F.; Ruipérez, V.; López, I.; Villanueva, J.; Gutierrez, L.M.; Davletov, B. α-Synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010, 11, 528–533. [Google Scholar] [CrossRef]
- Choi, B.-K.; Choi, M.-G.; Kim, J.-Y.; Yang, Y.; Lai, Y.; Kweon, D.-H.; Lee, N.K.; Shin, Y.-K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef]
- Yoo, G.; Yeou, S.; Son, J.B.; Shin, Y.-K.; Lee, N.K. Cooperative inhibition of SNARE-mediated vesicle fusion by α-synuclein monomers and oligomers. Sci. Rep. 2021, 11, 10955. [Google Scholar] [CrossRef]
- Alza, N.P.; Iglesias González, P.A.; Conde, M.A.; Uranga, R.M.; Salvador, G.A. Lipids at the Crossroad of α-Synuclein Function and Dysfunction: Biological and Pathological Implications. Front. Cell. Neurosci. 2019, 13, 175. [Google Scholar] [CrossRef]
- Medeiros, A.T.; Soll, L.G.; Tessari, I.; Bubacco, L.; Morgan, J.R. α-Synuclein Dimers Impair Vesicle Fission during Clathrin-Mediated Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2017, 11, 388. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The Role of Lipids in the Initiation of α-Synuclein Misfolding. Front. Cell. Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef]
- Tozzi, A.; Sciaccaluga, M.; Loffredo, V.; Megaro, A.; Ledonne, A.; Cardinale, A.; Federici, M.; Bellingacci, L.; Paciotti, S.; Ferrari, E.; et al. Dopamine-dependent early synaptic and motor dysfunctions induced by α-synuclein in the nigrostriatal circuit. Brain 2021, 1–34. [Google Scholar] [CrossRef]
- Matuskey, D.; Tinaz, S.; Wilcox, K.C.; Naganawa, M.; Toyonaga, T.; Dias, M.; Henry, S.; Pittman, B.; Ropchan, J.; Nabulsi, N.; et al. Synaptic Changes in Parkinson Disease Assessed with in vivo Imaging. Ann. Neurol. 2020, 87, 329–338. [Google Scholar] [CrossRef]
- Soukup, S.; Vanhauwaert, R.; Verstreken, P. Parkinson’s disease: Convergence on synaptic homeostasis. EMBO J. 2018, 37, 1–16. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Thüne, K.; Cramm, M.; Rabano, A.; Zafar, S.; Stoops, E.; Vanderstichele, H.; Villar-Pique, A.; Llorens, F.; et al. Effect of the micro-environment on α-synuclein conversion and implication in seeded conversion assays. Transl. Neurodegener. 2020, 9, 5. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef]
- McLean, P.J.; Kawamata, H.; Ribich, S.; Hyman, B.T. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 2000, 275, 8812–8816. [Google Scholar] [CrossRef]
- Ruf, V.C.; Nübling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Bötzel, K.; Kamp, F.; Giese, A. Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659. [Google Scholar] [CrossRef]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s disease: Proteinopathy or lipidopathy? NPJ Park. Dis. 2020, 6, 3. [Google Scholar] [CrossRef]
- Killinger, B.A.; Melki, R.; Brundin, P.; Kordower, J.H. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: Let’s talk about the lipids in the room. NPJ Park. Dis. 2019, 5, 23. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef]
- Reynolds, N.P.; Soragni, A.; Rabe, M.; Verdes, D.; Liverani, E.; Handschin, S.; Riek, R.; Seeger, S. Mechanism of Membrane Interaction and Disruption by α-Synuclein. J. Am. Chem. Soc. 2011, 133, 19366–19375. [Google Scholar] [CrossRef]
- Suzuki, M.; Sango, K.; Wada, K.; Nagai, Y. Pathological role of lipid interaction with α-synuclein in Parkinson’s disease. Neurochem. Int. 2018, 119, 97–106. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jin, H.K.; Bae, J.S. Sphingolipids in neuroinflammation: A potential target for diagnosis and therapy. BMB Rep. 2020, 53, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.-A.; Stokholm, K.; Zareba-Paslawska, J.; Jakobsen, S.; Vang, K.; Gjedde, A.; Landau, A.M.; Romero-Ramos, M. Early synaptic dysfunction induced by α-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 6363. [Google Scholar] [CrossRef] [PubMed]
- Takao, M.; Aoyama, M.; Ishikawa, K.; Sakiyama, Y.; Yomono, H.; Saito, Y.; Kurisaki, H.; Mihara, B.; Murayama, S. Spinocerebellar ataxia type 2 is associated with Parkinsonism and Lewy body pathology. BMJ Case Rep. 2011, 2011, bcr0120113685. [Google Scholar] [CrossRef]
- Sen, N.-E.; Arsovic, A.; Meierhofer, D.; Brodesser, S.; Oberschmidt, C.; Canet-Pons, J.; Kaya, Z.-E.; Halbach, M.-V.; Gispert, S.; Sandhoff, K.; et al. In Human and Mouse Spino-Cerebellar Tissue, Ataxin-2 Expansion Affects Ceramide-Sphingomyelin Metabolism. Int. J. Mol. Sci. 2019, 20, 5854. [Google Scholar] [CrossRef]
- Hogarth, P.; Gregory, A.; Kruer, M.C.; Sanford, L.; Wagoner, W.; Natowicz, M.R.; Egel, R.T.; Subramony, S.H.; Goldman, J.G.; Berry-Kravis, E.; et al. New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013, 80, 268–275. [Google Scholar] [CrossRef]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef]
- Chen, Y.P.; Song, W.; Huang, R.; Chen, K.; Zhao, B.; Li, J.; Yang, Y.; Shang, H.-F. GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population. J. Clin. Neurosci. 2013, 20, 880–883. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Bidinosti, M.; Shimshek, D.R.; Mollenhauer, B.; Marcellin, D.; Schweizer, T.; Lotz, G.P.; Schlossmacher, M.G.; Weiss, A. Novel one-step immunoassays to quantify α-synuclein: Applications for biomarker development and high-throughput screening. J. Biol. Chem. 2012, 287, 33691–33705. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Li, G.; Cui, S.; Du, J.; Liu, J.; Zhang, P.; Fu, Y.; He, Y.; Zhou, H.; Ma, J.; Chen, S. Association of GALC, ZNF184, IL1R2 and ELOVL7 With Parkinson’s Disease in Southern Chinese. Front. Aging Neurosci. 2018, 10, 402. [Google Scholar] [CrossRef]
- Marshall, M.S.; Issa, Y.; Heller, G.; Nguyen, D.; Bongarzone, E.R. AAV-Mediated GALC Gene Therapy Rescues Alpha-Synucleinopathy in the Spinal Cord of a Leukodystrophic Lysosomal Storage Disease Mouse Model. Front. Cell. Neurosci. 2020, 14, 619712. [Google Scholar] [CrossRef]
- Marshall, M.S.; Jakubauskas, B.; Bogue, W.; Stoskute, M.; Hauck, Z.; Rue, E.; Nichols, M.; DiAntonio, L.L.; van Breemen, R.B.; Kordower, J.H.; et al. Analysis of age-related changes in psychosine metabolism in the human brain. PLoS ONE 2018, 13, e0193438. [Google Scholar] [CrossRef]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.W.; Hardy, J.; Houlden, H.; Singleton, A.; Schneider, S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef]
- Sumi-Akamaru, H.; Beck, G.; Shinzawa, K.; Kato, S.; Riku, Y.; Yoshida, M.; Fujimura, H.; Tsujimoto, Y.; Sakoda, S.; Mochizuki, H. High expression of α-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol. Commun. 2016, 4, 27. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry. Sci. Rep. 2015, 4, 5797. [Google Scholar] [CrossRef]
- Chakroun, T.; Evsyukov, V.; Nykänen, N.-P.; Höllerhage, M.; Schmidt, A.; Kamp, F.; Ruf, V.C.; Wurst, W.; Rösler, T.W.; Höglinger, G.U. Alpha-synuclein fragments trigger distinct aggregation pathways. Cell Death Dis. 2020, 11, 84. [Google Scholar] [CrossRef]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.H.; Fanning, S.; Stirtz, G.; Kim, T.-E.; Levy, O.A.; Young-Pearse, T.L.; et al. Cell models of lipid-rich α-synuclein aggregation validate known modifiers of α-synuclein biology and identify stearoyl-CoA desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef]
- Iljina, M.; Tosatto, L.; Choi, M.L.; Sang, J.C.; Ye, Y.; Hughes, C.D.; Bryant, C.E.; Gandhi, S.; Klenerman, D. Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein. Sci. Rep. 2016, 6, 33928. [Google Scholar] [CrossRef]
- Itokazu, Y.; Fuchigami, T.; Morgan, J.C.; Yu, R.K. Intranasal infusion of GD3 and GM1 gangliosides down-regulates alpha-synuclein and controls tyrosine hydroxylase gene in a PD model mouse. Mol. Ther. 2021, in press. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R. V Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Cohen, S. Lipid Droplets as Organelles. Int. Rev. Cell Mol. Biol. 2018, 337, 83–110. [Google Scholar] [CrossRef]
- Girard, V.; Jollivet, F.; Knittelfelder, O.; Arsac, J.; Chatelain, G. A non-canonical lipid droplet metabolism regulates the conversion of alpha-Synuclein to proteolytic resistant forms in neurons of a Drosophila model of Parkinson disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Gérard, P. The crosstalk between the gut microbiota and lipids. OCL 2020, 27, 70. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox. Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.-C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef] [PubMed]
- Caló, L.; Hidari, E.; Wegrzynowicz, M.; Dalley, J.W.; Schneider, B.L.; Podgajna, M.; Anichtchik, O.; Carlson, E.; Klenerman, D.; Spillantini, M.G. CSPα reduces aggregates and rescues striatal dopamine release in α-synuclein transgenic mice. Brain 2021, 144, 1661–1669. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Model | Description | Principal Fields of Investigation |
|---|---|---|
| Vesicular systems | ||
| Micelles | Spherical and monolayer system of amphipathic molecules. Substantial difference with biological membranes. | To identify the conformational change of α-syn domains upon interaction with lipids [42]. |
| Liposomes | Spherical vesicles composed of at least one lipid bilayer and of different sizes and curvatures [43] (1) SUV* of 10–100 nm; (2) LUV* 100 nm; (3) GUV* 1 μm. | To investigate the effect of membrane curvature on α-syn oligomer–membrane interactions based on the size: (1) SUV interaction of α-syn with SV; (2) LUV mimicking cell membrane organelles; (3) GUV α-syn relationship with cell membrane [43]. |
| Planar systems | ||
| Lipid monolayer or bilayer | Planar structure composed of one or two layers. | To investigate the interaction between oligomers and membranes and to analyse the effect of α-syn oligomers on membrane disruption [43]. |
| Nanodisc | Planar bilayer structure composed of (1) phospholipids of artificial or cell membrane origin. (2) scaffolding proteins or polymers conferring stability to the system. Size variability from 7 to 50 nm. High similarity to biological membranes. | To allow structuring of disordered proteins, such as α-syn into non-toxic α-helical structures [44]. |
| Lipid Classes | Alterations in PD Patients | Effects on α-Syn |
|---|---|---|
| Phospholipids | ||
| Phosphatidylcholine (PC) | Decreased PC (34:5, 36:5, and 38:5) in the frontal cortex of PD brains [63]. Decreased PC species with polyunsaturated 3, 4, and 36 carbon in visual cortex of PD [63]. Increased PC 44:6 and 44:5 and decreased PC 35:6 in the plasma of PD patients [63]. Deregulated PC pathway across transcriptome data derived from SN and putamen of PD patients versus controls [64]. Increased PC in CSF of PD patients [65]. | POPC bilayer affects the α-syn aggregation [66]. |
| Phosphatidylethanolamine (PE) | Reduced PE in early PD but not in advanced PD [67]. Deregulated PE pathway across transcriptome data derived from SN and putamen of PD patients versus controls [64]. | Reduced levels of PE in the phosphatidylserine decarboxylase deletion mutant (psd1Δ) increase cytoplasmic α-syn inclusion and enhance toxicity in yeast [68]. |
| Phosphatidylinositol (PI) | Decreased PI in rat and human cortical neurons overexpressing α-syn [63]. Deregulated PI pathway across transcriptome data derived from SN and putamen of PD patients versus controls [64]. | Decreased PI species in yeast as well as rat or human cortical neurons overexpressing α-syn [63]. |
| Phosphatidylserine (PS) | Increased PS with 36:1, 36:2 and 38:3 fatty acyl side chains in PD frontal cortex [69]. Deregulated PS pathway across transcriptome data derived from SN and putamen of PD patients versus controls [64]. | Facilitation of SNARE complex formation and SNARE-dependent vesicles docking upon α-syn interaction with PS and v-SNARE [69]. Accelerated aggregation on POPS bilayers compared to POPC [66]. |
| Sphingolipids | ||
| Sphingomyelin | Reduced in PD anterior cingulate cortex compared to controls [70]. Deregulated sphingomyelin pathway across transcriptome data derived from SN and putamen of PD patients versus controls [64]. | Increased α-syn transcript and protein levels upon cell treatment with exogenous sphingomyelin [71]. |
| Gangliosides (GM) | Increased in lipid rafts [72]; 22% reduction in GM brain content in PD male patients, with no differences for PD female [60]. | Hypothesised to be involved in both inhibition or enhancement of the α-syn aggregation kinetics [73]. Accelerate α-syn aggregation in presence of high GM1 and GM3 ganglioside concentration in exosomes [74]. |
| Ceramides | Reduced total ceramides in PD anterior cingulate cortex compared to controls [70]. Increased in CSF of PD patients [72]. | Increased α-syn toxicity as well as α-syn oligomers formation are linked to alteration in ceramide content [75]. |
| Saturated fatty acids | ||
| Stearic acid | Increased in lipid rafts [72]. Increased in rat treated with 6-hydroxydopamine (6-OHDA) [76]. | Interaction with α-syn [77]. |
| Palmitic Acid (PA) | Increased in lipid rafts [72]. | Increased of α-syn expression levels in Thy1-α-syn mouse model after diet enriched in palmitic acid [78]. |
| Palmitoleic Acid | Decreased in CSF of PD patients [72]. | |
| Unsaturated fatty acids | ||
| α-linolenic acid | Decreased in CSF of PD patients [72]. | Promoted formation of α-syn oligomers and α-syn induced cytotoxicity [79]. |
| Oleic acid (OA) | Decreased in CFS of PD patients [72]. | Increased in response to increase concentration of α-syn monomers [63]. Decreased by stearoyl-CoA desaturase (SCD) inhibition reduced α-syn toxicity [80]. |
| Unsaturated fatty acids Omega-3 | ||
| Eicosapentaenoic acid (EPA) | Decreased EPA in lipid rafts [72]. | |
| Docosanoic acid (DHA) | Decreased DHA in lipid rafts of DLB brain [81]. Increased amount of DHA (22:6) in PD and DLB brains [79]. | Increased in α-syn oligomerisation in a DHA dose-dependent manner [79]. Increased accumulation of soluble and insoluble neuronal α-syn in A53T α-syn mice fed with an enriched DHA diet [82]. |
| Other lipids | ||
| Lipids with high solubility in aqueous solution and short hydrocarbon chains. | NI | Induced amyloid fibril formation of α-syn [83]. |
| Enzyme associated to lipid metabolism | ||
| Sphingomyelinase | Increased activity in PD brain and increased ceramide level [84]. Of note, acid sphingomyelinase (ASMase) encoded by SMPD1 is responsible for the hydrolysis of sphingomyelin into ceramide and phosphorylcholine and a reduced ASMase enzymatic activity was associated with an earlier age at onset SMPD1 variants in PD vs. controls. These genetic variants impair the traffic of acid-sphingomyelinase to the lysosomes [85]. | Increased α-syn levels in HeLa and BE(2)-M17 dopaminergic cells in SMPD1 KO and KD [85]. |
| Sphingosine kinase I | Reduced SPHKs activity under oxidative stress evoked by MPP+ [84]. | Induced of α-syn secretion and propagation upon SPHK inhibition [86]. |
| Phospholipase D1 enzyme (PLD1) | Reduced activity and expression level of PLD1 observed in DLB post-mortem brains [87]. | PLD1 prevents α-syn accumulation by autophagic flux activation [87]. |
| Glucocerebrosidase (GBA) | Reduced GCase activity in the SN and hippocampus of iPD patients [88]. | Misfolded GCase interacts with α-syn and induces α-syn accumulation and aggregation [89]. |
| Cathepsins D and E | Increased activity of cathepsin D in PRKN-PD-derived fibroblasts [78] or in iPSC-derived dopaminergic neurons from N370S-GBA PD [90]. Increased activity of cathepsin E in blood and CSF from PD patients (See for review [91,92]). | α-syn is degraded by lipid-associated cathepsin D [93]. |
| β-hexosaminidase | Decreased activity in blood and CSF from PD patients [91]. | Increased β-hexosaminidase activity rescues the neurodegeneration induced by α-syn in dopaminergic neurons of the rodent SN [94]. |
| β-galactosidase | Increased activity in blood and CSF from PD patients [91,92]. | NI |
| Protein | Model | Positive Effect | Negative Effect |
|---|---|---|---|
| Complexin | Mice model over-expressing α-syn | Reduction in complexin 2 level in brain extracts from α-syn transgenic mice compared to controls [129]. | |
| Mice α/β-syn double-KO | 30% increase in complexins in α/β-syn double-KO mice [141]. | ||
| CSPα | CSPα-KO mice | Reduction in SNAP25, Hsc70 and Hsp70 [141]. Impairment in SNARE complex formation [141]. | |
| Increase in SNAP25 ubiquitylation and proteasomal degradation [134]. Reduction in SNAP25 [134,142] and Hsc70 protein levels [134] Impairment in SNARE-complex assembly [142]. | |||
| Neurons overexpressing CSPα. | CSPα suppresses the degradation of SNAP25 and Hsc70 and increases their protein levels [134]. | ||
| CSPα-KO mice overexpressing WT or A30P α-syn. | Overexpression of WT α-syn but no A30P rescues the SNARE-complex assembly deficit induced by CSPα-KO [142]. | ||
| Mice expressing a truncated human α-syn (1–120) injected with viral CSPα. | Viral CSPα injection reduces α-syn aggregates [142]. | ||
| SNAP25 | Snap25S187A/S187A KI mice carrying an unphosphorylated form of SNAP25 (Ser/Ala phospho-dead mutation on position 187). | Increased number of endogenous α-syn aggregates associated with cytoplasmic side of the plasma membrane. Decreased ability of the SNARE complex assembly [143]. | |
| Synapsin III | AAV-human α-syn injections in synapsin III KO mice. | Reduction in α-syn aggregation [144]. Reduction in the α-syn S129 phosphorylation in synapsin III KO mice in the striatum ipsilateral of an unilateral injection of AAV-human α-syn and no difference in the contralateral striatum [144]. | |
| Primary rodent dopaminergic neurons synapsin III KO. | Prevention of α-syn aggregation [135]. | ||
| LB-enriched protein extracts from the SN of PD versus control brain samples. | LB-enriched fractions are immunopositive for both synapsin III and α-syn aggregates [145]. | ||
| VAMP2 | Rat cortical neurons treated with α-syn aggregates. | Direct binding of VAMP2 with α-syn aggregates. Reduction in VAMP2 and SNAP25 protein level, but no change in Syntaxin1A. 45% decrease in glutamate release [133]. |
| α-Synuclein Models | Positive Effect on Exocytosis | Negative Effect on Exocytosis |
|---|---|---|
| α-syn overexpression models | ||
| PC12 cells and chromaffin cells overexpressing α-syn. | Reduced catecholamine release in both PC12 and chromaffin cells. Accumulation of docked vesicles at the plasma membrane in PC12, but not in chromaffin cells Potential inhibition of the priming of neurosecretory vesicles in chromaffin cells [128]. | |
| Transgenic expression of α-syn in CSPα knockout mice. | Rescue the assembly and function of the exocytic SNARE29, preventing neurodegeneration [159]. | |
| Hippocampal neurons overexpressing α-syn. | Enhanced both spontaneous and evoked neurotransmitters release [160]. | |
| Primary rat hippocampal neurons overexpressing α-syn and endogenous α-syn. | Promoted dilation of the fusion pore [125]. | |
| α-syn KO/deletion models | ||
| α-syn KO mice obtained by deleting exons 1 and 2 of the SNCA gene. | No impairment in structure of synapse, release of neurotransmitters, mobilisation of SV [141]. | |
| α-syn KD by antisense oligonucleotides in hippocampal neurons. | Blocking the potentiation of synaptic transmission | |
| α-syn KO mice obtained by deleting exons 4 and 5 of the SNCA gene in embryonal stem cells. | Dramatic loss of reserve vesicles and an increase in synaptic depression [146]. | |
| Non-viral gene therapy based on a new indatraline-conjugated antisense oligonucleotide (IND-ASO) to disrupt the α-synuclein mRNA transcription selectively in monoamine neurons of a PD-like mouse model and elderly non-human primates. | Intracerebroventricular and intranasal IND-ASO administration for four weeks in a mouse model with AAV-mediated WT human α-syn overexpression in dopamine neurons prevented the synthesis and accumulation of α-syn in the connected brain regions, improving dopamine neurotransmission [161]. | |
| α-syn aggregates models and recombinant α-syn treatment | ||
| Introduction of α-syn aggregates into single dopaminergic neurons via the patch electrode. | Accumulation of α-syn aggregates may chronically activate KATP channels leading to loss of excitability and dopamine release [162]. | |
| Synapse treated with recombinant human α-syn-112. | α-syn-112 strongly inhibits SV recycling [163]. | |
| Giant Lamprey synapse injected with α-syn. | Accumulation of clathrin coated pits and clathrin coated vesicles [164]. | |
| In vitro studies | ||
| Immobilised α-syn on sepharose beads incubated with radioactive arachidonic acid. | α-syn inhibits both exocytosis and SNARE complex formation by decreasing the levels of free arachidonic acid available to the SNARE proteins [165]. | |
| Single-vesicle and bulk in vitro lipid-mixing assays with α-syn purified monomer. | The α-syn monomers promote SNARE complex formation [166]. | |
| Single-vesicle and bulk in vitro lipid-mixing assays with α-syn purified oligomers. | Interaction of large α-syn oligomers with VAMP2 Inhibition of SNARE complex formation Inhibition of docking vesicles [166]. | |
| In vitro lipid-mixing assay with monomers and oligomers. | Both α-syn monomers and α-syn oligomers induce the clustering of SV. The α-syn mutant T44P/A89P with reduced lipid-binding affinity reduces the clustering of SV by α-syn oligomers in vitro [167]. | |
| Genes | Genetic Determinants Associated with α-Syn Pathologies | Effect on Lipids | Effects on α-Syn |
|---|---|---|---|
| ATXN2 | Diseases associated with ATXN2 include Spinocerebellar Ataxia 2 and PD/parkinsonism with LB pathology [191]. | Ataxin-2 expansion affects ceramide-sphingomyelin metabolism [192]. | NI |
| C19orf12 | C19orf12 is associated with Neurodegeneration with Brain Iron Accumulation disorders with prominent widespread Lewy body pathology [193]. | Role in lipid homeostasis [194]. | NI |
| DGKQ | DGKQ emerged as PD risk factor in independent GWAS studies [195,196] . | Controls the cellular content of diglycerides. | DGKQ loss-of-function in PD might potentially leads to enhanced transcription of SNCA [197]. |
| ELOVL7 | ELOVL7 identified in GWAS studies as PD-associated gene [198]. | FA elongase 7 plays a role in synthesis of long-chain saturated fatty acids involved as precursors of membrane lipids and lipid mediators [199]. | Defects in very long chain fatty acid synthesis enhance the toxicity of α-syn WT, A53T and E46K toxicity in a yeast model of PD. The effect on α-syn A30P is inappreciable in a yeast model of PD [75]. |
| GALC | Mutations in the GALC gene are responsible for Krabbe disease, a demyelinating disorder characterised by the presence of neuronal aggregates, in part composed of α-syn [200]. | GALC catalyses the hydrolysis of substrates including galactosylceramide (GalC) and galactosylsphingosine. In PD patients, higher levels of galactosylsphingosine were found respect to controls [201]. | Galactosylceramidase treatment improves the survival and health of KD mice, prevents the formation of α-syn in spinal neurons [200] galactosylsphingosine accelerates aggregation of α-syn in a dose-dependent manner [202]. |
| GBA | PD risk factor confirmed in GWAS studies [203,204]. | Involved in glycolipid catabolism. | The decreased GCase activity identified in CSF and blood PD patients and the consequent increase in glucosylceramide level directly correlates with increased α-syn oligomer formation. |
| PLA2G6 | PLA2G6 is causative for PARK14 in patients with autosomal recessive dystonia-parkinsonism [205]. | PLA2G6 hydrolyses the sn-2 acyl chain of glycerophospholipids in free fatty acids and lysophospholipids [206]. | Pla2g6 KO mouse neurons show early increase in α-syn/phospho α-syn level [207]. Increased expression of α-syn in cell and animal model with PLA2G6 dysfunction [207]. |
| SCARB2 | SCARB2 locus identified in GWAS studies as PD-associated gene. This gene encodes LIMP2 [208]. | LIMP2 deficiency can lead to a decrease in GCase activity and α-syn degradation [209]. | LIMP2 deficiency can lead to a decrease in GCase activity and α-syn degradation [209]. |
| SREBF1 | SREBF1 locus identified as PD risk factor in GWAS [208]. | SREBF1 encodes SREBP-1 that regulates synthesis of sterol. | NI |
| VPS13C | VPS13C first mutation identified in a form of early-onset parkinsonism; the pathological features were reminiscent of diffuse Lewy body disease [210]. | VPS13C is a lipid transport proteins [211]. | NI |
| Type of α-Syn Modifications | Lipid Effect | Vesicle Trafficking | |||
|---|---|---|---|---|---|
| Docking | Fusion Pore | Exocytosis | Recycling | ||
| WT 140 | Increased α-helical multimers formation. | Increased cluster of VAMP2- vesicles and SNARE complex assembly. | |||
| A30P 140 | Decreased membrane binding. Decreased membrane curvature. Abolition of interaction with lipid-rafts. | Accumulation of docked vesicles at the plasma membrane. Decreased priming of neurosecretory vesicles. | Perturbation of fusion pore formation. | Decreased catecholamine release. No change in synaptic exocytosis. | |
| A53T 140 | Increased multimerisation long chain PUFA-mediated. Decreased multimerisation mediated by saturated fatty acids. No change in lipid binding. | Clustering of VAMP2 SV at the active zone. | Perturbation of fusion pore formation. | Perturbation of SV recycling. | |
| E46K 140 | Clustering of VAMP2 SV at the active zone. | ||||
| K O * | Decreased reserve pool. | Increased concentration of VMAT2 molecules per vesicle. | Increased dopamine release. | ||
| Overexpression | Decreased membrane curvature induction. Increased oleic acid and unsaturated fatty acid. | Decreased reserve vesicles. Decreased vesicle density. | Prevention the fusion pore closure. | Rescue the SNARE-complex assembly deficit in CSPα-KO mice. Decreased level of synapsins and complexins. | Increased cytosolic dopamine levels due to inhibition of VMAT2 activity. |
| Overexpression in CSPα-KO mice | Rescues the SNARE-complex assembly deficit. | Prevents neurodegeneration. | |||
| WT 112 | Increased phospholipid binding Increased tendency to oligomerisation. | Perturbation SV recycling. | |||
| Soluble aggregates | Increased aggregates by PUFA. | ||||
| Pathological aggregates | Increased aggregation by cholesterol, lipids with short saturated acyl chain, GM1, and GM3. | Perturbation of vesicles docking at the presynaptic terminal. | Increased VAMP2 binding affinity. | Alteration in neurotransmission. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarchione, A.; Marchand, A.; Taymans, J.-M.; Chartier-Harlin, M.-C. Alpha-Synuclein and Lipids: The Elephant in the Room? Cells 2021, 10, 2452. https://doi.org/10.3390/cells10092452
Sarchione A, Marchand A, Taymans J-M, Chartier-Harlin M-C. Alpha-Synuclein and Lipids: The Elephant in the Room? Cells. 2021; 10(9):2452. https://doi.org/10.3390/cells10092452
Chicago/Turabian StyleSarchione, Alessia, Antoine Marchand, Jean-Marc Taymans, and Marie-Christine Chartier-Harlin. 2021. "Alpha-Synuclein and Lipids: The Elephant in the Room?" Cells 10, no. 9: 2452. https://doi.org/10.3390/cells10092452
APA StyleSarchione, A., Marchand, A., Taymans, J.-M., & Chartier-Harlin, M.-C. (2021). Alpha-Synuclein and Lipids: The Elephant in the Room? Cells, 10(9), 2452. https://doi.org/10.3390/cells10092452