
Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Flow Cytometry Analysis
2.3. Isolation of Adipose Tissue Macrophages (ATMs)
2.4. Isolation of Islets
2.5. EV Purification and Pharacterization
2.6. In Vivo and In Vitro EV Treatment
2.7. Argonaut-RNA Co-Immunoprecipitation Assay
2.8. Differentiation of Bone Marrow-Derived Macrophages
2.9. Depletion of Tissue Macrophages
2.10. Transfection of miR-155 Mimics, miR-155 Hairpin Inhibitor, or siRNA
2.11. Co-Culture Assay
2.12. Immuno-Fluorescence Staining
2.13. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Analysis
2.14. Glucose-Stimulated Insulin Secretion Assays
2.15. Statistics
3. Results
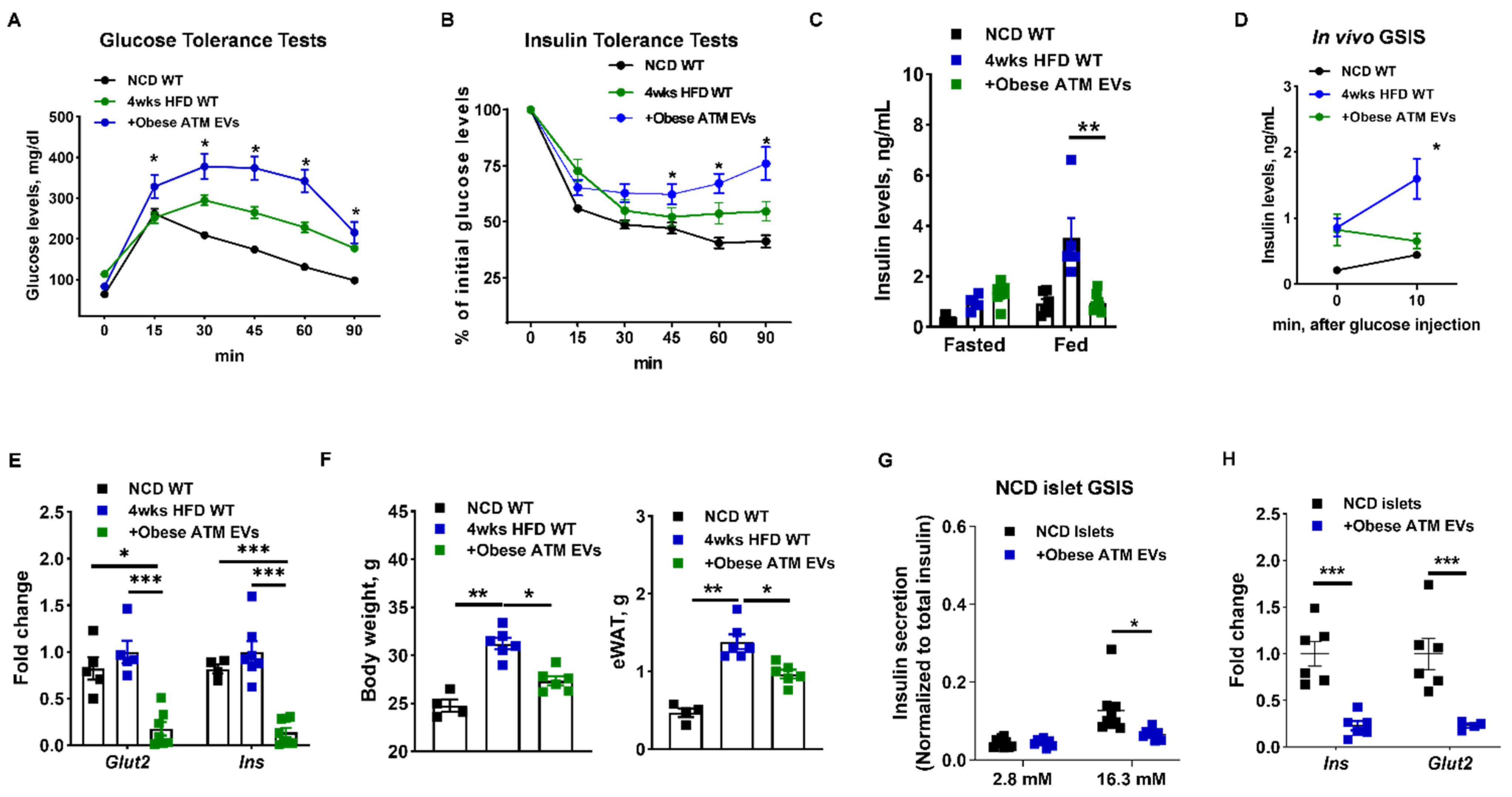
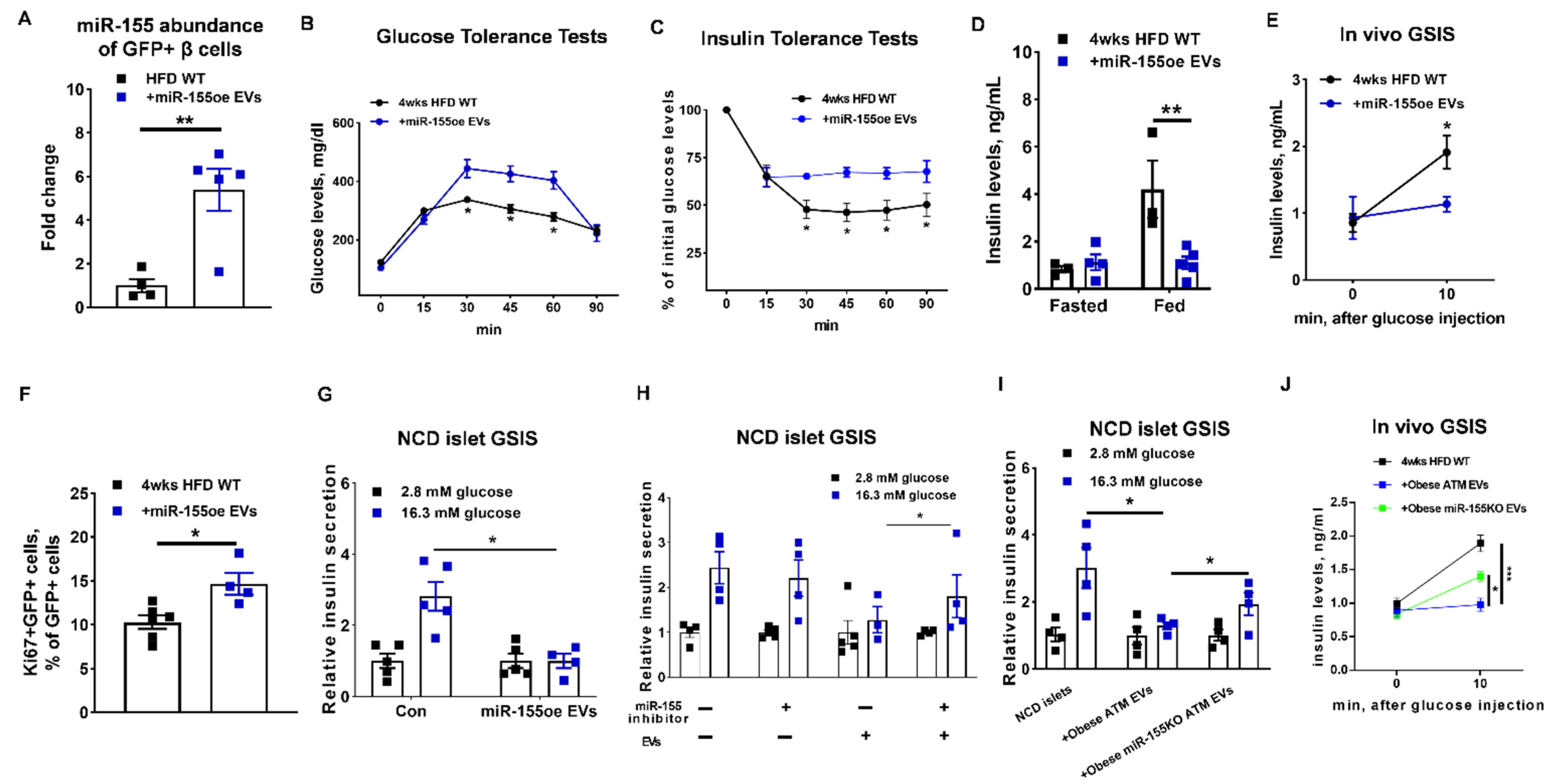
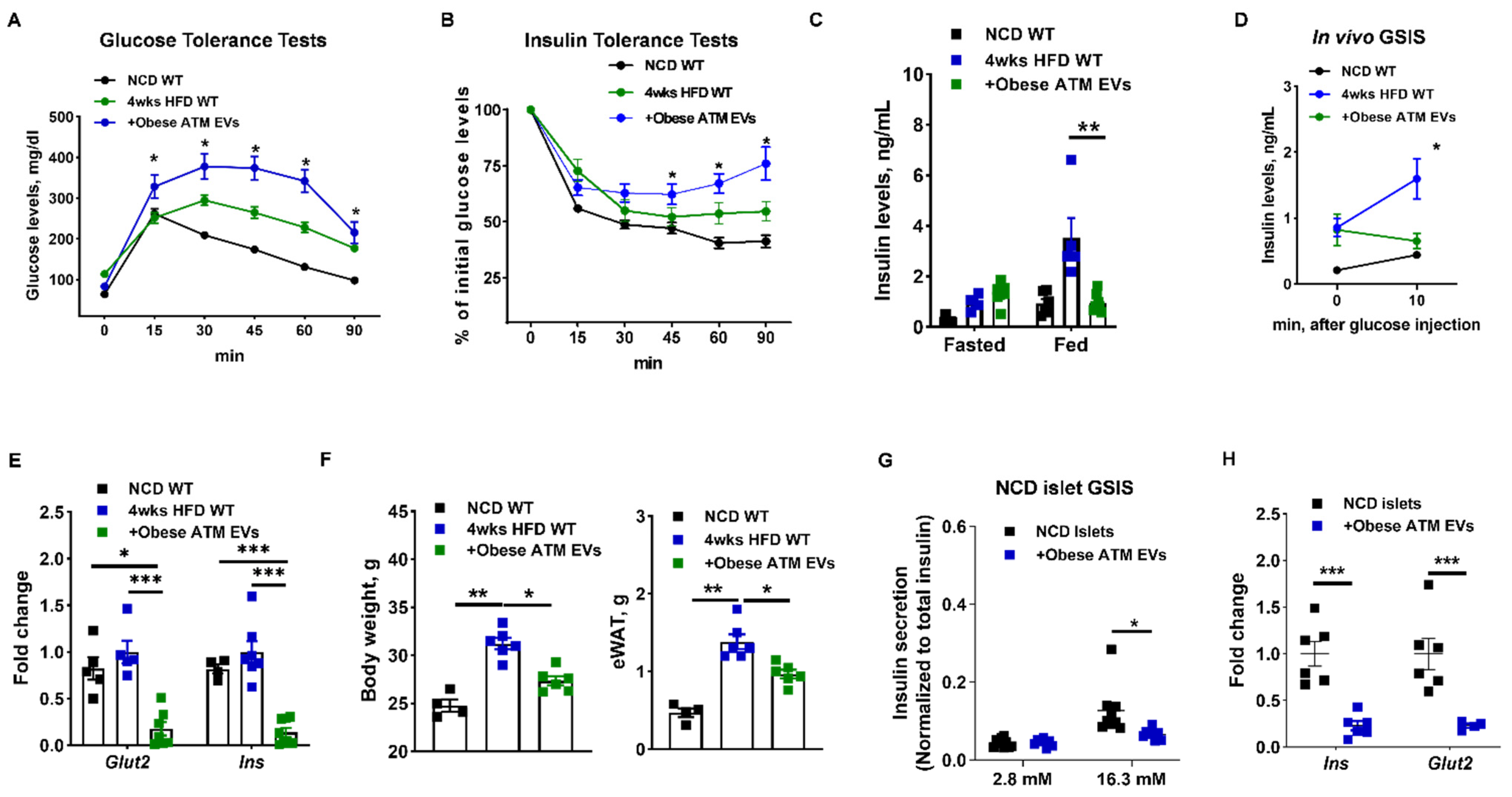
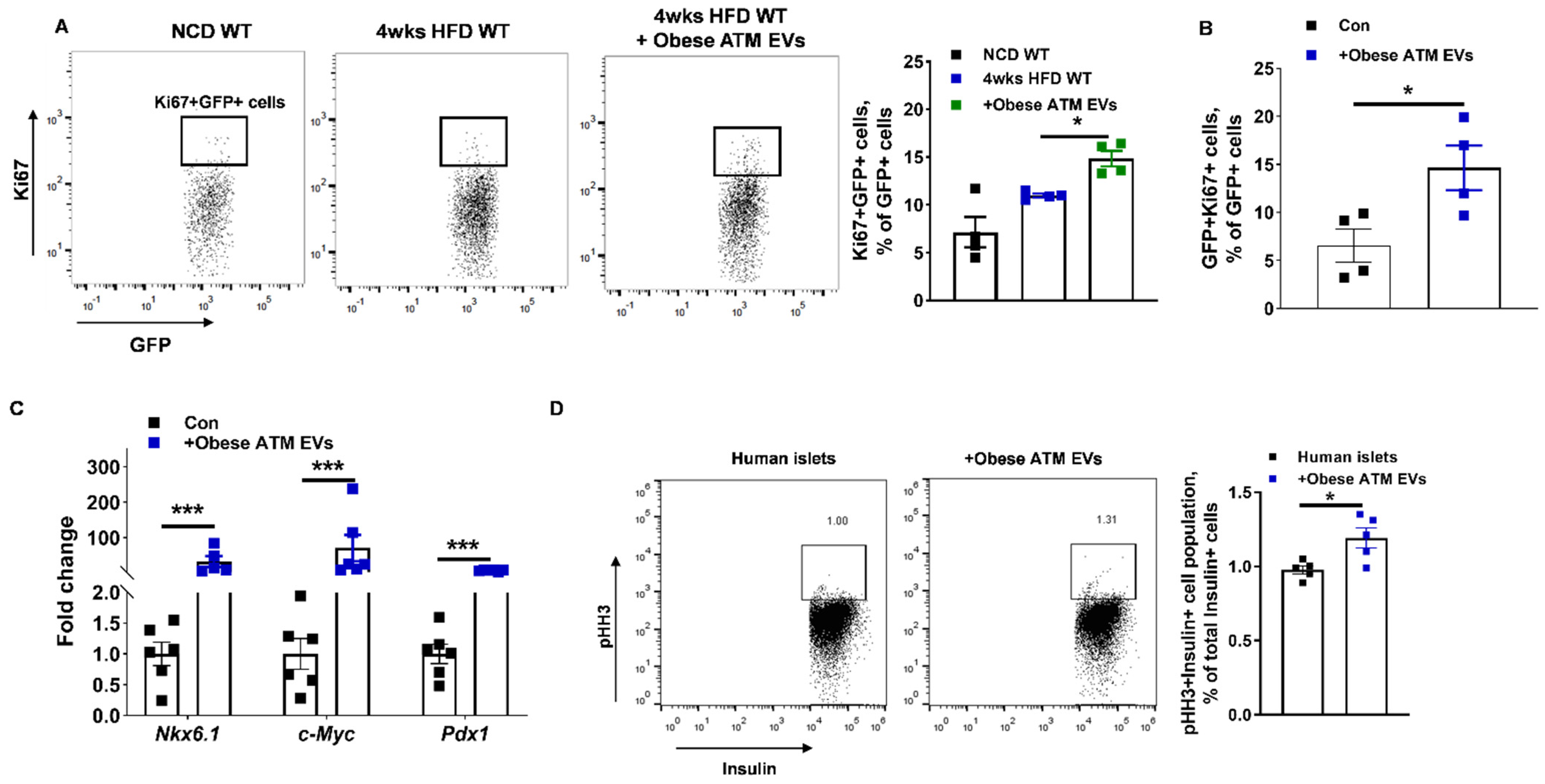
3.1. Adipose Tissue Macrophage-Produced EVs Are Pathogenic for Insulin Secretion in Obesity
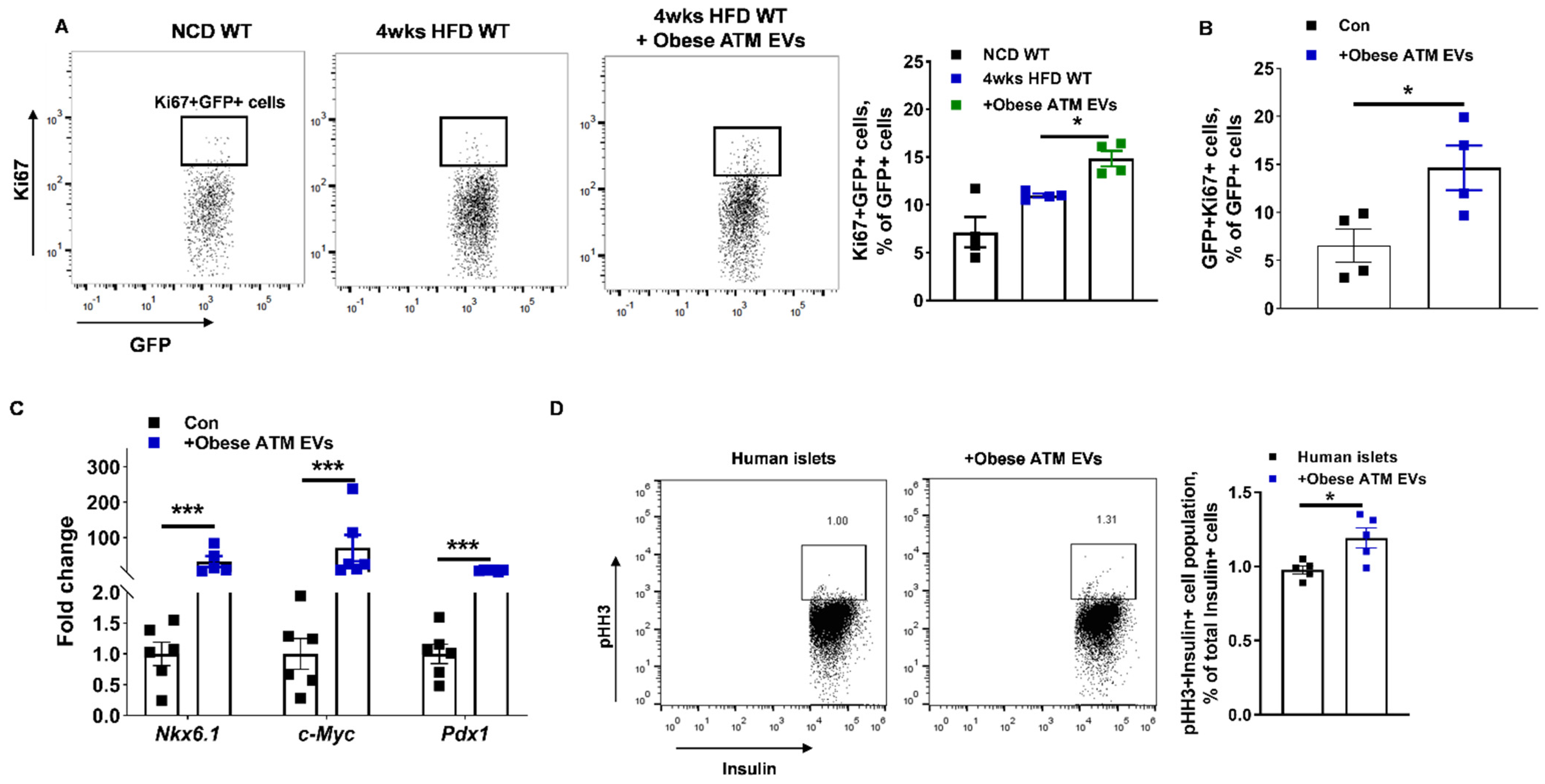
3.2. Obese ATM-Derived EVs Can Promote Obesity-Induced β Cell Proliferation
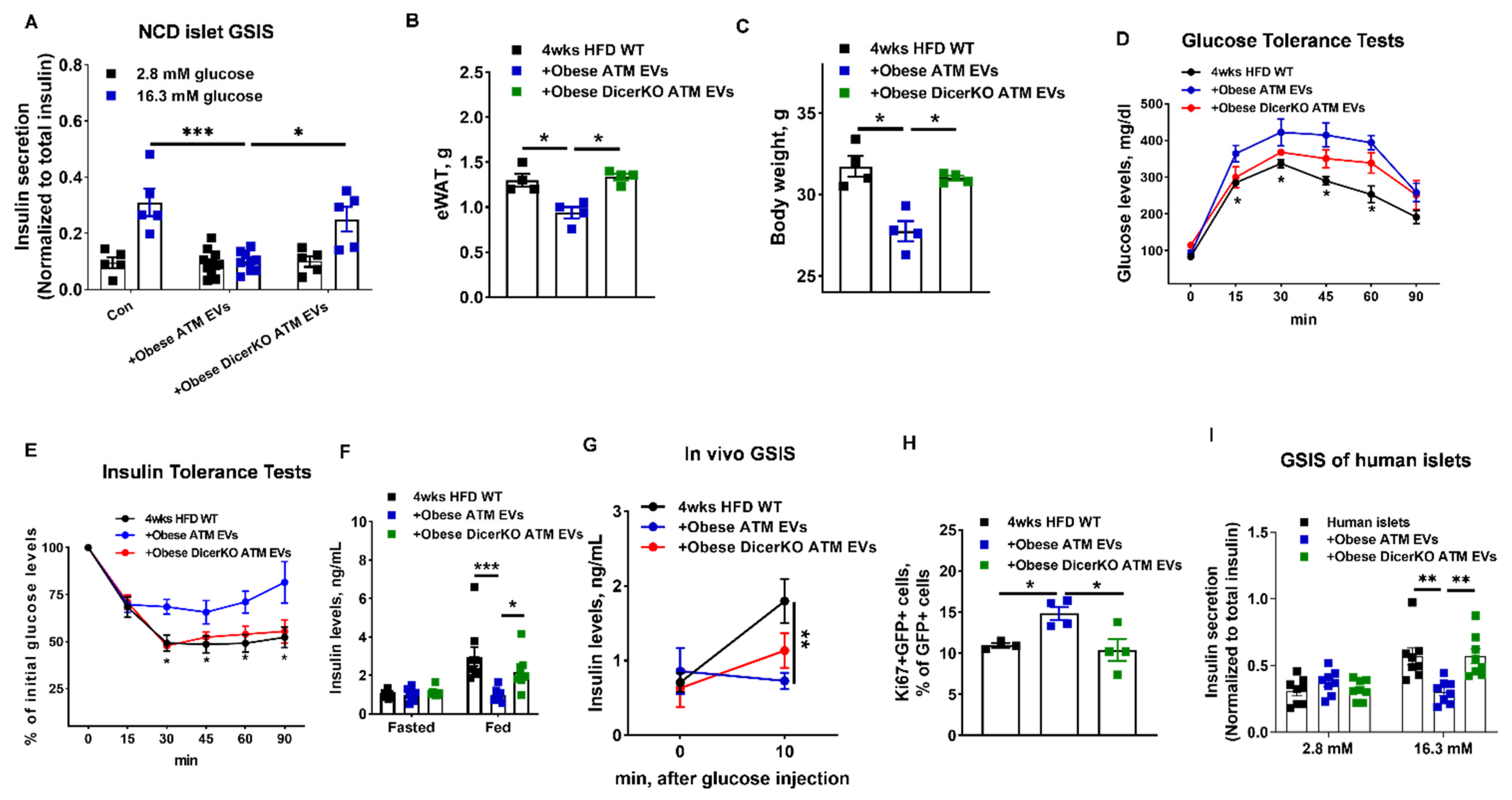
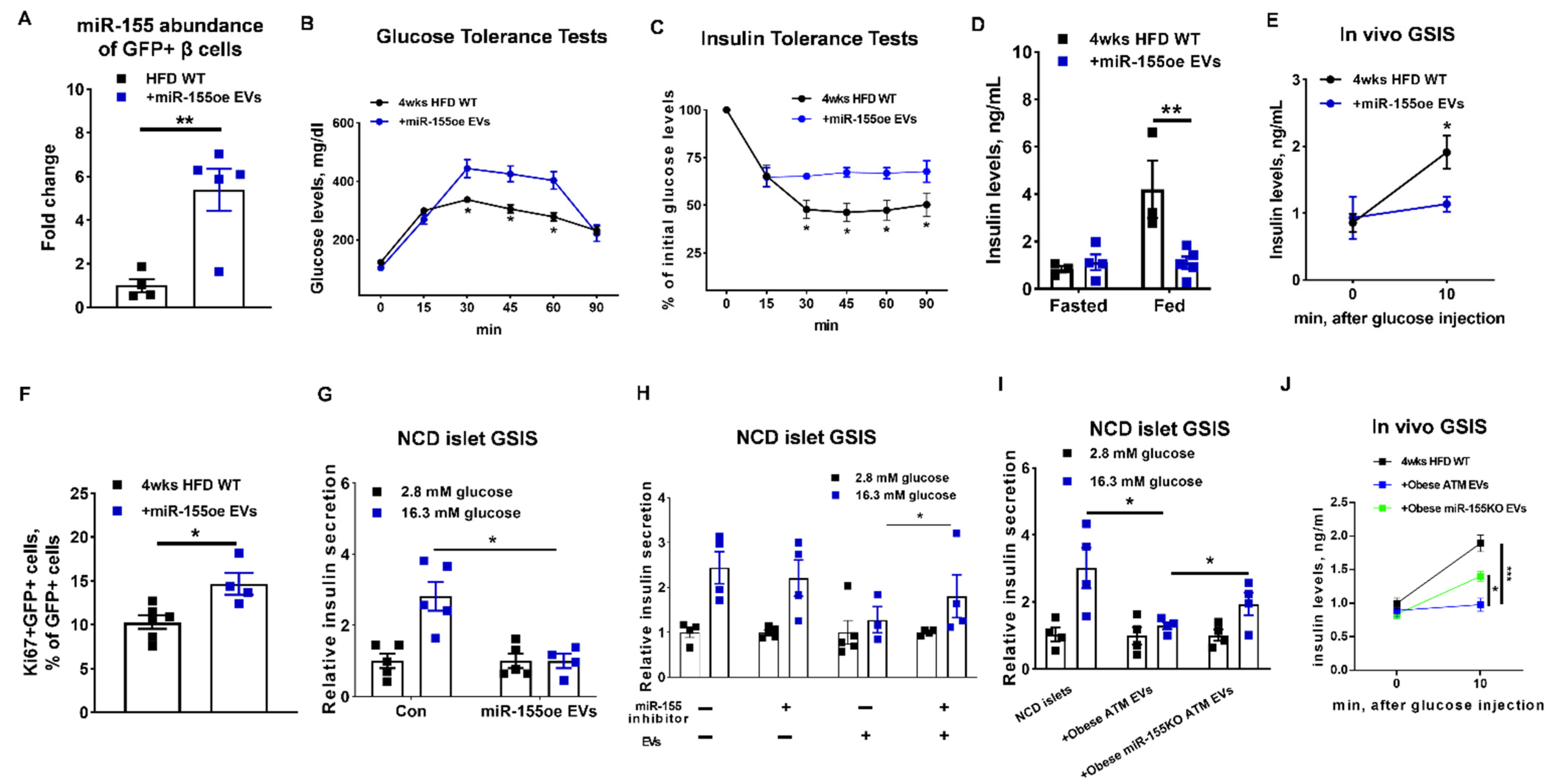
3.3. miRNAs Are Indispensable Components for the Regulation of Obese ATM-Derived EVs on β Cell Responses
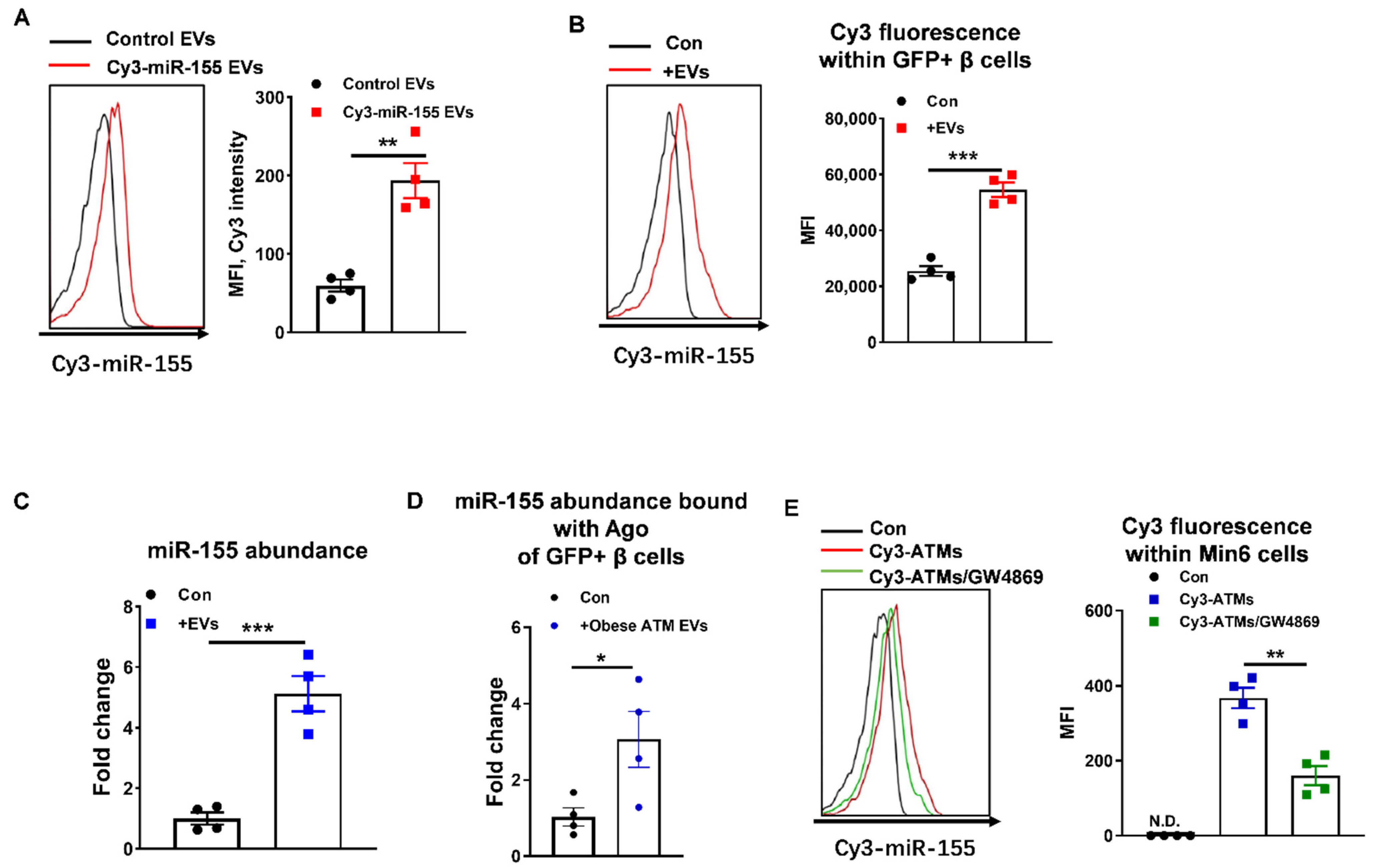
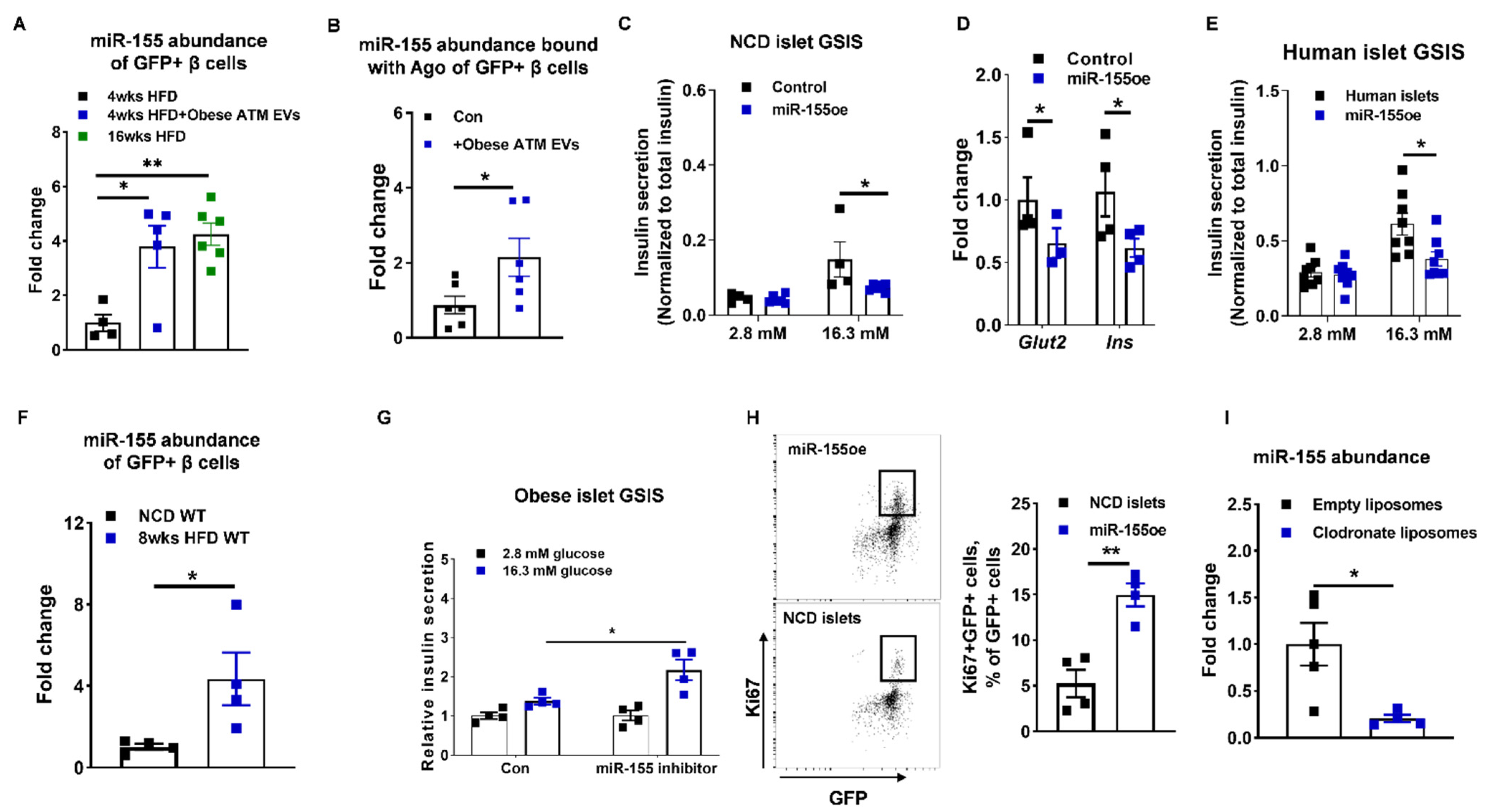
3.4. miR-155 Is a Functional Molecule within Obese ATM EVs Mediating β Cell Responses
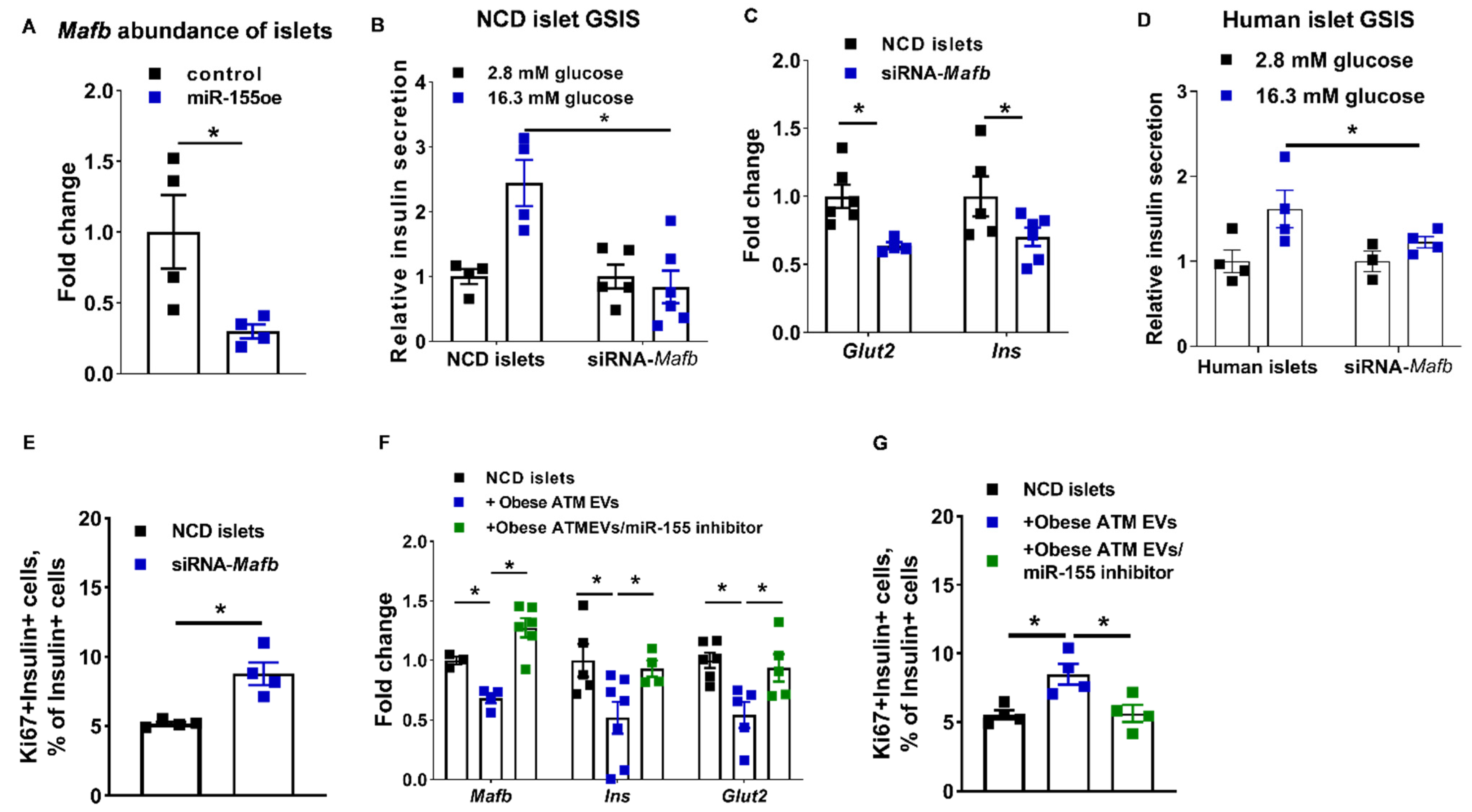
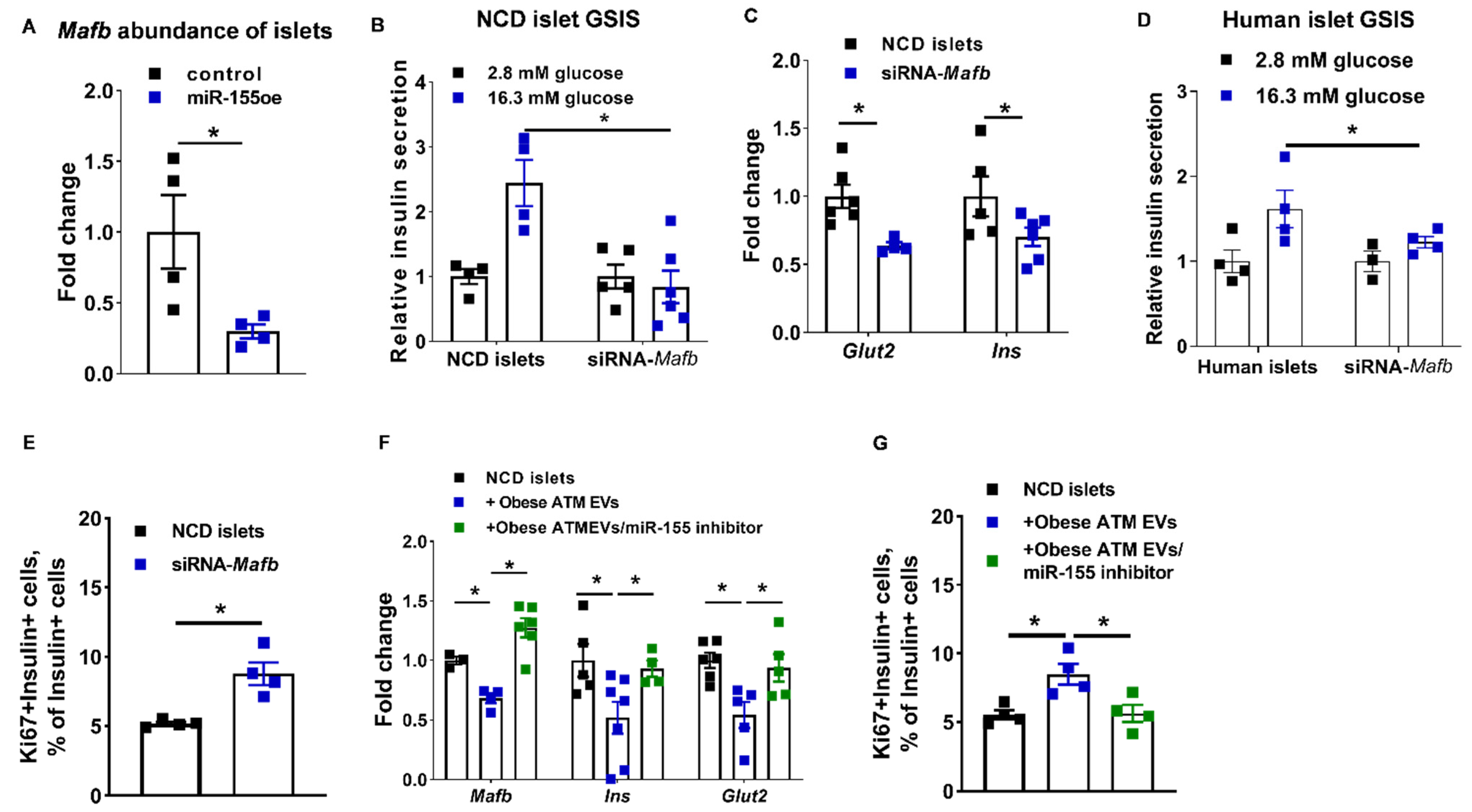
3.5. The miR-155-Mafb Axis Exerts Profound Regulation on the Insulin Secretion and Proliferation of β Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; Fernandes, J.D.d.R.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas. Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, G.R.; Lee, J.; Shoelson, S.E. Metabolic syndrome, insulin resistance, roles inflammation--mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1771–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Yuan, M. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. International journal of obesity and related metabolic disorders. J. Int. Assoc. Study Obes. 2003, 27, S49–S52. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Deyoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Maris, M.; Ofrecio, J.M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Deyoung, S.M.; Saltiel, A.R. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E166–E174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Liu, S.; Lu, M.; Bandyopadhyay, G.; Oh, D.; Imamura, T.; Johnson, A.M.F.; Sears, D.; Shen, Z.; Cui, B.; et al. Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 2016, 167, 973–984.e912. [Google Scholar] [CrossRef] [Green Version]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Patsouris, D.; Li, P.P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandao, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019, 30, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 2180–2181. [Google Scholar] [CrossRef]
- Das, S.; Extracellular, R.N.A.C.C.; Ansel, K.M.; Bitzer, M.; Breakefield, X.O.; Charest, A.; Galas, D.J.; Gerstein, M.B.; Gupta, M.; Milosavljevic, A.; et al. The Extracellular RNA Communication Consortium: Establishing Foundational Knowledge and Technologies for Extracellular RNA Research. Cell 2019, 177, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas Analysis Reveals Distinct Extracellular RNA Cargo Types and Their Carriers Present across Human Biofluids. Cell 2019, 177, 463–477.e415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, S.E., 3rd; Grijalva, A.; Xu, X.; Ables, E.; Nomani, A.; Ferrante, A.W., Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019, 363, 989–993. [Google Scholar] [CrossRef]
- Castano, C.; Kalko, S.; Novials, A.; Parrizas, M. Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12158–12163. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.B.; Poliakov, A.; Hardy, R.W.; Clements, R.; Liu, C.R.; Liu, Y.L.; Wang, J.H.; Xiang, X.Y.; Zhang, S.Q.; Zhuang, X.Y.; et al. Adipose Tissue Exosome-Like Vesicles Mediate Activation of Macrophage-Induced Insulin Resistance. Diabetes 2009, 58, 2498–2505. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, S.C.; Nadler, E.P.; Pillai, D.K.; Hubal, M.J.; Wang, Z.; Wang, J.M.; Gordish-Dressman, H.; Koeck, E.; Sevilla, S.; Wiles, A.A.; et al. Adipocyte-derived exosomal miRNAs: A novel mechanism for obesity-related disease. Pediatr Res. 2015, 77, 447–454. [Google Scholar] [CrossRef]
- Whitham, M.; Parker, B.L.; Friedrichsen, M.; Hingst, J.R.; Hjorth, M.; Hughes, W.E.; Egan, C.L.; Cron, L.; Watt, K.I.; Kuchel, R.P.; et al. Extracellular Vesicles Provide a Means for Tissue Crosstalk during Exercise. Cell Metab. 2018, 27, 237–251.e234. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e312. [Google Scholar] [CrossRef] [Green Version]
- Guay, C.; Kruit, J.K.; Rome, S.; Menoud, V.; Mulder, N.L.; Jurdzinski, A.; Mancarella, F.; Sebastiani, G.; Donda, A.; Gonzalez, B.J.; et al. Lymphocyte-Derived Exosomal MicroRNAs Promote Pancreatic beta Cell Death and May Contribute to Type 1 Diabetes Development. Cell Metab. 2019, 29, 348–361.e346. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Jhala, U.S.; Winnay, J.N.; Krajewski, S.; Montminy, M.; Kahn, C.R. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Investig. 2004, 114, 828–836. [Google Scholar] [CrossRef] [Green Version]
- Aamodt, K.I.; Powers, A.C. Signals in the pancreatic islet microenvironment influence beta-cell proliferation. Diabetes Obes. Metab. 2017, 19, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.N.; Mizrachi, E.B.; Ocana, A.G.; Stewart, A.F. Human beta-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef] [Green Version]
- Alonso, L.C.; Yokoe, T.; Zhang, P.; Scott, D.K.; Kim, S.K.; O’Donnell, C.P.; Garcia-Ocana, A. Glucose infusion in mice: A new model to induce beta-cell replication. Diabetes 2007, 56, 1792–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ocana, A.; Takane, K.K.; Syed, M.A.; Philbrick, W.M.; Vasavada, R.C.; Stewart, A.F. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J. Biol. Chem. 2000, 275, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Araujo, T.G.; Oliveira, A.G.; Carvalho, B.M.; Guadagnini, D.; Protzek, A.O.; Carvalheira, J.B.; Boschero, A.C.; Saad, M.J. Hepatocyte growth factor plays a key role in insulin resistance-associated compensatory mechanisms. Endocrinology 2012, 153, 5760–5769. [Google Scholar] [CrossRef] [PubMed]
- Assmann, A.; Ueki, K.; Winnay, J.N.; Kadowaki, T.; Kulkarni, R.N. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol. Cell Biol. 2009, 29, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and beta-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filios, S.R.; Shalev, A. Beta-Cell MicroRNAs. Small but Powerful. Diabetes 2015, 64, 3631–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Wei, Y.; Geissler, C.; Abschlag, K.; Campos, J.C.; Hristov, M.; Mollmann, J.; Lehrke, M.; Karshovska, E.; Schober, A. Hyperlipidemia-Induced MicroRNA-155-5p Improves beta-Cell Function by Targeting Mafb. Diabetes 2017, 66, 3072–3084. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Cheruku, P.S.; Bazer, F.W.; Safe, S.H.; Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp. 2013, 76, 50323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Canales, D.; Penko, D.; Min, K.K.M.; Parha, K.A.; Peiris, H.; Haberberger, R.V.; Pitson, S.M.; Drogemuller, C.; Keating, D.J.; Grey, S.T.; et al. Local Sphingosine Kinase 1 Activity Improves Islet Transplantation. Diabetes 2017, 66, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Wattez, J.S.; Lim, L.; Rozance, P.J.; Hay, W.W., Jr.; Shao, J. Prolonged Prepregnant Maternal High-Fat Feeding Reduces Fetal and Neonatal Blood Glucose Concentrations by Enhancing Fetal beta-Cell Development in C57BL/6 Mice. Diabetes 2019, 68, 1604–1613. [Google Scholar]
- Bartel, D.P. MicroRNAs. Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Crewe, C.; Joffin, N.; Rutkowski, J.M.; Kim, M.; Zhang, F.; Towler, D.A.; Gordillo, R.; Scherer, P.E. An Endothelial-to-Adipocyte Extracellular Vesicle Axis Governed by Metabolic State. Cell 2018, 175, 695–708.e613. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Jetton, T.L.; LaRock, K.; Monga, N.; Satish, B.; Lausier, J.; Peshavaria, M.; Leahy, J.L. Temporal characterization of beta cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J. Biol. Chem. 2017, 292, 12449–12459. [Google Scholar] [CrossRef] [Green Version]
- Hull, R.L.; Kodama, K.; Utzschneider, K.M.; Carr, D.B.; Prigeon, R.L.; Kahn, S.E. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: Evidence for specificity of impaired beta cell adaptation. Diabetologia 2005, 48, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.C.; Ljubicic, S.; Leibiger, B.; Kern, M.; Leibiger, I.B.; Moede, T.; Kelly, M.E.; Bhowmick, D.C.; Murano, I.; Cohen, P.; et al. Adipsin is an adipokine that improves beta cell function in diabetes. Cell 2014, 158, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Banoy, N.; Guseh, J.S.; Li, G.; Rubio-Navarro, A.; Chen, T.; Poirier, B.; Putzel, G.; Rosselot, C.; Pabon, M.A.; Camporez, J.P.; et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat. Med. 2019, 25, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Corbalan-Campos, J.; Gurung, R.; Natarelli, L.; Zhu, M.; Exner, N.; Erhard, F.; Greulich, F.; Geissler, C.; Uhlenhaut, N.H.; et al. Dicer in Macrophages Prevents Atherosclerosis by Promoting Mitochondrial Oxidative Metabolism. Circulation 2018, 138, 2007–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.K.; Son, Y.; Kim, S.N.; Song, H.D.; Kim, M.; Park, J.H.; Jung, Y.S.; Ahn, S.Y.; Saha, A.; Granneman, J.G.; et al. MicroRNA-10a-5p regulates macrophage polarization and promotes therapeutic adipose tissue remodeling. Mol. Metab. 2019, 29, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Siegel, F.; Kipschull, S.; Haas, B.; Frohlich, H.; Meister, G.; Pfeifer, A. miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat. Commun. 2013, 4, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, J.P.; Lu, Y.; Loeb, G.B.; Leslie, C.S.; Rudensky, A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018, 19, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Roccaro, A.M.; Rombaoa, C.; Flores, L.; Obad, S.; Fernandes, S.M.; Sacco, A.; Liu, Y.; Ngo, H.; Quang, P.; et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood 2012, 120, 1678–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.C.; Jung, Y.; Ugboma, C.M.; Shimbo, M.; Kuno, A.; Basha, W.A.; Kudo, T.; Oishi, H.; Takahashi, S. MafB Is Critical for Glucagon Production and Secretion in Mouse Pancreatic alpha Cells In Vivo. Mol. Cell Biol. 2018, 38, e00504–e00517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The MAFB transcription factor impacts islet alpha-cell function in rodents and represents a unique signature of primate islet beta-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E91–E102. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Luo, Z.; Jin, Z.; Ji, Y.; Ying, W. Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles. Cells 2021, 10, 2451. https://doi.org/10.3390/cells10092451
Gao H, Luo Z, Jin Z, Ji Y, Ying W. Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles. Cells. 2021; 10(9):2451. https://doi.org/10.3390/cells10092451
Chicago/Turabian StyleGao, Hong, Zhenlong Luo, Zhongmou Jin, Yudong Ji, and Wei Ying. 2021. "Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles" Cells 10, no. 9: 2451. https://doi.org/10.3390/cells10092451
APA StyleGao, H., Luo, Z., Jin, Z., Ji, Y., & Ying, W. (2021). Adipose Tissue Macrophages Modulate Obesity-Associated β Cell Adaptations through Secreted miRNA-Containing Extracellular Vesicles. Cells, 10(9), 2451. https://doi.org/10.3390/cells10092451