
Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
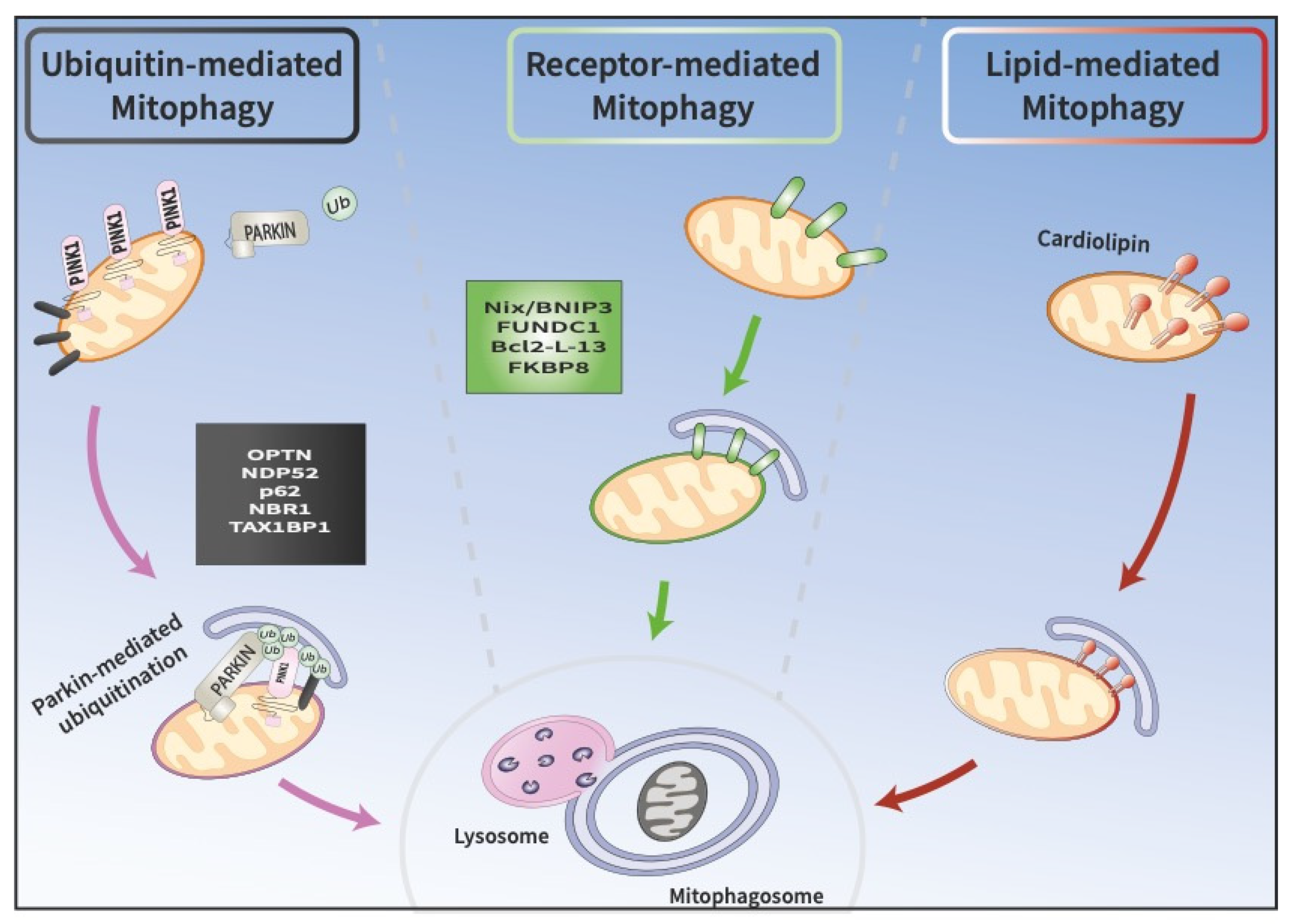
2. Molecular Pathways of Mitophagy
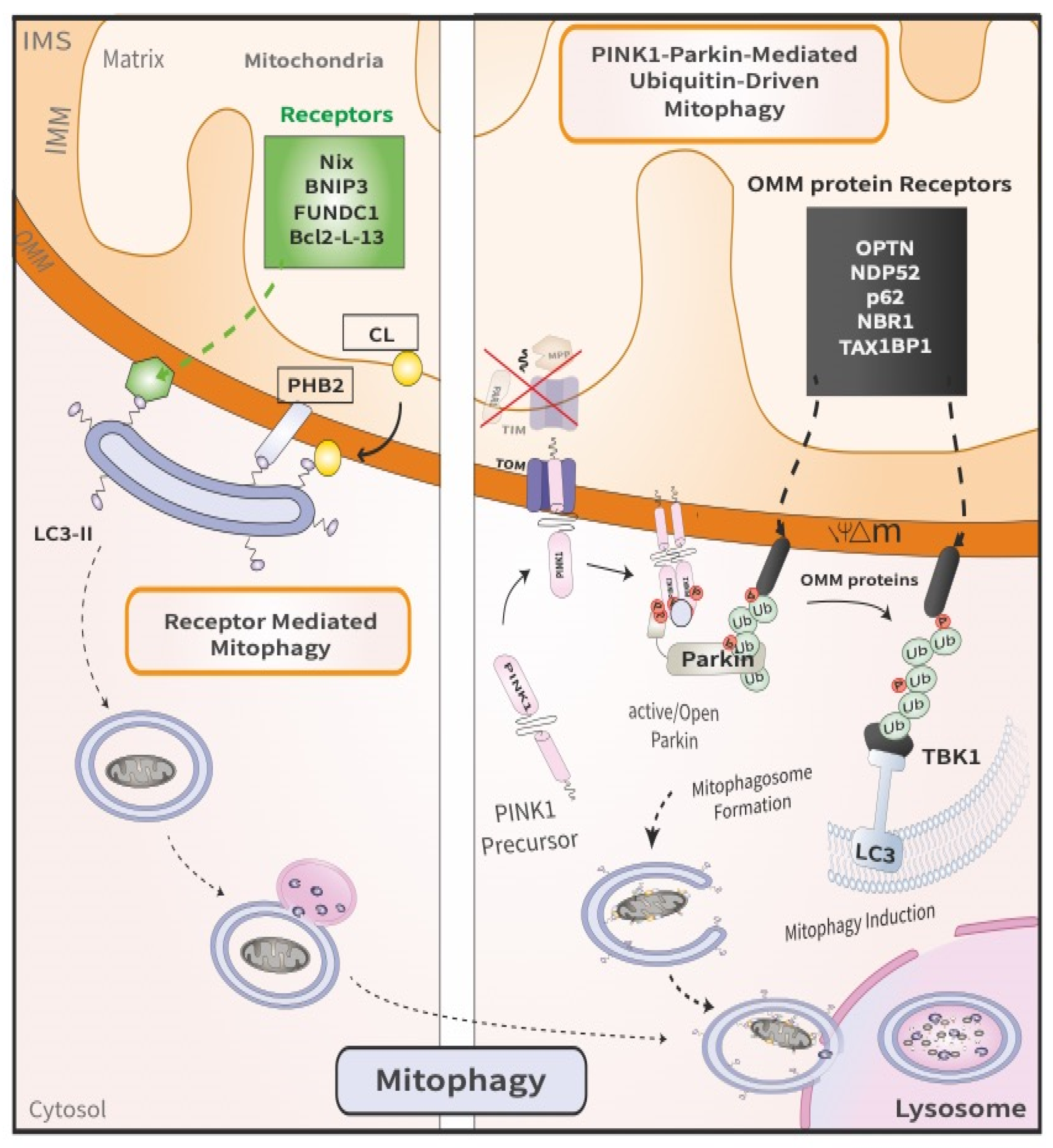
2.1. PINK1/Parkin-Mediated Ubiquitin-Driven Mitophagy
2.2. Receptor Mediated Mitophagy
3. Mitochondrial Defects in PD
3.1. Environmental Toxins as Risk Factors for PD
3.2. Genetic Links to PD Risk
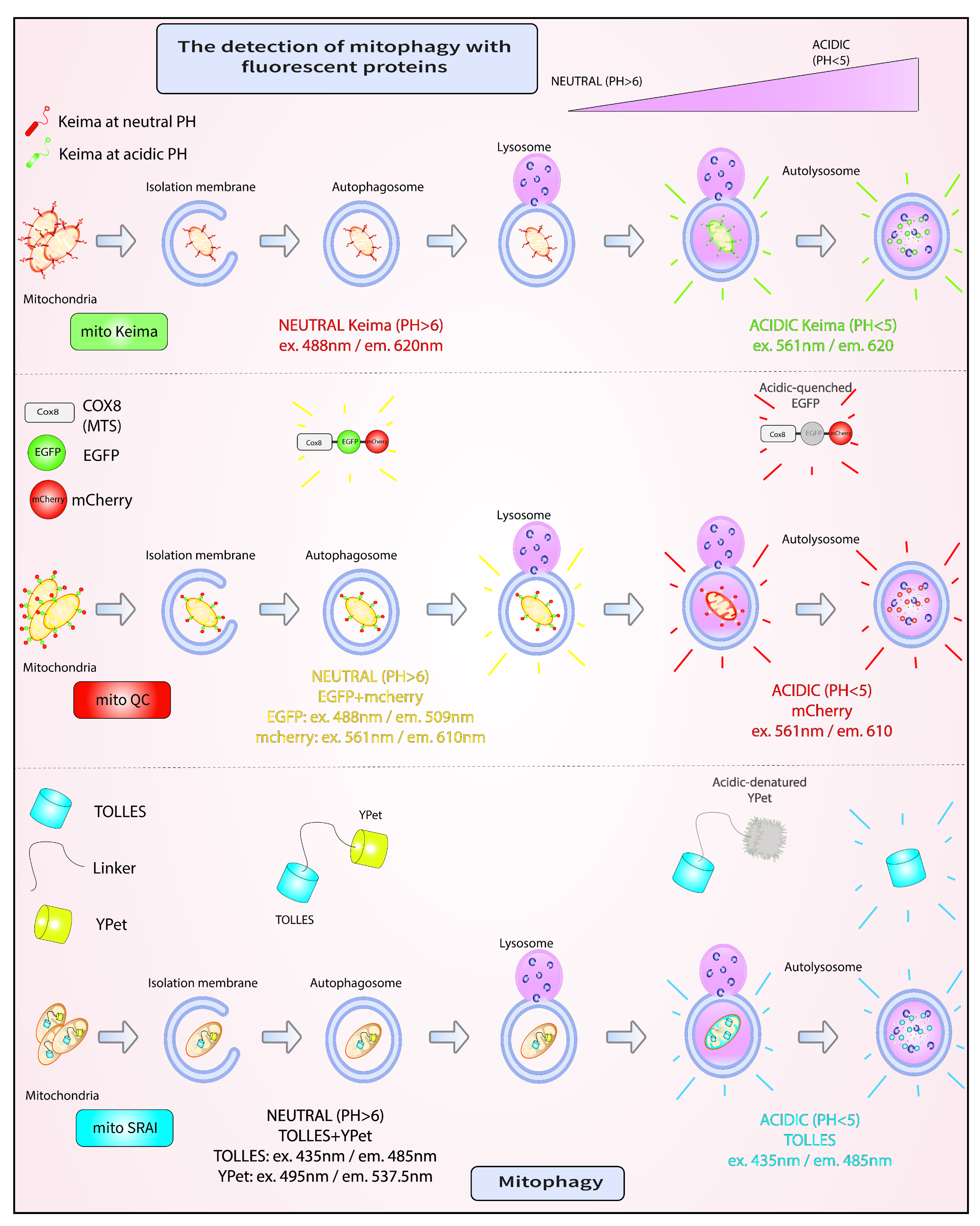
4. Mitophagy Assays
4.1. mito-Keima
4.2. mito-QC
4.3. mito-SRAI
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The Protein Import Machinery of Mitochondria—A Regulatory Hub in Metabolism, Stress, and Disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Hou, J.; Eldeeb, M.; Wang, X. Beyond Deubiquitylation: USP30-Mediated Regulation of Mitochondrial Homeostasis. Mitochondrial DNA Dis. 2017, 1038, 133–148. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Aki, T.; Unuma, K.; Uemura, K. Chemically Induced Models of Parkinson’s Disease: History and Perspectives for the Involvement of Ferroptosis. Front. Cell. Neurosci. 2020, 14, 581191. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Pezzoli, G.; Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 2013, 80, 2035–2041. [Google Scholar] [CrossRef]
- Polito, L.; Greco, A.; Seripa, D. Genetic Profile, Environmental Exposure, and Their Interaction in Parkinson’s Disease. Park. Dis. 2016, 2016, 6465793. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Marchetti, S.; Ricci, J.-E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef]
- Chu, T.C.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [Green Version]
- Springer, M.Z.; MacLeod, K.F. In Brief: Mitophagy: Mechanisms and role in human disease. J. Pathol. 2016, 240, 253–255. [Google Scholar] [CrossRef]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, C.B.; Abbas, N.; Dürr, A.; Bonifati, V.; Bonnet, A.M.; De Broucker, T.; De Michele, G.; Wood, N.W.; Agid, Y.; Brice, A.; et al. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. Lancet 1998, 352, 1355–1356. [Google Scholar] [CrossRef]
- Abbas, N.; Lücking, C.B.; Ricard, S.; Dürr, A.; Bonifati, V.; De Michele, G.; Bouley, S.; Vaughan, J.R.; Gasser, T.; Marconi, R.; et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the european consortium on genetic susceptibility in Parkinson’s Disease. Hum. Mol. Genet 1999, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Arkinson, C.; Walden, H. Parkin function in Parkinson’s disease. Science 2018, 360, 267–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristova, V.A.; Beasley, S.A.; Rylett, R.J.; Shaw, G.S. Identification of a Novel Zn2+-binding Domain in the Autosomal Recessive Juvenile Parkinson-related E3 Ligase Parkin. J. Biol. Chem. 2009, 284, 14978–14986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trempe, J.-F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Muqit, M.M. Ubiquitin and Parkinson’s disease through the looking glass of genetics. Biochem. J. 2017, 474, 1439–1451. [Google Scholar] [CrossRef] [Green Version]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization mechanism to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2010, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A Dimeric PINK1-containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V. PINK1 Kinase Catalytic Activity Is Regulated by Phosphorylation on Serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J. PINK 1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Arano, T.; Matsumoto, G.; Inoshita, T.; Yoshida, S.; Ishihama, Y.; Ryu, K.-Y.; Nukina, N.; Hattori, N.; Imai, Y. Phosphorylation of Mitochondrial Polyubiquitin by PINK1 Promotes Parkin Mitochondrial Tethering. PLoS Genet. 2014, 10, e1004861. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Heo, J.-M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskaite, A.; Martinez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Kumar, A.; Aguirre, J.; Condos, T.E.C.; Martinez-Torres, R.J.; Chaugule, V.; Toth, R.; Sundaramoorthy, R.; Mercier, P.; Knebel, A.; Spratt, D.; et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 2015, 34, 2506–2521. [Google Scholar] [CrossRef]
- Sauvé, V.; Lilov, A.; Seirafi, M.; Vranas, M.; Rasool, S.; Kozlov, G.; Sprules, T.; Wang, J.; Trempe, J.; Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015, 34, 2492–2505. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.; Skujat, D.; Fiesel, F.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Sarraf, S.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nature 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-Anchored Receptor Atg32 Mediates Degradation of Mitochondria via Selective Autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lewis, W.; Diwan, A.; Cheng, E.H.-Y.; Matkovich, S.J.; Dorn, G.W. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 9035–9042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.; Schaeper, U.; Elangovan, B.; D’Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Matsushima, M.; Fujiwara, T.; Takahashi, E.I.; Minaguchi, T.; Eguchi, Y.; Tsujimoto, Y.; Suzumori, K.; Nakamura, Y. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 1998, 21, 230–235. [Google Scholar] [CrossRef]
- Yasuda, M.; Theodorakis, P.; Subramanian, T.; Chinnadurai, G. Adenovirus E1B-19K/BCL-2 Interacting Protein BNIP3 Contains a BH3 Domain and a Mitochondrial Targeting Sequence. J. Biol. Chem. 1998, 273, 12415–12421. [Google Scholar] [CrossRef] [Green Version]
- Imazu, T.; Shimizu, S.; Tagami, S.; Matsushima, M.; Nakamura, Y.; Miki, T.; Okuyama, A.; Tsujimoto, Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 1999, 18, 4523–4529. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2009, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif-crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.-M. Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.E.; Lee, H.J.; Chae, C.W.; Cho, J.H.; Jung, Y.H.; Kim, J.S.; Kim, S.Y.; Lim, J.R.; Han, H.J. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat. Commun. 2021, 12, 487. [Google Scholar] [CrossRef]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at theER–mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Holt, I.J.; Sweeney, M.; Harding, A.E.; Jenner, P.; Marsden, C.D. Mitochondrial DNA analysis in Parkinson’s disease. Mov. Disord. 1990, 5, 294–297. [Google Scholar] [CrossRef]
- Janetzky, B.; Hauck, S.; Youdim, M.B.H.; Riederer, P.; Jellinger, K.; Pantucek, F.; Zöchling, R.; Boissl, K.W.; Reichmann, H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 1994, 169, 126–128. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Jr, W.D.P.; Boyson, S.J.; Ba, J.K.P. Abnormalities of the electron transport chain in idiopathic parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Benecke, R.; Strümper, P.; Weiss, H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain 1993, 116, 1451–1463. [Google Scholar] [CrossRef]
- Bindoff, L.; Birch-Machin, M.; Cartlidge, N.; Parker, W.; Turnbull, D. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef]
- Blin, O.; Desnuelle, C.; Rascol, O.; Borg, M.; Peyro Saint Paul, H.; Azulay, J.-P.; Billè, F.; Figarella, D.; Coulom, F.; Pellissier, J.F.; et al. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci. 1994, 125, 95–101. [Google Scholar] [CrossRef]
- Cardellach, F.; Martí, M.J.; Fernández-Solá, J.; Marín, C.; Hoek, J.B.; Tolosa, E.; Urbano-Márquez, A. Mitochondria1 respiratory chain activity in skeletal muscle from patients with Parkinson’s disease. Neurology 1993, 43, 2258–2262. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H.V.; (The Royal Kings Queens Parkinson Disease Research Group). Platelet mitochondria function in Parkinson’s disease. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s Disease Brain Mitochondrial Complex I Has Oxidatively Damaged Subunits and Is Functionally Impaired and Misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M.; Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Heikkila, R.E.; Manzino, L.; Cabbat, F.S.; Duvoisin, R.C. Protection against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors. Nature 1984, 311, 467–469. [Google Scholar] [CrossRef]
- Gainetdinov, R.; Fumagalli, F.; Jones, S.; Caron, M.G. Dopamine Transporter Is Required for In Vivo MPTP Neurotoxicity: Evidence from Mice Lacking the Transporter. J. Neurochem. 2002, 69, 1322–1325. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Kowal, A.T.; Johnson, M.K.; Salach, J.I.; Singer, T.P. The inhibition site of MPP+, the neurotoxic bioactivation product of 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine is near the Q-binding site of NADH dehydrogenase. Arch. Biochem. Biophys. 1987, 259, 645–649. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone Model of Parkinson Disease. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklas, W.J.; Vyas, I.; Heikkila, R.E. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985, 36, 2503–2508. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Salach, J.I.; Singer, T.P. Uptake of the neurotoxin 1-methyl-4-phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+-linked substrates by MPP+. Biochem. Biophys. Res. Commun. 1986, 134, 743–748. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef] [Green Version]
- Perier, C.; Bove, J.; Wu, D.-C.; Dehay, B.; Choi, D.-K.; Jackson-Lewis, V.; Rathke-Hartlieb, S.; Bouillet, P.; Strasser, A.; Schulz, J.B.; et al. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 8161–8166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perier, C.; Tieu, K.; Guégan, C.; Caspersen, C.; Jackson-Lewis, V.; Carelli, V.; Martinuzzi, A.; Hirano, M.; Przedborski, S.; Vila, M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc. Natl. Acad. Sci. USA 2005, 102, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabre, E.; Monserrat, J.; Herrero, A.; Barja, G.; Leret, M.L. Effect of MPTP on brain mitochondrial H2O2 and ATP production and on dopamine and DOPAC in the striatum. J. Physiol. Biochem. 1999, 55, 325–331. [Google Scholar]
- Chan, P.; DeLanney, L.E.; Irwin, I.; Langston, J.W.; Di Monte, D. Rapid ATP Loss Caused by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in Mouse Brain. J. Neurochem. 1991, 57, 348–351. [Google Scholar] [CrossRef]
- Burté, F.; De Girolamo, L.A.; Hargreaves, A.J.; Billett, E.E. Alterations in the Mitochondrial Proteome of Neuroblastoma Cells in Response to Complex 1 Inhibition. J. Proteome Res. 2011, 10, 1974–1986. [Google Scholar] [CrossRef]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.; Choi, Y.; Park, J.; Nah, W.; Park, J.; Jung, Y.; Lee, J.; Lee, H.; Park, S.; Hwang, S.; et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological α-synuclein. Sci. Adv. 2020, 6, eaba1193. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: Molecular mechanisms and implications for human disease. Cell Death Dis. 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Zilocchi, M.; Finzi, G.; Lualdi, M.; Sessa, F.; Fasano, M.; Alberio, T. Mitochondrial alterations in Parkinson’s disease human samples and cellular models. Neurochem. Int. 2018, 118, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease. Neurology 1997, 48, 1583–1588. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.; Goldman, S.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. NeuroToxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Lapointe, N.; St-Hilaire, M.; Martinoli, M.; Blanchet, J.; Gould, P.; Rouillard, C.; Cicchetti, F. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J. 2004, 18, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.; Féger, J.; Prigent, A.; Michel, P.P.; Parain, K.; Champy, P.; Ruberg, M.; Oertel, W.H.; Hirsch, E. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem. 2003, 84, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and Wild-Type α-Synuclein Interact with Mitochondrial Cytochrome C Oxidase. J. Mol. Neurosci. 2002, 18, 229–238. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [Green Version]
- Robotta, M.; Gerding, H.R.; Vogel, A.; Hauser, K.; Schildknecht, S.; Karreman, C.; Leist, M.; Subramaniam, V.; Drescher, M. Alpha-Synuclein Binds to the Inner Membrane of Mitochondria in an α-Helical Conformation. ChemBioChem 2014, 15, 2499–2502. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Reeve, A.; Meagher, M.; Lax, N.; Simcox, E.; Hepplewhite, P.; Jaros, E.; Turnbull, D. The Impact of Pathogenic Mitochondrial DNA Mutations on Substantia Nigra Neurons. J. Neurosci. 2013, 33, 10790–10801. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; da Cruz, A.B.; Burbulla, L.F.; Lawrence, E.S.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Singh, F.; Prescott, A.R.; Rosewell, P.; Ball, G.; Reith, A.D.; Ganley, I.G. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. eLife 2021, 10, e67604. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Vilas, D.; Taylor, P.; Shaw, L.M.; Tolosa, E.; Trullas, R. Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 2016, 94, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. CCF-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anichtchik, O.; Diekmann, H.; Fleming, A.; Roach, A.; Goldsmith, P.; Rubinsztein, D.C. Loss of PINK1 Function Affects Development and Results in Neurodegeneration in Zebrafish. J. Neurosci. 2008, 28, 8199–8207. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lei, L.; Song, Y.; Liu, M.; Lu, S.; Lou, D.; Shi, Y.; Wang, Z.; He, D. Mutation of hop-1 and pink-1 attenuates vulnerability of neurotoxicity in C. elegans: The role of mitochondria-associated membrane proteins in Parkinsonism. Exp. Neurol. 2018, 309, 67–78. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- von Coelln, R.; Thomas, B.; Savitt, J.M.; Lim, K.L.; Sasaki, M.; Hess, E.J.; Dawson, V.L.; Dawson, T.M. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc. Natl. Acad. Sci. USA 2004, 101, 10744–10749. [Google Scholar] [CrossRef] [Green Version]
- Itier, J.-M.; Ibáñez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient Mice Exhibit Nigrostriatal Deficits but Not Loss of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [Green Version]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2019, 136, 104717. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.; Ottemann, B.M.; Emanuel, K.; Fox, H.S. Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: Concomitant complex I respiratory defects and increased complex II-mediated respiration. Proteom. Clin. Appl. 2016, 10, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.S.; Lee, C.-Y.J.; Lim, E.C.H.; Tan, J.J.H.; Quek, A.M.L.; Chong, W.-L.; Looi, W.-F.; Huang, S.-H.; Wang, H.; Chan, Y.-H. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic. Biol. Med. 2010, 48, 560–566. [Google Scholar] [CrossRef]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and α-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2018, 177, 73–93. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Krohn, L.; Öztürk, T.N.; Vanderperre, B.; Bencheikh, B.O.A.; Msc, J.A.R.; Laurent, S.B.; Spiegelman, D.; Postuma, R.B.; Arnulf, I.; Hu, M.T.M.; et al. Genetic, Structural, and Functional Evidence Link TMEM175 to Synucleinopathies. Ann. Neurol. 2019, 87, 139–153. [Google Scholar] [CrossRef]
- Bras, J.; Guerreiro, R.; Hardy, J. SnapShot: Genetics of Parkinson’s Disease. Cell 2015, 160, 570–570.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Papkovskaia, T.D.; Chau, K.; Inesta-Vaquera, F.; Papkovsky, D.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.; Hardy, J.; Schapira, A.; et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.K.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2011, 21, 1725–1743. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease–associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Apicco, D.J.; Shlevkov, E.; Nezich, C.L.; Tran, D.T.; Guilmette, E.; Nicholatos, J.W.; Bantle, C.M.; Chen, Y.; Glajch, K.E.; Abraham, N.A.; et al. The Parkinson’s disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc. Natl. Acad. Sci. USA 2020, 118, e2006476118. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef] [PubMed]
- Um, J.-H.; Yun, A.J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.H.; de la Torre, A.V.; Hoshikawa, T.; Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 2021, 296, 100209. [Google Scholar] [CrossRef]
- Dolman, N.J.; Chambers, K.M.; Mandavilli, B.; Batchelor, R.H.; Janes, M.S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 2013, 9, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Rice, M. Protocols for Assessing Mitophagy in Neuronal Cell Lines and Primary Neurons. Tech. Investig. Mitochondrial Funct. Neurons 2017, 123, 249–277. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Bonora, M.; Bouhamida, E.; Danese, A.; Marchi, S.; Morciano, G.; Previati, M.; Pedriali, G.; Rimessi, A.; Anania, G.; et al. Methods to Monitor Mitophagy and Mitochondrial Quality: Implications in Cancer, Neurodegeneration, and Cardiovascular Diseases. Methods Mol. Biol. 2021, 2310, 113–159. [Google Scholar] [CrossRef] [PubMed]
- Mauro-Lizcano, M.; Esteban-Martinez, L.; Seco, E.; Serrano-Puebla, A.; Garcia-Ledo, L.; Figueiredo-Pereira, C.; Vieira, H.L.; Boya, P. New method to assess mitophagy flux by flow cytometry. Autophagy 2015, 11, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.H.; Tang, A.M.Y.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-T.; Sliter, D.A.; Shammas, M.K.; Huang, X.; Wang, C.; Calvelli, H.; Maric, D.S.; Narendra, D.P. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 2021, 17, 3753–3762. [Google Scholar] [CrossRef]
- Suzuki, S.; Akamatsu, W.; Kisa, F.; Sone, T.; Ishikawa, K.-I.; Kuzumaki, N.; Katayama, H.; Miyawaki, A.; Hattori, N.; Okano, H. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 2017, 483, 88–93. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.Y.; Um, J.; Yoon, J.; Kim, H.; Lee, D.; Lee, Y.J.; Jee, H.J.; Jang, J.S.; Jang, Y.; Chung, J.; et al. Assessment of mitophagy in mt-Keima Drosophila revealed an essential role of the PINK1-Parkin pathway in mitophagy induction in vivo. FASEB J. 2019, 33, 9742–9751. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A Sensitive and Quantitative Technique for Detecting Autophagic Events Based on Lysosomal Delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Malide, D.; Liu, J.; Rovira, I.I.; A Combs, C.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef]
- Winsor, N.J.; Killackey, S.A.; Philpott, D.J.; Girardin, S.E. An optimized procedure for quantitative analysis of mitophagy with the mtKeima system using flow cytometry. BioTechniques 2020, 69, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- Katayama, H.; Hama, H.; Nagasawa, K.; Kurokawa, H.; Sugiyama, M.; Ando, R.; Funata, M.; Yoshida, N.; Homma, M.; Nishimura, T.; et al. Visualizing and Modulating Mitophagy for Therapeutic Studies of Neurodegeneration. Cell 2020, 181, 1176–1187.e16. [Google Scholar] [CrossRef] [PubMed]
- Khmelinskii, A.; Meurer, M.; Ho, C.-T.; Besenbeck, B.; Fuller, J.; Lemberg, M.K.; Bukau, B.; Mogk, A.; Knop, M. Incomplete proteasomal degradation of green fluorescent proteins in the context of tandem fluorescent protein timers. Mol. Biol. Cell 2016, 27, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; McQuibban, G.A. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Ganley, I.G.; Whitworth, A.J.; McWilliams, T.G. Comment on “mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC”. Autophagy 2021, 17, 4477–4479. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Lin, T.-H.; Bis-Brewer, D.M.; Sheehan, A.E.; Townsend, L.N.; Maddison, D.C.; Züchner, S.; Smith, G.A.; Freeman, M.R. TSG101 negatively regulates mitochondrial biogenesis in axons. Proc. Natl. Acad. Sci. USA 2021, 118, e2018770118. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 2020, 9, e50260. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Hees, J.T.; Wanderoy, S.; Segura, I.; Gibbs, W.; Cheng, Y.; Ordonez, M.; Cai, Z.; Cartoni, R.; Ashrafi, G.; et al. Neuronal mitochondria transport Pink1 mRNA via synaptojanin 2 to support local mitophagy. Neuron 2022, 110, 1516–1531.e9. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Lavie, J.; Bénard, G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 2015, 1853, 2812–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2020, 17, 1232–1243. [Google Scholar] [CrossRef]
- Jiang, W.-X.; Dong, X.; Jiang, J.; Yang, Y.-H.; Yang, J.; Lu, Y.-B.; Fang, S.-H.; Wei, E.-Q.; Tang, C.; Zhang, W.-P. Specific cell surface labeling of GPCRs using split GFP. Sci. Rep. 2016, 6, 20568. [Google Scholar] [CrossRef] [Green Version]
- Koker, T.; Fernandez, A.; Pinaud, F. Characterization of Split Fluorescent Protein Variants and Quantitative Analyses of Their Self-Assembly Process. Sci. Rep. 2018, 8, 5344. [Google Scholar] [CrossRef] [Green Version]
- Cevheroğlu, O.; Kumaş, G.; Hauser, M.; Becker, J.M.; Son, D. The yeast Ste2p G protein-coupled receptor dimerizes on the cell plasma membrane. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 698–711. [Google Scholar] [CrossRef]
- Bader, G.; Enkler, L.; Araiso, Y.; Hemmerle, M.; Binko, K.; Baranowska, E.; De Craene, J.-O.; Ruer-Laventie, J.; Pieters, J.; Tribouillard-Tanvier, D.; et al. Assigning mitochondrial localization of dual localized proteins using a yeast Bi-Genomic Mitochondrial-Split-GFP. eLife 2020, 9, e56649. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goiran, T.; Eldeeb, M.A.; Zorca, C.E.; Fon, E.A. Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease. Cells 2022, 11, 2097. https://doi.org/10.3390/cells11132097
Goiran T, Eldeeb MA, Zorca CE, Fon EA. Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease. Cells. 2022; 11(13):2097. https://doi.org/10.3390/cells11132097
Chicago/Turabian StyleGoiran, Thomas, Mohamed A. Eldeeb, Cornelia E. Zorca, and Edward A. Fon. 2022. "Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease" Cells 11, no. 13: 2097. https://doi.org/10.3390/cells11132097
APA StyleGoiran, T., Eldeeb, M. A., Zorca, C. E., & Fon, E. A. (2022). Hallmarks and Molecular Tools for the Study of Mitophagy in Parkinson’s Disease. Cells, 11(13), 2097. https://doi.org/10.3390/cells11132097