
T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Immunosenescence as a Function
2.1. Immunosenescence Changes with Aging
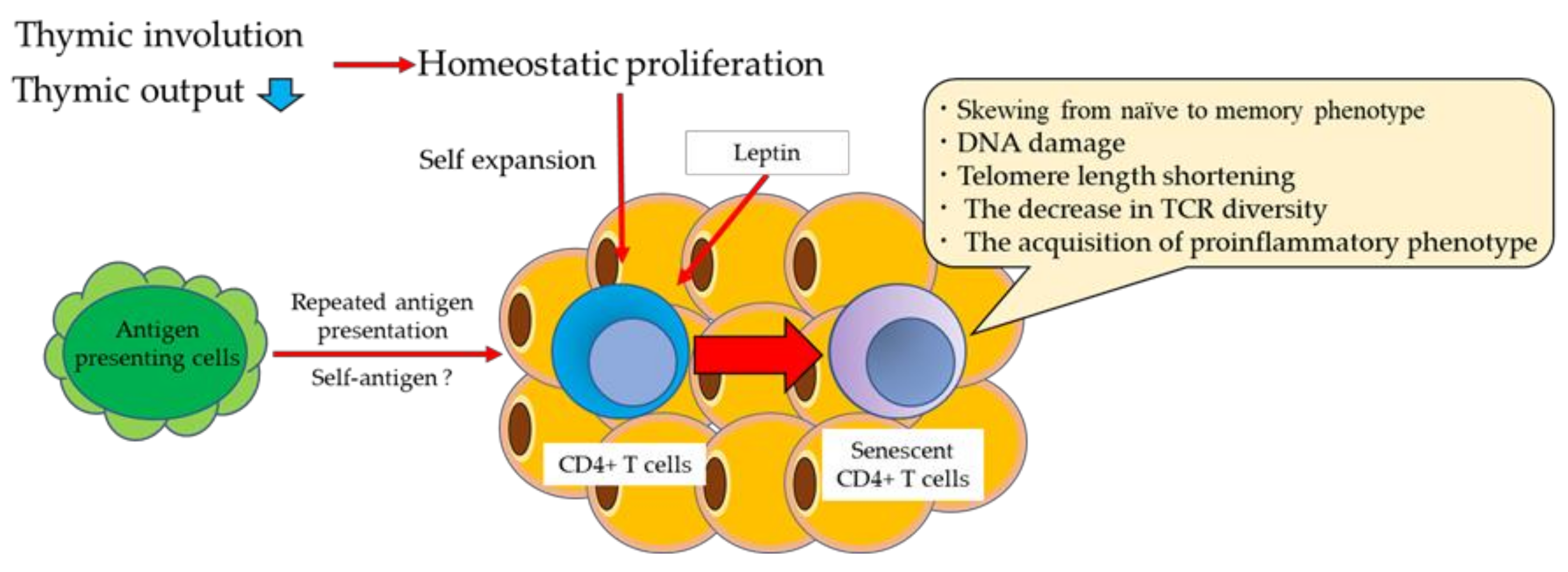
2.2. Thymic Involution with Aging
2.3. Homeostatic Proliferation with Aging
2.4. Phenotypic Changes in T Cells with Aging
2.5. T Cell Subset Found to Increase with Age in Mice
3. Obesity and T Cell Senescence
3.1. Visceral Obesity and Immune Function
3.2. Visceral Obesity and Cellular Senescence
3.3. T Cell Senescence in VAT
3.4. Senescence-Associated CD4+ T Cells in VAT with Obesity
3.5. Effect of Weight Loss on CVD and T Cell Senescence
3.6. Thymic Involution and Homeostatic Proliferation with Obesity
3.7. Metabolic Dysfunction and Continuous Antigen Stimulation with Obesity
3.8. Obesity and B Cell Senescence
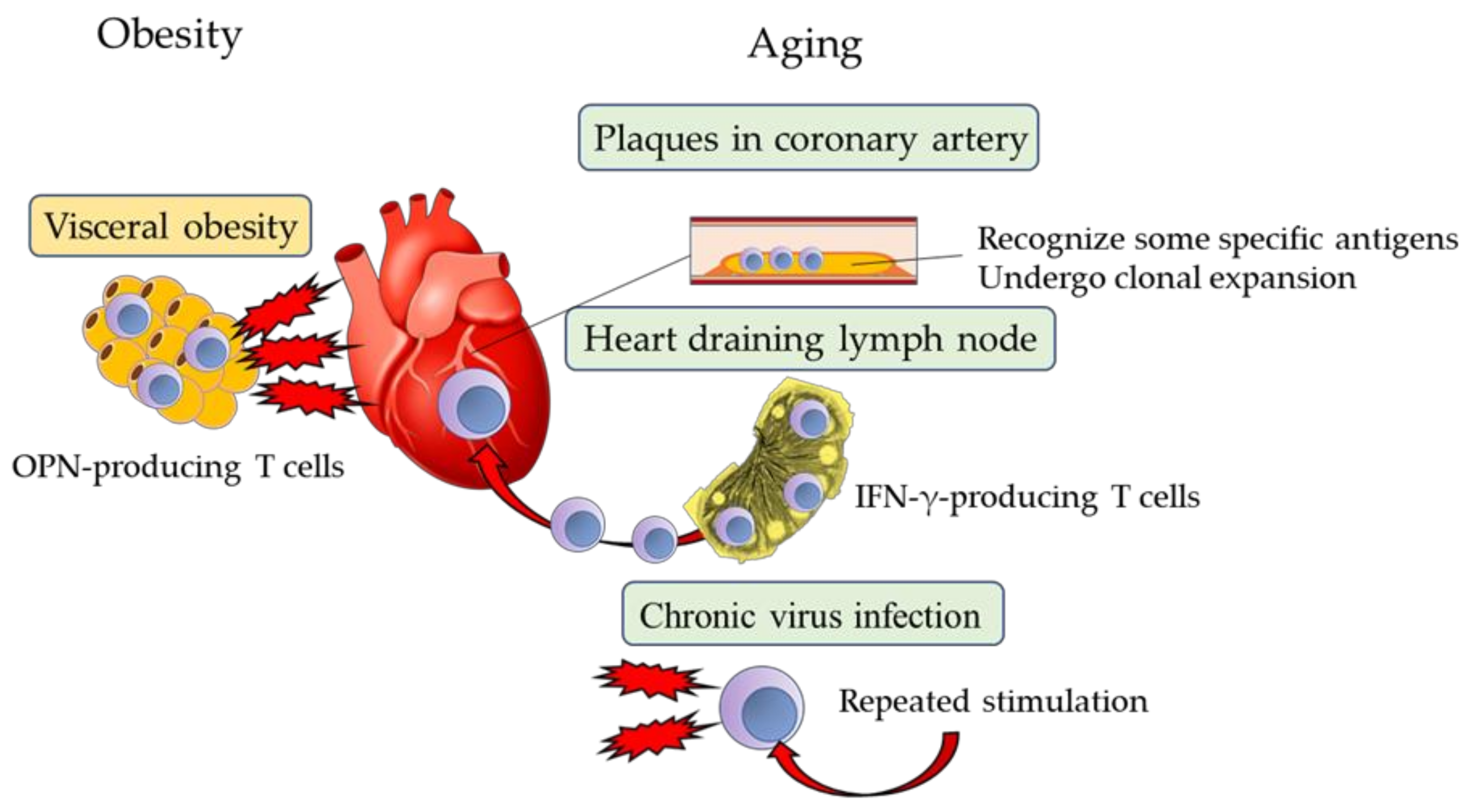
4. Immunosenescence in Cardiovascular Diseases
4.1. Senescence of Human CD4+ T Cells
4.2. Senescence of CD8+ T Cells
4.3. Plasma OPN Levels Predict Prognosis in Patients with Cardiovascular Diseases
4.4. Possible Mechanism of Exacerbation of CVD by OPN
5. Translational Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 1–7. [Google Scholar] [CrossRef]
- Lutgens, E.; Atzler, D.; Döring, Y.; Duchene, J.; Steffens, S.; Weber, C. Immunotherapy for cardiovascular disease. Eur. Heart J. 2019, 40, 3937–3946. [Google Scholar] [CrossRef]
- Hervas, S.M.; González-Navarro, H. Anti-inflammatory Therapies for Cardiovascular Disease: Signaling Pathways and Mecanisms. Rev. Española Cardiol. (Engl. Ed.) 2019, 72, 767–773. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Ridker, P.M. Clinical Application of C-Reactive Protein for Cardiovascular Disease Detection and Prevention. Circulation 2003, 107, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nat. Cell Biol. 2019, 566, 383–387. [Google Scholar] [CrossRef]
- Frodermann, V.; Rohde, D.; Courties, G.; Severe, N.; Schloss, M.J.; Amatullah, H.; McAlpine, C.S.; Cremer, S.; Hoyer, F.F.; Ji, F.; et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019, 25, 1761–1771. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Minamino, T. Role of Cellular Senescence in Lifestyle-Related Disease. Circ. J. 2010, 74, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The role of senescent cells in ageing. Nat. Cell Biol. 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Cavanagh, M.M.; Weyand, C.M.; Goronzy, J.J. Chronic inflammation and aging: DNA damage tips the balance. Curr. Opin. Immunol. 2012, 24, 488–493. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory Cytokines, Aging, and Age-Related Diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Akbar, A.N.; Fletcher, J.M. Memory T cell homeostasis and senescence during aging. Curr. Opin. Immunol. 2005, 17, 480–485. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Successful and Maladaptive T Cell Aging. Immunity 2017, 46, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.A.; Kaouache, M.; Rempel, P.; Joseph, L.; Dawes, M.; Lau, D.C.; Lowensteyn, I. Years of life lost and healthy life-years lost from diabetes and cardiovascular disease in overweight and obese people: A modelling study. Lancet Diabetes Endocrinol. 2015, 3, 114–122. [Google Scholar] [CrossRef]
- Lim, S.; Meigs, J.B. Links Between Ectopic Fat and Vascular Disease in Humans. Arter. Thromb. Vasc. Biol. 2014, 34, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’Ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity 2015, 23, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D. Immunological Goings-on in Visceral Adipose Tissue. Cell Metab. 2013, 17, 851–859. [Google Scholar] [CrossRef]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Tahir, S.; Fukushima, Y.; Sakamoto, K.; Sato, K.; Fujita, H.; Inoue, J.; Uede, T.; Hamazaki, Y.; Hattori, M.; Minato, N. A CD153+CD4+ T Follicular Cell Population with Cell-Senescence Features Plays a Crucial Role in Lupus Pathogenesis via Osteopontin Production. J. Immunol. 2015, 194, 5725–5735. [Google Scholar] [CrossRef]
- Fukushima, Y.; Minato, N.; Hattori, M. The impact of senescence-associated T cells on immunosenescence and age-related disorders. Inflamm. Regen. 2018, 38, 24. [Google Scholar] [CrossRef]
- Minato, N.; Hattori, M.; Hamazaki, Y. Physiology and pathology of T-cell aging. Int. Immunol. 2020, 32, 223–231. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Pawelec, G. Human T Cell Aging and the Impact of Persistent Viral Infections. Front. Immunol. 2013, 4, 271. [Google Scholar] [CrossRef]
- Montecino-Rodriguez, E.; Berent-Maoz, B.; Dorshkind, K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar] [CrossRef]
- Braber, I.D.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mögling, R.; de Boer, A.B.; Willems, N.; Schrijver, E.H.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of Peripheral Naive T Cells Is Sustained by Thymus Output in Mice but Not Humans. Immun. 2012, 36, 288–297. [Google Scholar] [CrossRef]
- Hamazaki, Y. Adult thymic epithelial cell (TEC) progenitors and TEC stem cells: Models and mechanisms for TEC development and maintenance. Eur. J. Immunol. 2015, 45, 2985–2993. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, Y.; Fujita, H.; Kobayashi, T.; Choi, Y.; Scott, H.; Matsumoto, M.; Minato, N. Medullary thymic epithelial cells expressing Aire represent a unique lineage derived from cells expressing claudin. Nat. Immunol. 2007, 8, 304–311. [Google Scholar] [CrossRef]
- Sekai, M.; Hamazaki, Y.; Minato, N. Medullary Thymic Epithelial Stem Cells Maintain a Functional Thymus to Ensure Lifelong Central T Cell Tolerance. Immun. 2014, 41, 753–761. [Google Scholar] [CrossRef]
- Sprent, J.; Surh, C.D. Normal T cell homeostasis: The conversion of naive cells into memory-phenotype cells. Nat. Immunol. 2011, 12, 478–484. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nat. Rev. Immunol. 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Seddon, B.; Yates, A.J. The natural history of naive T cells from birth to maturity. Immunol. Rev. 2018, 285, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Surh, C.D.; Sprent, J. Homeostasis of Naive and Memory T Cells. Immunity 2008, 29, 848–862. [Google Scholar] [CrossRef]
- Dowling, M.; Hodgkin, P.D. Why does the thymus involute? A selection-based hypothesis. Trends Immunol. 2009, 30, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019, 19, 573–583. [Google Scholar] [CrossRef]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.-Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef]
- Rufer, N.; Brümmendorf, T.H.; Kolvraa, S.; Bischoff, C.; Christensen, K.; Wadsworth, L.; Schulzer, M.; Lansdorp, P.M. Telomere Fluorescence Measurements in Granulocytes and T Lymphocyte Subsets Point to a High Turnover of Hematopoietic Stem Cells and Memory T Cells in Early Childhood. J. Exp. Med. 1999, 190, 157–168. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.M.; Correia-Melo, C.; Hardy, T.L.D.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P.; Levine, B.L.; June, C.; Hodes, R.J. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc. Natl. Acad. Sci. USA 1995, 92, 11091–11094. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. Aging of the T Cell Compartment in Mice and Humans: From No Naive Expectations to Foggy Memories. J. Immunol. 2014, 193, 2622–2629. [Google Scholar] [CrossRef]
- Van Kaer, L. Innate and virtual memory T cells in man. Eur. J. Immunol. 2015, 45, 1916–1920. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Eling, N.; Chen, H.-C.; Vallejos, C.A.; Kolodziejczyk, A.A.; Connor, F.; Stojic, L.; Rayner, T.F.; Stubbington, M.J.T.; Teichmann, S.A.; et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 2017, 355, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kato, A.; Sekai, M.; Hamazaki, Y.; Minato, N. Physiologic Thymic Involution Underlies Age-Dependent Accumulation of Senescence-Associated CD4+ T Cells. J. Immunol. 2017, 199, 138–148. [Google Scholar] [CrossRef]
- Groom, L.A.; Sneddon, A.A.; Alessi, D.R.; Dowd, S.; Keyse, S.M. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996, 15, 3621–3632. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Gadeau, A.-P.; Hasan, A.; Abdulrahman, N.; Mraiche, F. Osteopontin: A Promising Therapeutic Target in Cardiac Fibrosis. Cells 2019, 8, 1558. [Google Scholar] [CrossRef] [PubMed]
- Lok, Z.S.Y.; Lyle, A. Osteopontin in Vascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef]
- Waller, A.H.; Sanchez-Ross, M.; Kaluski, E.; Klapholz, M. Osteopontin in Cardiovascular Disease. Cardiol. Rev. 2010, 18, 125–131. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lamort, A.-S.; Giopanou, I.; Psallidas, I.; Stathopoulos, G.T. Osteopontin as a Link between Inflammation and Cancer: The Thorax in the Spotlight. Cells 2019, 8, 815. [Google Scholar] [CrossRef]
- Shimatani, K.; Nakashima, Y.; Hattori, M.; Hamazaki, Y.; Minato, N. PD-1+ memory phenotype CD4+ T cells expressing C/EBPα underlie T cell immunodepression in senescence and leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 15807–15812. [Google Scholar] [CrossRef]
- Lim, S.; Meigs, J.B. Ectopic fat and cardiometabolic and vascular risk. Int. J. Cardiol. 2013, 169, 166–176. [Google Scholar] [CrossRef] [PubMed]
- DiSpirito, J.R.; Mathis, D. Immunological contributions to adipose tissue homeostasis. Semin. Immunol. 2015, 27, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Beck, M.A. The impact of obesity on the immune response to infection. In Proceedings of the Proceedings of the Nutrition Society; Cambridge University Press, 2012; Volume 71, pp. 298–306. [Google Scholar]
- Frebel, H.; Richter, K.; Oxenius, A. How chronic viral infections impact on antigen-specific T-cell responses. Eur. J. Immunol. 2010, 40, 654–663. [Google Scholar] [CrossRef]
- Klenerman, P.; Hill, A.B. T cells and viral persistence: Lessons from diverse infections. Nat. Immunol. 2005, 6, 873–879. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.L.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Mundstock, E.; Sarria, E.E.; Zatti, H.; Louzada, F.M.; Grun, L.K.; Jones, M.; Guma, F.T.C.R.; Memoriam, J.M.; Epifanio, M.; Stein, R.; et al. Effect of obesity on telomere length: Systematic review and meta-analysis. Obesity 2015, 23, 2165–2174. [Google Scholar] [CrossRef]
- Kim, S.; Parks, C.; DeRoo, L.A.; Chen, H.; Taylor, J.; Cawthon, R.M.; Sandler, D.P. Obesity and Weight Gain in Adulthood and Telomere Length. Cancer Epidemiol. Biomark. Prev. 2009, 18, 816–820. [Google Scholar] [CrossRef]
- Lee, M.; Martin, H.; Firpo, M.A.; Demerath, E.W. Inverse association between adiposity and telomere length: The Fels longitudinal study. Am. J. Hum. Biol. 2011, 23, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.; Andrew, T.; Gardner, J.; Kimura, M.; Oelsner, E.; Cherkas, L.; Aviv, A.; Spector, T. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Kaushik, P.; Anderson, J.T. Obesity: Epigenetic aspects. Biomol. Concepts 2016, 7, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, H.; Snieder, H.; Su, S.; Munn, D.; Harshfield, G.; Maria, B.L.; Dong, Y.; Treiber, F.; Gutin, B.; et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010, 8, 1–8. [Google Scholar] [CrossRef]
- Nevalainen, T.; Kananen, L.; Marttila, S.; Jylhävä, J.; Mononen, N.; Kähönen, M.; Raitakari, O.T.; Hervonen, A.; Jylhä, M.; Lehtimäki, T.; et al. Obesity accelerates epigenetic aging in middle-aged but not in elderly individuals. Clin. Epigenetics 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Cho, K.W.; Morris, D.; DelProposto, J.L.; Geletka, L.; Zamarron, B.; Martinez-Santibanez, G.; Meyer, K.A.; Singer, K.; O’Rourke, R.; Lumeng, C.N. An MHC II-Dependent Activation Loop between Adipose Tissue Macrophages and CD4+ T Cells Controls Obesity-Induced Inflammation. Cell Rep. 2014, 9, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Cantrell, D.A. Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 2011, 11, 109–117. [Google Scholar] [CrossRef]
- Strissel, K.J.; DeFuria, J.; Shaul, M.E.; Bennett, G.; Greenberg, A.S.; Obin, M.S. T-Cell Recruitment and Th1 Polarization in Adipose Tissue During Diet-Induced Obesity in C57BL/6 Mice. Obesity 2010, 18, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Youm, Y.-H.; Vandanmagsar, B.; Ravussin, A.; Gimble, J.M.; Greenway, F.; Stephens, J.M.; Mynatt, R.L.; Dixit, V.D. Obesity Increases the Production of Proinflammatory Mediators from Adipose Tissue T Cells and Compromises TCR Repertoire Diversity: Implications for Systemic Inflammation and Insulin Resistance. J. Immunol. 2010, 185, 1836–1845. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- The Look AHEAD Research Group; Wing, R.R.; Bolin, P.; Brancati, F.L.; Bray, G.A.; Clark, J.M.; Coday, M.; Crow, R.S.; Curtis, J.M.; Egan, C.M.; et al. Cardiovascular Effects of Intensive Lifestyle Intervention in Type 2 Diabetes. N. Engl. J. Med. 2013, 369, 145–154. [Google Scholar] [CrossRef]
- Romeo, S.; Maglio, C.; Burza, M.A.; Pirazzi, C.; Sjöholm, K.; Jacobson, P.; Svensson, P.-A.; Peltonen, M.; Sjöström, L.; Carlsson, L.M. Cardiovascular Events After Bariatric Surgery in Obese Subjects With Type 2 Diabetes. Diabetes Care 2012, 35, 2613–2617. [Google Scholar] [CrossRef] [PubMed]
- Harrington, M.; Gibson, S.; Cottrell, R.C. A review and meta-analysis of the effect of weight loss on all-cause mortality risk. Nutr. Res. Rev. 2009, 22, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Endo, J.; Katsumata, Y.; Yamamoto, T.; Kataoka, M.; Isobe, S.; Yoshida, N.; Fukuda, K.; Sano, M. Negative legacy of obesity. PLoS ONE 2017, 12, e0186303. [Google Scholar] [CrossRef]
- Yang, H.; Youm, Y.-H.; Vandanmagsar, B.; Rood, J.; Kumar, K.G.; Butler, A.; Dixit, V.D. Obesity accelerates thymic aging. Blood 2009, 114, 3803–3812. [Google Scholar] [CrossRef]
- Van Der Weerd, K.; Dik, W.A.; Schrijver, B.; Schweitzer, D.H.; Langerak, A.W.; Drexhage, H.A.; Kiewiet-Kemper, R.; Van Aken, M.O.; Van Huisstede, A.; Van Dongen, J.; et al. Morbidly Obese Human Subjects Have Increased Peripheral Blood CD4+ T Cells With Skewing Toward a Treg- and Th2-Dominated Phenotype. Diabetes 2012, 61, 401–408. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Saucillo, D.C.; Gerriets, V.A.; Sheng, J.; Rathmell, J.C.; Maciver, N.J. Leptin Metabolically Licenses T Cells for Activation To Link Nutrition and Immunity. J. Immunol. 2014, 192, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nat. Cell Biol. 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab. 2018, 28, 922–934.e4. [Google Scholar] [CrossRef]
- Morris, D.; Cho, K.W.; DelProposto, J.L.; Oatmen, K.E.; Geletka, L.M.; Martinez-Santibanez, G.; Singer, K.; Lumeng, C.N. Adipose Tissue Macrophages Function As Antigen-Presenting Cells and Regulate Adipose Tissue CD4 + T Cells in Mice. Diabetes 2013, 62, 2762–2772. [Google Scholar] [CrossRef]
- Weltevrede, M.; Eilers, R.; de Melker, H.E.; van Baarle, D. Cytomegalovirus persistence and T-cell immunosenescence in people aged fifty and older: A systematic review. Exp. Gerontol. 2016, 77, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Vazquez, T.; Blomberg, B.B. Obesity induces pro-inflammatory B cells and impairs B cell function in old mice. Mech. Ageing Dev. 2017, 162, 91–99. [Google Scholar] [CrossRef]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Leptin induces immunosenescence in human B cells. Cell. Immunol. 2020, 348, 103994. [Google Scholar] [CrossRef]
- Saule, P.; Trauet, J.; Dutriez, V.; Lekeux, V.; Dessaint, J.-P.; Labalette, M. Accumulation of memory T cells from childhood to old age: Central and effector memory cells in CD4+ versus effector memory and terminally differentiated memory cells in CD8+ compartment. Mech. Ageing Dev. 2006, 127, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yao, D.; Zeng, X.; Kasakovski, D.; Zhang, Y.; Chen, S.; Zha, X.; Li, Y.; Xu, L. Age related human T cell subset evolution and senescence. Immun. Ageing 2019, 16, 24. [Google Scholar] [CrossRef]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Boultby, R.; Butler, R.; Thompson, J.R.; Goodall, A. Telomere shortening in atherosclerosis. Lancet 2001, 358, 472–473. [Google Scholar] [CrossRef]
- Brouilette, S.; Singh, R.K.; Thompson, J.R.; Goodall, A.; Samani, N.J. White Cell Telomere Length and Risk of Premature Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2003, 23, 842–846. [Google Scholar] [CrossRef]
- Liuzzo, G.; Goronzy, J.J.; Yang, H.; Kopecky, S.L.; Holmes, D.R.; Frye, R.L.; Weyand, C.M. Monoclonal T-Cell Proliferation and Plaque Instability in Acute Coronary Syndromes. Circ. 2000, 101, 2883–2888. [Google Scholar] [CrossRef] [PubMed]
- Giubilato, S.; Liuzzo, G.; Brugaletta, S.; Pitocco, D.; Graziani, F.; Smaldone, C.; Montone, R.A.; Pazzano, V.; Pedicino, D.; Biasucci, L.M.; et al. Expansion of CD4+CD28null T-lymphocytes in diabetic patients: Exploring new pathogenetic mechanisms of increased cardiovascular risk in diabetes mellitus. Eur. Heart J. 2011, 32, 1214–1226. [Google Scholar] [CrossRef]
- Liuzzo, G.; Kopecky, S.L.; Frye, R.L.; Fallon, W.M.O.; Maseri, A.; Goronzy, J.J.; Weyand, C.M. Perturbation of the T-Cell Repertoire in Patients With Unstable Angina. Circulation 1999, 100, 2135–2139. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Trotta, G.; Brugaletta, S.; Pinnelli, M.; Di Giannuario, G.; Rizzello, V.; Rebuzzi, A.G.; Rumi, C.; Maseri, A.; et al. Unusual CD4+CD28nullT Lymphocytes and Recurrence of Acute Coronary Events. J. Am. Coll. Cardiol. 2007, 50, 1450–1458. [Google Scholar] [CrossRef]
- Ramos, G.C.; Berg, A.V.D.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeßer, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell–mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed]
- Delgobo, M.; Heinrichs, M.; Hapke, N.; Ashour, D.; Appel, M.; Srivastava, M.; Heckel, T.; Spyridopoulos, I.; Hofmann, U.; Frantz, S.; et al. Terminally Differentiated CD4+ T Cells Promote Myocardial Inflammaging. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.D.; Battistini, L.; Akbar, A.N. Reversible Senescence in Human CD4+CD45RA+CD27− Memory T Cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Henson, S.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- Lanna, A.; Gomes, D.C.O.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.; Lee, J.H.; Karin, M.; Akbar, A.N. A sestrin-dependent Erk–Jnk–p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A.H. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.T.; Youn, J.-C.; Lee, J.; Park, S.; Chi, H.-S.; Lee, J.; Choi, C.; Park, S.; Choi, D.; Ha, J.-W.; et al. Characterization of CD8+CD57+ T cells in patients with acute myocardial infarction. Cell. Mol. Immunol. 2014, 12, 466–473. [Google Scholar] [CrossRef]
- Kaplan, R.C.; Sinclair, E.; Landay, A.L.; Lurain, N.; Sharrett, A.R.; Gange, S.J.; Xue, X.; Hunt, P.; Karim, R.; Kern, D.M.; et al. T Cell Activation and Senescence Predict Subclinical Carotid Artery Disease in HIV-Infected Women. J. Infect. Dis. 2011, 203, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.R.; Wolf, J.; Brunner, S.; Herndler-Brandstetter, D.; Grubeck-Loebenstein, B. Gain and Loss of T Cell Subsets in Old Age—Age-Related Reshaping of the T Cell Repertoire. J. Clin. Immunol. 2011, 31, 137–146. [Google Scholar] [CrossRef]
- Brunner, S.; Herndler-Brandstetter, D.; Weinberger, B.; Grubeck-Loebenstein, B. Persistent viral infections and immune aging. Ageing Res. Rev. 2011, 10, 362–369. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Hoffmann, J.; Aicher, A.; Brümmendorf, T.H.; Doerr, H.W.; Zeiher, A.M.; Dimmeler, S. Accelerated Telomere Shortening in Leukocyte Subpopulations of Patients With Coronary Heart Disease. Circulation 2009, 120, 1364–1372. [Google Scholar] [CrossRef]
- Lachmann, R.; Bajwa, M.; Vita, S.; Smith, H.; Cheek, E.; Akbar, A.; Kern, F. Polyfunctional T Cells Accumulate in Large Human Cytomegalovirus-Specific T Cell Responses. J. Virol. 2011, 86, 1001–1009. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Lin, C.-H.; Sung, B.-Y.; Chuang, Y.-F.; Schneck, J.P.; Kern, F.; Pawelec, G.; Wang, G.C. Cytotoxic polyfunctionality maturation of cytomegalovirus-pp65-specific CD4 + and CD8 + T-cell responses in older adults positively correlates with response size. Sci. Rep. 2016, 6, 19227. [Google Scholar] [CrossRef]
- Makedonas, G.; Banerjee, P.P.; Pandey, R.; Hersperger, A.R.; Sanborn, K.B.; Hardy, G.A.D.; Orange, J.S.; Betts, M.R. Rapid Up-Regulation and Granule-Independent Transport of Perforin to the Immunological Synapse Define a Novel Mechanism of Antigen-Specific CD8+ T Cell Cytotoxic Activity. J. Immunol. 2009, 182, 5560–5569. [Google Scholar] [CrossRef]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.A.; Mraiche, F. Targeting Osteopontin, the Silent Partner of Na+/H+ Exchanger Isoform 1 in Cardiac Remodeling. J. Cell. Physiol. 2015, 230, 2006–2018. [Google Scholar] [CrossRef]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound healing and tissue remodeling after injury. J. Burn. Care Res. 2012, 33, 311–318. [Google Scholar] [CrossRef]
- Singh, M.; Foster, C.R.; Dalal, S.; Singh, K. Osteopontin: Role in extracellular matrix deposition and myocardial remodeling post-MI. J. Mol. Cell. Cardiol. 2010, 48, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Sano, M. Sodium-Glucose Co-Transporter 2 Inhibitors Correct Metabolic Maladaptation of Proximal Tubular Epithelial Cells in High-Glucose Conditions. Int. J. Mol. Sci. 2020, 21, 7676. [Google Scholar] [CrossRef]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10–STAT3–Galectin-3 Axis Is Essential for Osteopontin-Producing Reparative Macrophage Polarization After Myocardial Infarction. Circulation 2018, 138, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Anzai, A.; Moriyama, H.; Kitakata, H.; Hiraide, T.; Ko, S.; Goto, S.; et al. MerTK Expression and ERK Activation Are Essential for the Functional Maturation of Osteopontin-Producing Reparative Macrophages After Myocardial Infarction. J. Am. Heart Assoc. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kaleta, B. The role of osteopontin in kidney diseases. Inflamm. Res. 2018, 68, 93–102. [Google Scholar] [CrossRef]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction Through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef]
- Klingel, K.; Kandolf, R. Osteopontin: A Biomarker to Predict the Outcome of Inflammatory Heart Disease. Semin. Thromb. Hemost. 2010, 36, 195–202. [Google Scholar] [CrossRef]
- Abdalrhim, A.D.; Marroush, T.S.; Austin, E.E.; Gersh, B.J.; Solak, N.; Rizvi, S.A.; Bailey, K.R.; Kullo, I.J. Plasma Osteopontin Levels and Adverse Cardiovascular Outcomes in the PEACE Trial. PLoS ONE 2016, 11, e0156965. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Shingai, M.; Aso, N.; Hazuku, T.; Nasu, M. Osteopontin is Released From the Heart Into the Coronary Circulation in Patients With a Previous Anterior Wall Myocardial Infarction. Circ. J. 2003, 67, 742–744. [Google Scholar] [CrossRef][Green Version]
- Subramanian, V.; Krishnamurthy, P.; Singh, K.; Singh, M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am. J. Physiol. Circ. Physiol. 2007, 292, H673–H683. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Balligand, J.-L.; Fischer, T.A.; Smith, T.W.; Kelly, R.A. Glucocorticoids Increase Osteopontin Expression in Cardiac Myocytes and Microvascular Endothelial Cells. J. Biol. Chem. 1995, 270, 28471–28478. [Google Scholar] [CrossRef]
- Maniatis, K.; Siasos, G.; Oikonomou, E.; Vavuranakis, M.; Zaromytidou, M.; Mourouzis, K.; Paraskevopoulos, T.; Charalambous, G.; Papavassiliou, A.G.; Tousoulis, D. Osteoprotegerin and Osteopontin Serum Levels are Associated with Vascular Function and Inflammation in Coronary Artery Disease Patients. Curr. Vasc. Pharmacol. 2020, 18, 523–530. [Google Scholar] [CrossRef]
- Kitagori, K.; Yoshifuji, H.; Oku, T.; Sasaki, C.; Miyata, H.; Mori, K.P.; Nakajima, T.; Ohmura, K.; Kawabata, D.; Yukawa, N.; et al. Cleaved Form of Osteopontin in Urine as a Clinical Marker of Lupus Nephritis. PLoS ONE 2016, 11, e0167141. [Google Scholar] [CrossRef]
- Rosenberg, M.; Zugck, C.; Nelles, M.; Juenger, C.; Frank, D.; Remppis, A.; Giannitsis, E.; Katus, H.A.; Frey, N. Osteopontin, a New Prognostic Biomarker in Patients With Chronic Heart Failure. Circ. Heart Fail. 2008, 1, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Suezawa, C.; Kusachi, S.; Murakami, T.; Toeda, K.; Hirohata, S.; Nakamura, K.; Yamamoto, K.; Koten, K.; Miyoshi, T.; Shiratori, Y. Time-dependent changes in plasma osteopontin levels in patients with anterior-wall acute myocardial infarction after successful reperfusion: Correlation with left-ventricular volume and function. J. Lab. Clin. Med. 2005, 145, 33–40. [Google Scholar] [CrossRef]
- Strobescu-Ciobanu, C.; Giuşcă, S.E.; Căruntu, I.-D.; Amălinei, C.; Rusu, A.; Cojocaru, E.; Popa, R.F.; Lupaşcu, C.D. Osteopontin and osteoprotegerin in atherosclerotic plaque – are they significant markers of plaque vulnerability? Rom. J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2021, 61, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.W.; Lee, S.J.; Ye, B.H.; Kim, Y.W.; Bae, S.S.; Kim, C.D. Mechanical stretch enhances the expression and activity of osteopontin and MMP-2 via the Akt1/AP-1 pathways in VSMC. J. Mol. Cell. Cardiol. 2015, 85, 13–24. [Google Scholar] [CrossRef]
- Toemen, L.; Santos, S.; Roest, A.A.W.; Vernooij, M.W.; Helbing, W.A.; Gaillard, R.; Jaddoe, V.W.V. Pericardial adipose tissue, cardiac structures, and cardiovascular risk factors in school-age children. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 307–313. [Google Scholar] [CrossRef]
- Kenchaiah, S.; Ding, J.; Carr, J.J.; Allison, M.A.; Budoff, M.J.; Tracy, R.P.; Burke, G.L.; McClelland, R.L.; Arai, A.E.; Bluemke, D.A. Pericardial Fat and the Risk of Heart Failure. J. Am. Coll. Cardiol. 2021, 77, 2638–2652. [Google Scholar] [CrossRef]
- Hirata, Y.; Yamada, H.; Sata, M. Epicardial Fat and Pericardial Fat Surrounding the Heart Have Different Characteristics. Circ. J. 2018, 82, 2475–2476. [Google Scholar] [CrossRef]
- Luna-Luna, M.; Criales-Vera, S.; Medina-Leyte, D.; Díaz-Zamudio, M.; Flores-Zapata, A.; Cruz-Robles, D.; López-Meneses, M.; Olvera-Cruz, S.; Ramírez-Marroquín, S.; Flores-Castillo, C.; et al. Bone Morphogenetic Protein-2 and Osteopontin Gene Expression in Epicardial Adipose Tissue from Patients with Coronary Artery Disease Is Associated with the Presence of Calcified Atherosclerotic Plaques. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1943–1951. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Schauerte, C.; Hübner, A.; Kölling, M.; Martino, F.; Scherf, K.; Batkai, S.; Zimmer, K.; Foinquinos, A.; Kaucsar, T.; et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur. Heart J. 2015, 36, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Mann, D.; Entman, M.L.; Spinale, F.G. Extracellular matrix remodeling following myocardial injury. Ann. Med. 2003, 35, 316–326. [Google Scholar] [CrossRef]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-Regulation and Profibrotic Role of Osteopontin in Human Idiopathic Pulmonary Fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef]
- Xie, Z.; Singh, M.; Siwik, D.A.; Joyner, W.L.; Singh, K. Osteopontin Inhibits Interleukin-1β-stimulated Increases in Matrix Metalloproteinase Activity in Adult Rat Cardiac Fibroblasts. J. Biol. Chem. 2003, 278, 48546–48552. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Goverman, J. Osteopontin-induced survival of T cells. Nat. Immunol. 2007, 8, 19–20. [Google Scholar] [CrossRef]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Ramirez, E.; Florian, M.C. Understanding intrinsic hematopoietic stem cell aging. Haematologica 2019, 105, 22–37. [Google Scholar] [CrossRef]
- Guidi, N.; Sacma, M.; Ständker, L.; Soller, K.; Marka, G.; Eiwen, K.; Weiss, J.; Kirchhoff, F.; Weil, T.; Cancelas, J.A.; et al. Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 2017, 36, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An Overview and Update on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Macaulay, R.; Riddell, N.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8+T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirakawa, K.; Sano, M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells 2021, 10, 2435. https://doi.org/10.3390/cells10092435
Shirakawa K, Sano M. T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells. 2021; 10(9):2435. https://doi.org/10.3390/cells10092435
Chicago/Turabian StyleShirakawa, Kohsuke, and Motoaki Sano. 2021. "T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease" Cells 10, no. 9: 2435. https://doi.org/10.3390/cells10092435
APA StyleShirakawa, K., & Sano, M. (2021). T Cell Immunosenescence in Aging, Obesity, and Cardiovascular Disease. Cells, 10(9), 2435. https://doi.org/10.3390/cells10092435