
Strategic Development of an Immunotoxin for the Treatment of Glioblastoma and Other Tumours Expressing the Calcitonin Receptor


{kind=link}
Abstract
:1. Introduction
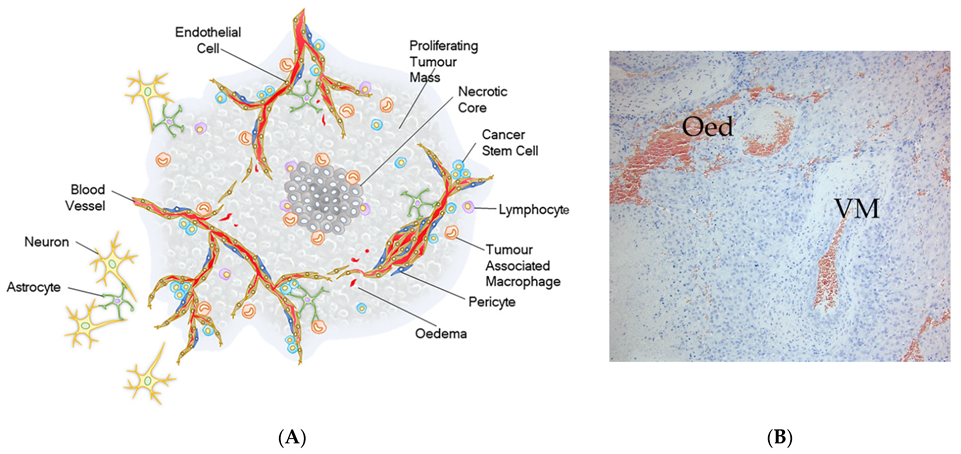
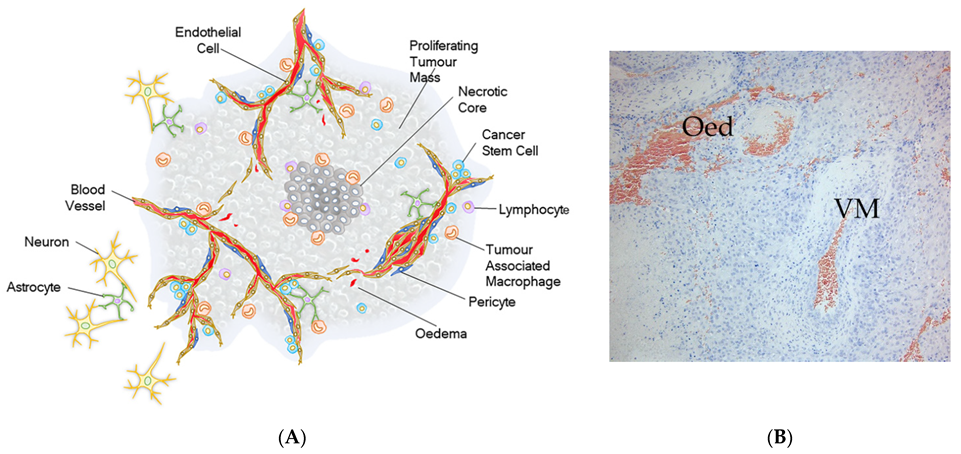
2. Tumour Hierarchy of Glioblastoma
3. Cancer Stem Cells
4. Quiescent Brain Tumour Stem Cells
5. The Elephant in the Room: Targeting Quiescent Glioma Stem Cells (GSCs) and High-Grade Glioma (HGG) Cell Lines
6. Expression of Calcitonin (CT) Receptor, a G Protein-Coupled Receptor, in HGG Cell Lines
7. An Immunotoxin That Binds CT Receptor
8. Further Challenges of Solid Tumours, in Particular, Glioblastoma
8.1. Nanobodies
8.2. Designed Ankyrin Repeat Proteins (DARPins)
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Sun, D.; Chen, Y.-J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e8. [Google Scholar] [CrossRef]
- Noble, M.; Gutowski, N.; Bevan, K.; Engel, U.; Linskey, M.; Urenjak, J.; Bhakoo, K.; Williams, S. From rodent glial precursor cell to human glial neoplasia in the oligodendrocyte-type-2-astrocyte lineage. Glia 1995, 15, 222–230. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.; Scott, M.P. The Developmental Biology of Brain Tumors. Annu. Rev. Neurosci. 2001, 24, 385–428. [Google Scholar] [CrossRef] [PubMed]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Furness, S.G.B.; Bittencourt, L.; Hare, D.L.; Wookey, P.J. Building the case for the calcitonin receptor as a viable target for the treatment of glioblastoma. Ther. Adv. Med. Oncol. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Lee, F.-T.; Tebbutt, N.; Herbertson, R.; Gill, S.S.; Liu, Z.; Skrinos, E.; Murone, C.; Saunder, T.; Chappell, B.; et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 4071–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Strasser, A. Is Tumor Growth Sustained by Rare Cancer Stem Cells or Dominant Clones? Cancer Res. 2008, 68, 4018–4021. [Google Scholar] [CrossRef] [Green Version]
- Dore-Duffy, P. Pericytes: Pluripotent Cells of the Blood Brain Barrier. Curr. Pharm. Des. 2008, 14, 1581–1593. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Bruce, W.R.; Van Der Gaag, H. A Quantitative Assay for the Number of Murine Lymphoma Cells capable of Proliferation in vivo. Nature 1963, 199, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E.; et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Wang, J.; Dick, J. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef]
- Lutz, C.; Woll, P.S.; Hall, G.; Castor, A.; Dreau, H.; Cazzaniga, G.; Zuna, J.; Jensen, C.; Clark, S.A.; Biondi, A.; et al. Quiescent leukaemic cells account for minimal residual disease in childhood lymphoblastic leukaemia. Leukemia 2012, 27, 1204–1207. [Google Scholar] [CrossRef]
- Dick, J.E. Breast cancer stem cells revealed. Proc. Natl. Acad. Sci. USA 2003, 100, 3547–3549. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Wookey, P.J.; McLean, C.A.; Hwang, P.; Furness, S.G.B.; Nguyen, S.; Kourakis, A.; Hare, D.L.; Rosenfeld, J.V. The expression of calcitonin receptor detected in malignant cells of the brain tumour glioblastoma multiforme and functional properties in the cell line A172. Histopathology 2012, 60, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Read, T.-A.; Fogarty, M.P.; Markant, S.L.; McLendon, R.E.; Wei, Z.; Ellison, D.W.; Febbo, P.G.; Wechsler-Reya, R.J. Identification of CD15 as a Marker for Tumor-Propagating Cells in a Mouse Model of Medulloblastoma. Cancer Cell 2009, 15, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.J.; Lee, L.; Graham, K.; Satkunendran, T.; Yoshikawa, K.; Ling, E.; Harper, L.; Austin, R.; Nieuwenhuis, E.; Clarke, I.D.; et al. Multipotent CD15+ Cancer Stem Cells in Patched-1–Deficient Mouse Medulloblastoma. Cancer Res. 2009, 69, 4682–4690. [Google Scholar] [CrossRef] [Green Version]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem. Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; O’Brien, C.A.; Van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.K.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.S. Molecular Pathways: Targeting Cancer Stem Cells Awakened by Chemotherapy to Abrogate Tumor Repopulation. Clin. Cancer Res. 2016, 22, 802–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly Teamwork: Neural Cancer Stem Cells and the Tumor Microenvironment. Cell Stem. Cell 2011, 8, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes Enhance the Invasion Potential of Glioblastoma Stem-Like Cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Parada, L.F.; Dirks, P.B.; Wechsler-Reya, R.J. Brain Tumor Stem Cells Remain in Play. J. Clin. Oncol. 2017, 35, 2428–2431. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; DiMeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Day, B.W.; Stringer, B.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, B.W.; Day, B.W.; D’Souza, R.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgul, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wookey, P.; Zulli, A.; Lo, C.; Hare, D.; Schwarer, A.; Darby, I.; Leung, A. Calcitonin receptor (CTR) expression in embryonic, foetal and adult tissues: Developmental and pathophysiological implications. In The Calcitonin Gene-Related Peptide Family: Form, Function and Future Perspectives; Hay, D., Dickerson, I., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 199–233. [Google Scholar]
- Dal Maso, E.; Glukhova, A.; Zhu, Y.; Garcia-Nafria, J.; Tate, C.G.; Atanasio, S.; Reynolds, C.A.; Ramírez-Aportela, E.; Carazo, J.-M.; Hick, C.A.; et al. The Molecular Control of Calcitonin Receptor Signaling. ACS Pharmacol. Transl. Sci. 2019, 2, 31–51. [Google Scholar] [CrossRef]
- Dal Maso, E.; Just, R.; Hick, C.; Christopoulos, A.; Sexton, P.; Wootten, D.; Furness, S.G. Characterization of signalling and regulation of common calcitonin receptor splice variants and polymorphisms. Biochem. Pharmacol. 2018, 148, 111–129. [Google Scholar] [CrossRef]
- Furness, S.G.B.; Liang, Y.-L.; Nowell, C.; Halls, M.L.; Wookey, P.; Maso, E.D.; Inoue, A.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Ligand-Dependent Modulation of G Protein Conformation Alters Drug Efficacy. Cell 2016, 167, 739–749.e11. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Pondel, M.D.; Partington, G.A.; Mould, R. Tissue-specific activity of the proximal human calcitonin receptor promoter is mediated by Sp1 and an epigenetic phenomenon. FEBS Lett. 2003, 554, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Gilabert-Oriol, R.; Furness, S.G.B.; Stringer, B.W.; Weng, A.; Fuchs, H.; Day, B.W.; Kourakis, A.; Boyd, A.W.; Hare, D.L.; Thakur, M.; et al. Dianthin-30 or gelonin versus monomethyl auristatin E, each configured with an anti-calcitonin receptor antibody, are differentially potent in vitro in high-grade glioma cell lines derived from glioblastoma. Cancer Immunol. Immunother. 2017, 66, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Ostrovskaya, A.; Hick, C.; Hutchinson, D.S.; Stringer, B.W.; Wookey, P.J.; Wootten, D.; Sexton, P.M.; Furness, S.G.B. Expression and activity of the calcitonin receptor family in a sample of primary human high-grade gliomas. BMC Cancer 2019, 19, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Akhurst, T.; Lee, F.T.; Ciprotti, M.; Davis, I.D.; Weickhardt, A.J.; Gan, H.K.; Hicks, R.J.; Lee, S.T.; Kocovski, P.; et al. First clinical study of a pegylated diabody (124)I-labeled PEG-AVP0458 in patients with tumor-associated glycoprotein 72 positive cancers. Theranostics 2020, 10, 11404–11415. [Google Scholar] [CrossRef]
- Fernandes, C.F.C.; Pereira, S.D.S.; Luiz, M.B.; Zuliani, J.P.; Furtado, G.P.; Stabeli, R.G. Camelid Single-Domain Antibodies As an Alternative to Overcome Challenges Related to the Prevention, Detection, and Control of Neglected Tropical Diseases. Front. Immunol. 2017, 8, 653. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharm. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Boersma, Y.L. Advances in the Application of Designed Ankyrin Repeat Proteins (DARPins) as Research Tools and Protein Therapeutics. Methods Mol. Biol. 2018, 1798, 307–327. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, P.; Hare, D.L.; Wookey, P.J. Strategic Development of an Immunotoxin for the Treatment of Glioblastoma and Other Tumours Expressing the Calcitonin Receptor. Cells 2021, 10, 2347. https://doi.org/10.3390/cells10092347
Gupta P, Hare DL, Wookey PJ. Strategic Development of an Immunotoxin for the Treatment of Glioblastoma and Other Tumours Expressing the Calcitonin Receptor. Cells. 2021; 10(9):2347. https://doi.org/10.3390/cells10092347
Chicago/Turabian StyleGupta, Pragya, David L. Hare, and Peter J. Wookey. 2021. "Strategic Development of an Immunotoxin for the Treatment of Glioblastoma and Other Tumours Expressing the Calcitonin Receptor" Cells 10, no. 9: 2347. https://doi.org/10.3390/cells10092347
APA StyleGupta, P., Hare, D. L., & Wookey, P. J. (2021). Strategic Development of an Immunotoxin for the Treatment of Glioblastoma and Other Tumours Expressing the Calcitonin Receptor. Cells, 10(9), 2347. https://doi.org/10.3390/cells10092347