
D-2-Hydroxyglutarate in Glioma Biology



Abstract
:1. Introduction
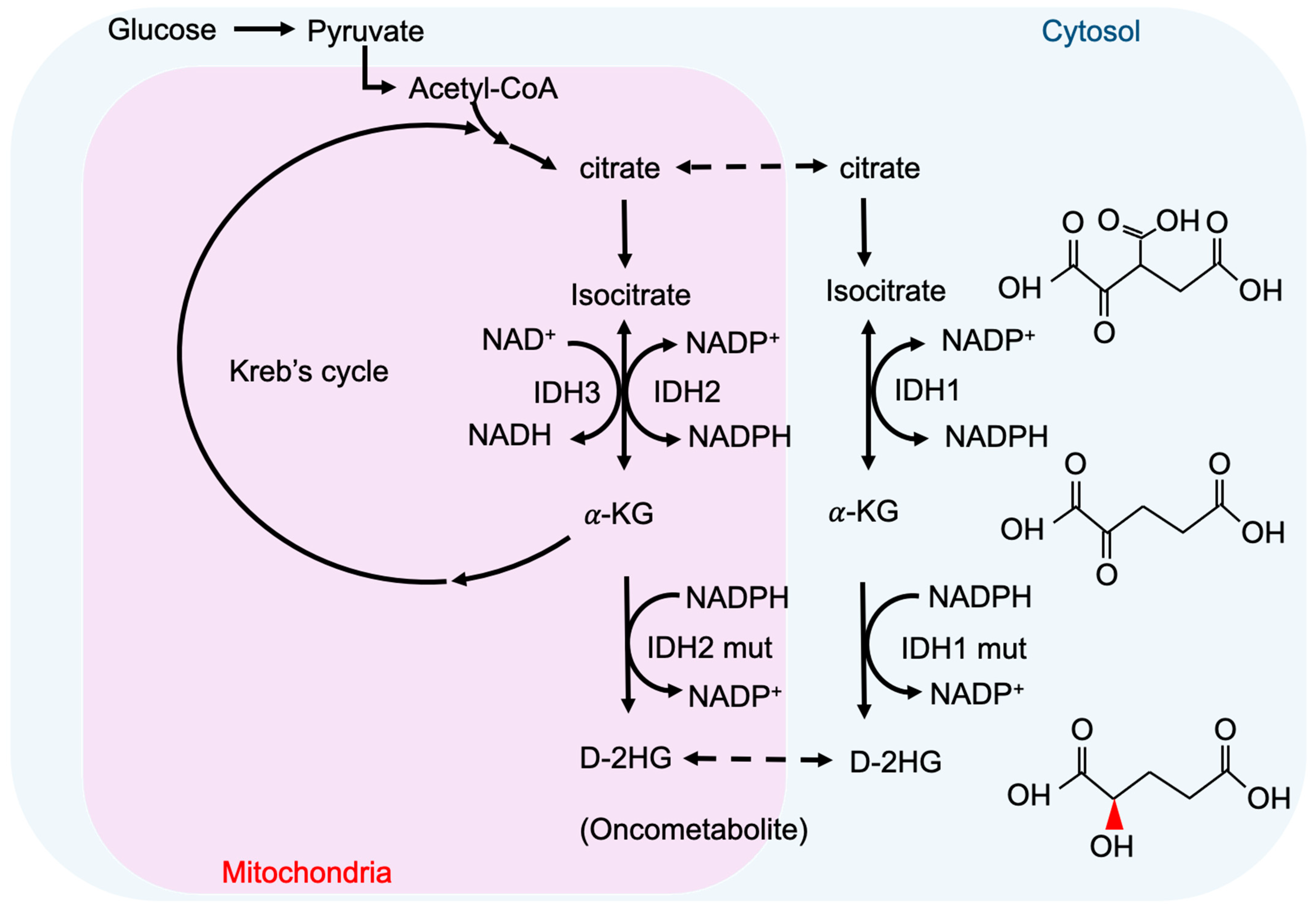
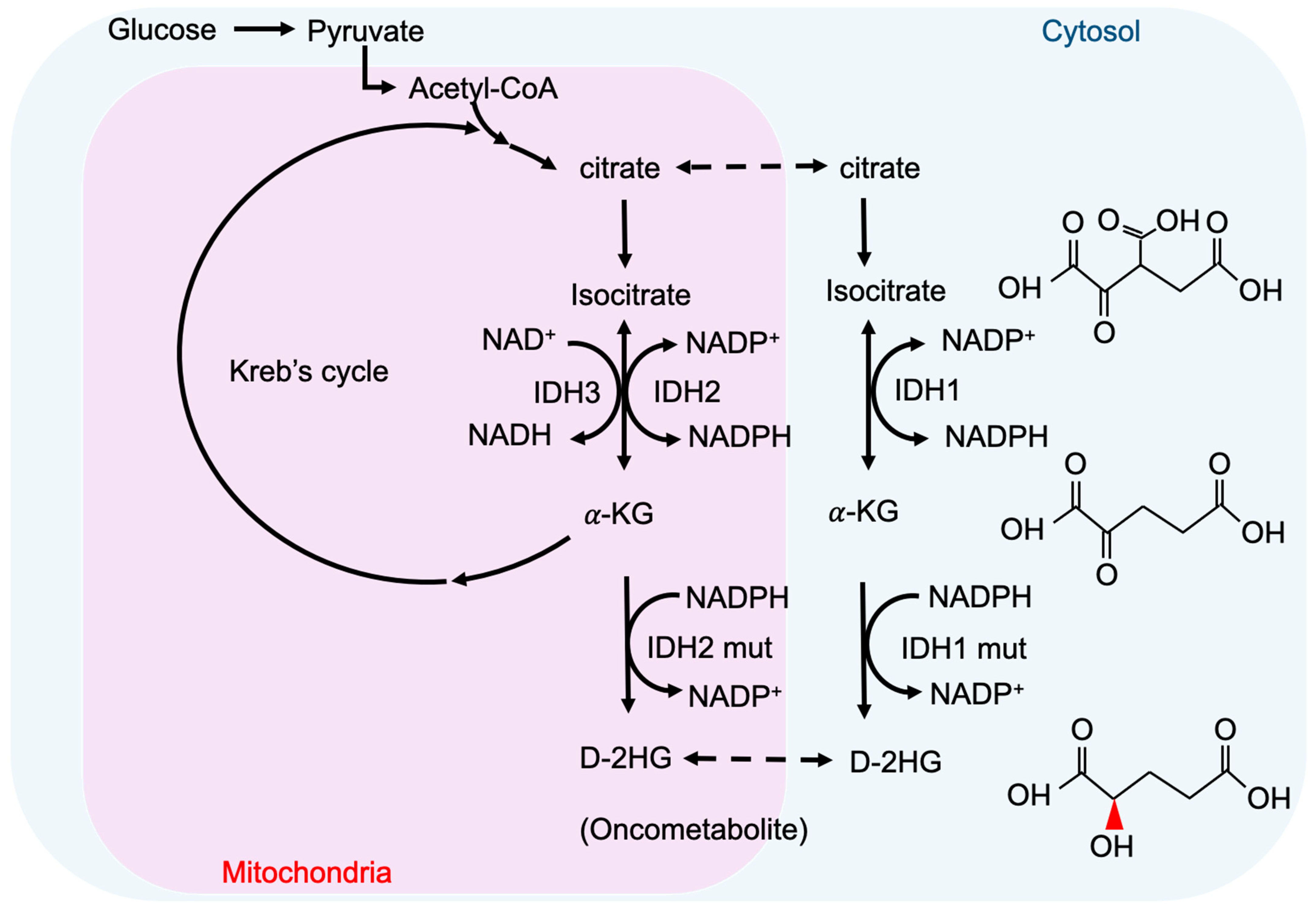
2. Metabolism and Oncometabolites
3. Cancer-Associated IDH Mutation and D-2-HG
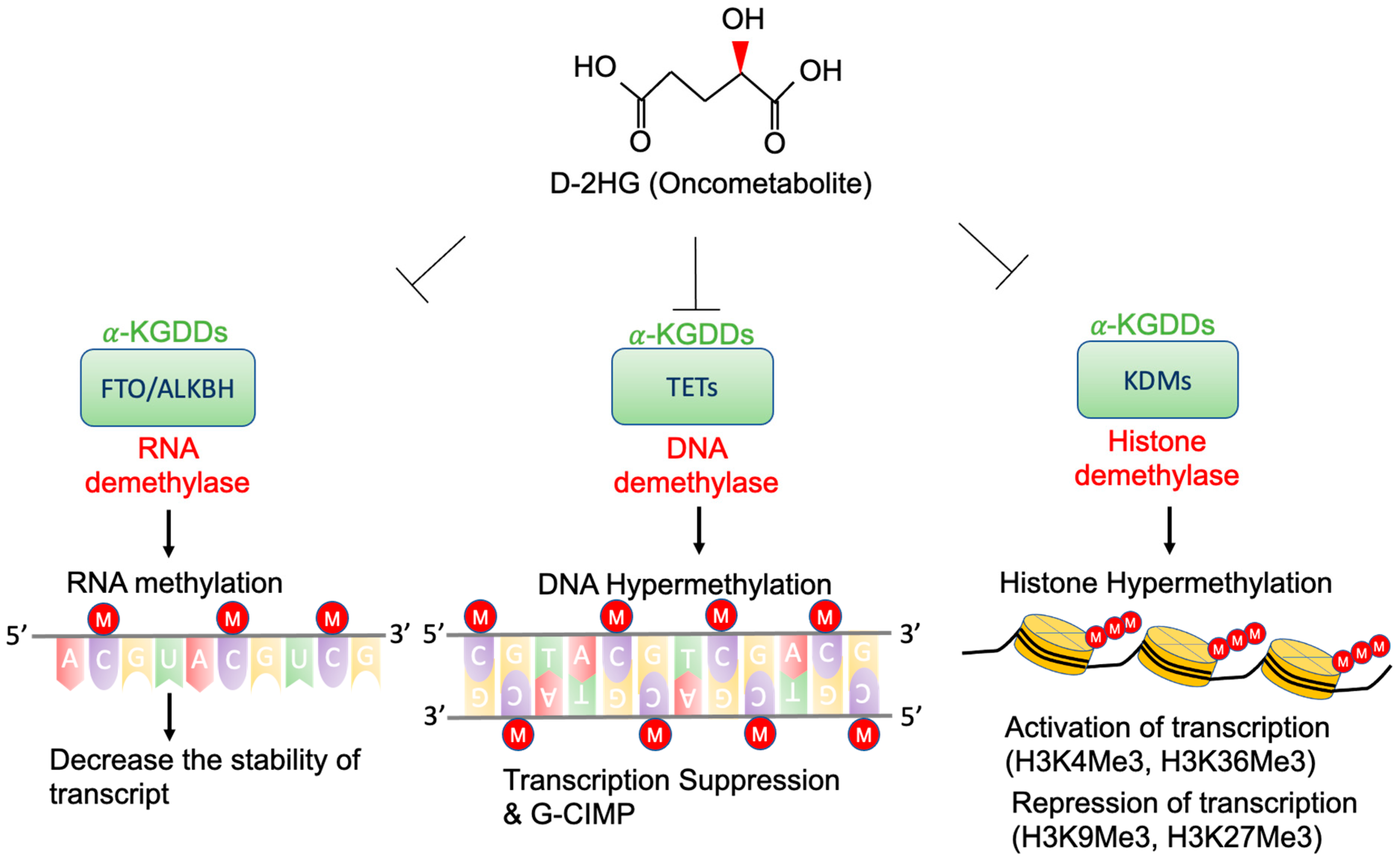
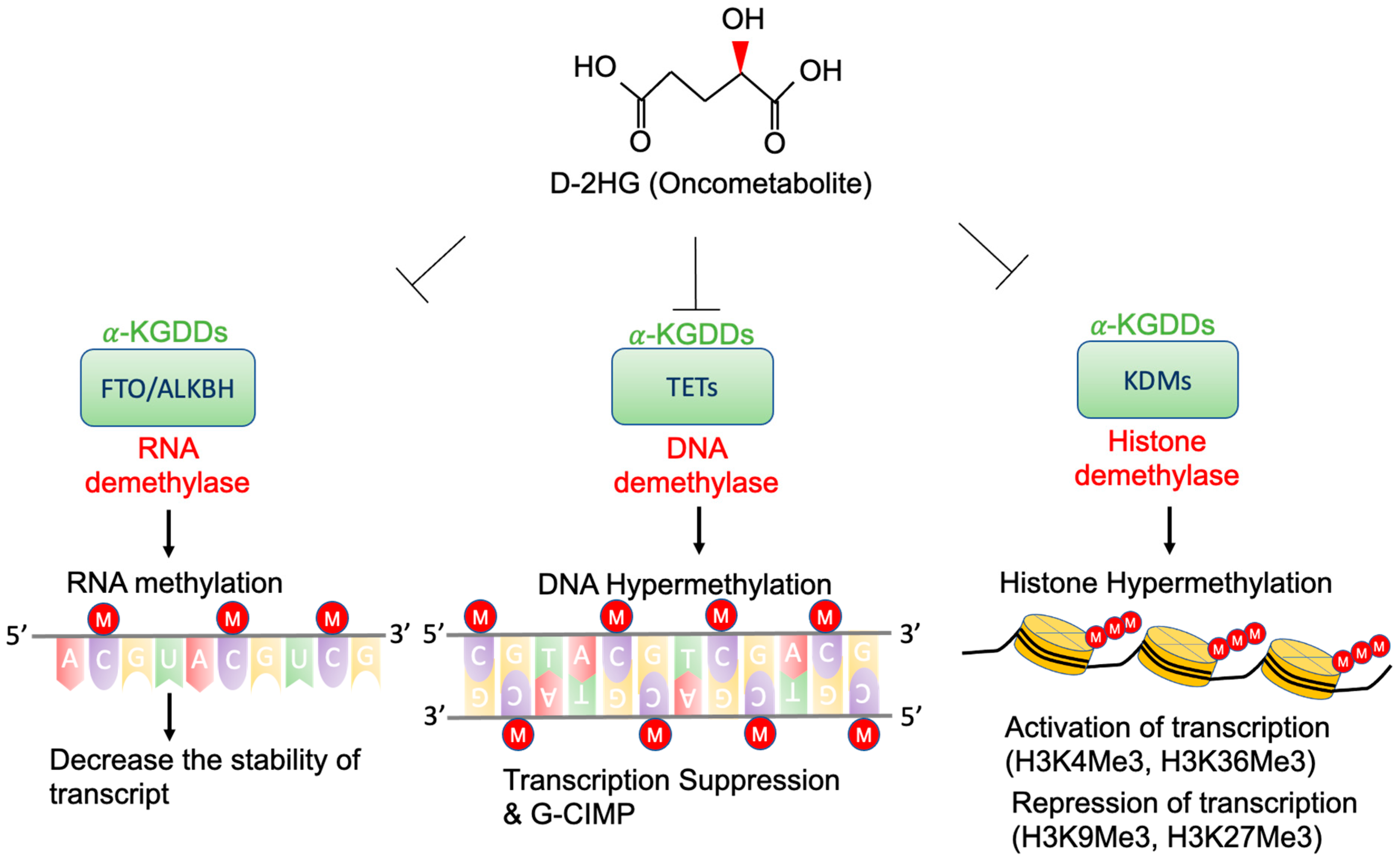
4. Epigenetic Regulation by D-2-HG
4.1. TETs and G-CIMP
4.2. KDMs and Histone Methylation
4.3. FTO and RNA Methylation
5. Signaling Pathway Alterations and D-2-HG
5.1. HIF-1 Signaling Pathway
5.2. RTK and mTOR Signaling Pathway
5.3. DNA Repair Pathways
5.4. Redox Homeostasis and Anti-Oxidative Pathways
6. Targeting D-2-HG in Cancers with IDH Mutation Inhibitors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Sanson, M.; Marie, Y.; Paris, S.; Idbaih, A.; Laffaire, J.; Ducray, F.; El Hallani, S.; Boisselier, B.; Mokhtari, K.; Hoang-Xuan, K.; et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J. Clin. Oncol. 2009, 27, 4150–4154. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.; von Felten, S.; Frank, S.; Boulay, J.-L.; Mariani, L. IDH mutation is associated with higher risk of malignant transformation in low-grade glioma. J. Neuro-Oncol. 2016, 127, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.R.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A new paradigm for oncology, metabolism, and the clinical laboratory. Clin. Chem. 2017, 63, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2, 85. [Google Scholar] [CrossRef] [Green Version]
- Ritthausen, H. Ueber die glutansäure, das zersetzungsproduct der glutaminsäure durch salpetrige säure. J. Für Prakt. Chem. 1868, 103, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Duran, M.; Kamerling, J.; Bakker, H.; Van Gennip, A.; Wadman, S. L-2-Hydroxyglutaric aciduria: An inborn error of metabolism? J. Inherit. Metab. Dis. 1980, 3, 109–112. [Google Scholar] [CrossRef]
- Chalmers, R.; Lawson, A.; Watts, R.; Tavill, A.; Kamerling, J.; Hey, E.; Ogilvie, D. d-2-Hydroxyglutaric aciduria: Case report and biochemical studies. J. Inherit. Metab. Dis. 1980, 3, 11–15. [Google Scholar] [CrossRef]
- Struys, E.A.; Salomons, G.S.; Achouri, Y.; Van Schaftingen, E.; Grosso, S.; Craigen, W.J.; Verhoeven, N.M.; Jakobs, C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2005, 76, 358–360. [Google Scholar] [CrossRef] [Green Version]
- Kranendijk, M.; Struys, E.A.; Van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; Van Der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; De Klerk, J.B. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 2010, 330, 336. [Google Scholar] [CrossRef]
- Abbas, S.; Lugthart, S.; Kavelaars, F.G.; Schelen, A.; Koenders, J.E.; Zeilemaker, A.; van Putten, W.J.; Rijneveld, A.W.; Löwenberg, B.; Valk, P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood J. Am. Soc. Hematol. 2010, 116, 2122–2126. [Google Scholar] [CrossRef]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p. R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 2010, 163, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011, 43, 1262–1265. [Google Scholar] [CrossRef]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef]
- Juratli, T.A.; Kirsch, M.; Robel, K.; Soucek, S.; Geiger, K.; von Kummer, R.; Schackert, G.; Krex, D. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J. Neuro-Oncol. 2012, 108, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, E.; Wilding, C.; Engelman, B.; Huang, P.; Jones, R.L. Is the IDH Mutation a Good Target for Chondrosarcoma Treatment? Curr. Mol. Biol. Rep. 2020, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Bigner, D.D.; Velculescu, V.; Parsons, D.W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009, 69, 9157–9159. [Google Scholar] [CrossRef] [Green Version]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R (−)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 2015, 18, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Shapira, S.N.; Christofk, H.R. Metabolic Regulation of Stem Cell Fate and Function. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Struys, E.A.; Jansen, E.E.; Verhoeven, N.M.; Jakobs, C. Measurement of urinary D-and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography–tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin. Chem. 2004, 50, 1391–1395. [Google Scholar] [CrossRef] [Green Version]
- Sahm, F.; Capper, D.; Pusch, S.; Balss, J.; Koch, A.; Langhans, C.D.; Okun, J.G.; von Deimling, A. Detection of 2-hydroxyglutarate in formalin-fixed paraffin-embedded glioma specimens by gas chromatography/mass spectrometry. Brain Pathol. 2012, 22, 26–31. [Google Scholar] [CrossRef]
- Yuan, B.-F. Quantitative Analysis of Oncometabolite 2-Hydroxyglutarate. In Cancer Metabolomics. Advances in Experimental Medicine and Biology; Hu, S., Ed.; Springer: Cham, Switzerland, 2021; Volume 1280, pp. 161–172. [Google Scholar]
- Kalinina, J.; Ahn, J.; Devi, N.S.; Wang, L.; Li, Y.; Olson, J.J.; Glantz, M.; Smith, T.; Kim, E.L.; Giese, A.; et al. Selective detection of the D-enantiomer of 2-hydroxyglutarate in the CSF of glioma patients with mutated isocitrate dehydrogenase. Clin. Cancer Res. 2016, 22, 6256–6265. [Google Scholar] [CrossRef] [Green Version]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1474. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Laino, M.E.; Young, R.; Beal, K.; Haque, S.; Mazaheri, Y.; Corrias, G.; Bitencourt, A.G.; Karimi, S.; Thakur, S.B. Magnetic resonance spectroscopic imaging in gliomas: Clinical diagnosis and radiotherapy planning. BJR Open 2020, 2, 20190026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietze, A.; Choi, C.; Mickey, B.; Maher, E.A.; Ulhøi, B.P.; Sangill, R.; Lassen-Ramshad, Y.; Lukacova, S.; Østergaard, L.; Von Oettingen, G. Noninvasive assessment of isocitrate dehydrogenase mutation status in cerebral gliomas by magnetic resonance spectroscopy in a clinical setting. J. Neurosurg. 2017, 128, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol. 2018, 22, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 2018, 172, 90–105.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef]
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Levine, R.L. TET2 in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Med. 2017, 7, a026518. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517. [Google Scholar] [CrossRef]
- Mahfoudhi, E.; Talhaoui, I.; Cabagnols, X.; Della Valle, V.; Secardin, L.; Rameau, P.; Bernard, O.A.; Ishchenko, A.A.; Abbes, S.; Vainchenker, W.; et al. TET2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Repair 2016, 43, 78–88. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Kraus, T.F.; Greiner, A.; Steinmaurer, M.; Dietinger, V.; Guibourt, V.; Kretzschmar, H.A. Genetic characterization of ten-eleven-translocation methylcytosine dioxygenase alterations in human glioma. J. Cancer 2015, 6, 832. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Jiang, W.; Liu, J.; Zhao, S.; Xiong, J.; Mao, Y.; Wang, Y. IDH1 mutations inhibit multiple α-ketoglutarate-dependent dioxygenase activities in astroglioma. J. Neuro-Oncol. 2012, 109, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [Green Version]
- Duncan, C.G.; Barwick, B.G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.-T.; Bigner, D.D.; Vertino, P.M.; Yan, H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.-K.; Brenner, C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014, 9, 1827–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-Oncology 2018, 20, 608–620. [Google Scholar] [CrossRef]
- Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Venneti, S.; Akalin, A.; Fang, F.; Ward, P.S.; DeMatteo, R.G.; Intlekofer, A.M.; Chen, C.; Ye, J.; Hameed, M.; et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013, 27, 1986–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Höpfl, G.; Ogunshola, O.; Gassmann, M. HIFs and tumors—Causes and consequences. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 286, R608–R623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251. [Google Scholar]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N 6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Patiar, S.; Harris, A.L. Role of hypoxia-inducible factor-1α as a cancer therapy target. Endocr.-Relat. Cancer 2006, 13, S61–S75. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8, S62–S67. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cao, P. Clinical and prognostic significance of HIF-1α in glioma patients: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 22073–22083. [Google Scholar]
- Shaw, K. Environmental cues like hypoxia can trigger gene expression and cancer development. Nat. Educ. 2008, 1, 198. [Google Scholar]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1α stability in aerobic conditions. Cell Metab. 2016, 24, 740–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhao, Y.; Shi, J.; Zhang, J.; Yuan, Y.; Gu, Y.; Zhang, F.; Gao, X.; Wang, C.; Wang, Y.; et al. Isocitrate dehydrogenase1 mutation reduces the pericyte coverage of microvessels in astrocytic tumours. J. Neuro-Oncol. 2019, 143, 187–196. [Google Scholar] [CrossRef]
- Holroyd, A.K.; Michie, A.M. The role of mTOR-mediated signaling in the regulation of cellular migration. Immunol. Lett. 2018, 196, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-Oncology 2010, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Shahcheraghi, S.H.; Tchokonte-Nana, V.; Lotfi, M.; Lotfi, M.; Ghorbani, A.; Sadeghnia, H.R. Wnt/beta-catenin and PI3K/Akt/mtor Signaling Pathways in Glioblastoma: Two main targets for drug design: A Review. Curr. Pharm. Des. 2020, 26, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.-E.; Germain, M.-A.; Motorina, A.; Guiot, M.-C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700. [Google Scholar] [CrossRef]
- Caron, A.; Baraboi, E.D.; Laplante, M.; Richard, D. DEP domain-containing mTOR-interacting protein in the rat brain: Distribution of expression and potential implication. J. Comp. Neurol. 2015, 523, 93–107. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.; Briscoe, D.M.; Richard, D.; Laplante, M. DEPTOR at the Nexus of Cancer, Metabolism, and Immunity. Physiol. Rev. 2018, 98, 1765–1803. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, Y.; Li, A.; Celiku, O.; Han, S.; Qian, M.; Yang, C. Mtorc2/rac1 pathway predisposes cancer aggressiveness in idh1-mutated glioma. Cancers 2020, 12, 787. [Google Scholar] [CrossRef] [Green Version]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA repair in mammalian cells. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Qi, S.; Yu, L.; Gui, S.; Ding, Y.; Han, H.; Zhang, X.; Wu, L.; Yao, F. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Houillier, C.; Wang, X.; Kaloshi, G.; Mokhtari, K.; Guillevin, R.; Laffaire, J.; Paris, S.; Boisselier, B.; Idbaih, A.; Laigle-Donadey, F.; et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 2010, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chou, A.P.; Chen, W.; Chen, R.; Deng, Y.; Phillips, H.S.; Selfridge, J.; Zurayk, M.; Lou, J.J.; Everson, R.G.; et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro-Oncology 2013, 15, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohba, S.; Kuwahara, K.; Yamada, S.; Abe, M.; Hirose, Y. Correlation between IDH, ATRX, and TERT promoter mutations in glioma. Brain Tumor Pathol. 2020, 37, 33–40. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [Green Version]
- Khurshed, M.; Aarnoudse, N.; Hulsbos, R.; Hira, V.V.; van Laarhoven, H.W.; Wilmink, J.W.; Molenaar, R.J.; van Noorden, C.J. IDH1-mutant cancer cells are sensitive to cisplatin and an IDH1-mutant inhibitor counteracts this sensitivity. FASEB J. 2018, 32, 6344–6352. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.-T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The alkylating chemotherapeutic temozolomide induces metabolic stress in IDH1-mutant cancers and potentiates NAD+ depletion–mediated cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, T.-C.A.; Prestegarden, L.; Grudic, A.; Hegi, M.E.; Tysnes, B.B.; Bjerkvig, R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro-Oncology 2013, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d-and l-2-hydroxyglutarate inhibit the AlkB family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.-A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of glutathione metabolism in IDH1-mutated glioma. Mol. Cancer Ther. 2020, 19, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A. [44] Glutathione peroxidase. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1981; Volume 77, pp. 325–333. [Google Scholar]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol. 2014, 127, 221–233. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumor Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Liu, Y.; Zhou, Y.; Ruiz-Rodado, V.; Larion, M.; Xu, G.; Yang, C. Triptolide suppresses IDH1-mutated malignancy via Nrf2-driven glutathione metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 9964–9972. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yu, D.; Cai, S.; Yang, C. Targeting Nrf2 driven glutathione de novo synthesis as a novel strategy to suppresses IDH1-mutated glioma. Mol. Cell. Biol. 2019, 79, 875. [Google Scholar]
- Biedermann, J.; Preussler, M.; Conde, M.; Peitzsch, M.; Richter, S.; Wiedemuth, R.; Abou-El-Ardat, K.; Krüger, A.; Meinhardt, M.; Schackert, G.; et al. Mutant IDH1 differently affects redox state and metabolism in glial cells of normal and tumor origin. Cancers 2019, 11, 2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.Z.; Zhao, R.; Dong, Y.; Chen, X.Q.; Yu, A.C.H. Astrocyte and neuron intone through glutamate. Neurochem. Res. 2008, 33, 2480–2486. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Rahimpour, S.; Albert, C.; Lonser, R.R.; Zhuang, Z. Regulation and dysregulation of astrocyte activation and implications in tumor formation. Cell. Mol. Life Sci. 2013, 70, 4201–4211. [Google Scholar] [CrossRef]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Papathanassiu, A.E.; Ko, J.-H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8, 16040. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [Green Version]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171. [Google Scholar] [PubMed] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; De La Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in isocitrate dehydrogenase 1–mutated advanced glioma. Chin. Ger. J. Clin. Oncol. 2020, 38, 3398–3406. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.-C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Wakabayashi, T.; Miyakita, Y.; Narita, Y.; Mineharu, Y.; Arakawa, Y.; Yamasaki, F.; Sugiyama, K.; Hata, N.; Muragaki, Y.; et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. Chin. Ger. J. Clin. Oncol. 2019, 37, 2004. [Google Scholar] [CrossRef]
- Konteatis, Z.; Artin, E.; Nicolay, B.; Straley, K.; Padyana, A.K.; Jin, L.; Chen, Y.; Narayaraswamy, R.; Tong, S.; Wang, F.; et al. Vorasidenib (AG-881): A first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 2020, 11, 101–107. [Google Scholar] [CrossRef]
- Ma, R.; Yun, C.-H. Crystal structures of pan-IDH inhibitor AG-881 in complex with mutant human IDH1 and IDH2. Biochem. Biophys. Res. Commun. 2018, 503, 2912–2917. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Van Den Bent, M.J.; Clarke, J.L.; Maher, E.A.; Peters, K.B.; Touat, M.; De Groot, J.F.; De La Fuente, M.I.; Arrillaga-Romany, I.; Wick, W.; et al. INDIGO: A global, randomized, double-blind, phase III study of vorasidenib (VOR; AG-881) vs placebo in patients (pts) with residual or recurrent grade II glioma with an isocitrate dehydrogenase 1/2 (IDH1/2) mutation. Chin. Ger. J. Clin. Oncol. 2020, 38, TPS2574. [Google Scholar] [CrossRef]
- Kernytsky, A.; Wang, F.; Hansen, E.; Schalm, S.; Straley, K.; Gliser, C.; Yang, H.; Travins, J.; Murray, S.; Dorsch, M.; et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 2015, 125, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Kopinja, J.; Sevilla, R.S.; Levitan, D.; Dai, D.; Vanko, A.; Spooner, E.; Ware, C.; Forget, R.; Hu, K.; Kral, A.; et al. A brain penetrant mutant IDH1 inhibitor provides in vivo survival benefit. Sci. Rep. 2017, 7, 13853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.M.; Baer, M.R.; Lee, S.; Yang, J.; Dinner, S.N.; Prebet, T.; Schiller, G.J.; Seiter, K.; Ferrell, P.B.; Kelly, P.F.; et al. A phase 1 dose escalation study of the IDH1 m inhibitor, FT-2102, in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Blood 2018, 36, 7009. [Google Scholar]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schimmer, A.D.; Yee, K.W.; Hochhaus, A.; Kraemer, A.; Carvajal, R.D.; Janku, F.; Bedard, P.; Carpio, C.; Wick, A.; et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood 2016, 128, 1073. [Google Scholar] [CrossRef]
- Megías-Vericat, J.E.; Ballesta-López, O.; Barragán, E.; Montesinos, P. IDH1-mutated relapsed or refractory AML: Current challenges and future prospects. Blood Lymphat. Cancer Targets Ther. 2019, 9, 19–32. [Google Scholar] [CrossRef] [Green Version]
- McMurry, H.; Fletcher, L.; Traer, E. IDH Inhibitors in AML—Promise and Pitfalls. Curr. Hematol. Malig. Rep. 2021, 16, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Birendra, K.; DiNardo, C.D. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant IDH1 inhibitor, AG-120. Clin. Lymphoma Myeloma Leuk. 2016, 16, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; AG221-C-001 Study Investigators. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: Analysis of a phase 1/2 study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Mulkey, F.; Ward, A.F.; Przepiorka, D.; Deisseroth, A.B.; Farrell, A.T.; Pazdur, R. Incidence of differentiation syndrome with ivosidenib (IVO) and enasidenib (ENA) for treatment of patients with relapsed or refractory (R/R) isocitrate dehydrogenase (IDH) 1-or IDH2-mutated acute myeloid leukemia (AML): A systematic analysis by the US Food and Drug Administration (FDA). Blood 2018, 132, 288. [Google Scholar]
- Tiburcio, P.D.; Gillespie, D.L.; Jensen, R.L.; Huang, L.E. Extracellular glutamate and IDH1 R132H inhibitor promote glioma growth by boosting redox potential. J. Neuro-Oncol. 2020, 146, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E. Friend or foe—IDH1 mutations in glioma 10 years on. Carcinogenesis 2019, 40, 1299–1307. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Term | Full Name; Biological Function |
|---|---|
| Metabolic enzymes, mutations, and biomarker | |
| α-KG | α-ketoglutarate; the product of oxidative decarboxylation of isocitrate |
| IDH | Isocitrate dehydrogenase; catalyze the conversion of isocitrate to α-KG |
| D-2-HG | D-2-hydroxyglutarate, metabolite of IDH1 or 2 mutations; acts as an antagonist of α-KG |
| IDH1 mutation | Including R132H, R132C, R132G, R132L, and R132S; gain-of-function mutation; |
| result in D-2-HG abnormal accumulation | |
| IDH2 mutation | Including R140Q and R172K; gain-of-function mutation; result in D-2-HG abnormal accumulation |
| G-CIMP | Glioma CpG island methylator phenotype; a classification standard and diagnosis indicator |
| α-KG/Fe(II)-dependent dioxgenases (α-KGDDs) | |
| TET | Ten-eleven translocation enzymes; DNA demethylation |
| JmjC-KDMs | Jumonji (JmjC) domain-containing lysine-specific histone demethylases; histone demethylation |
| FTO | Fat mass and obesity-associated protein; RNA demethylation |
| PHDs | Prolyl hydroxylases domain proteins; prolyl hydroxylases; negative regulator of HIF |
| FIHs | Factor inhibiting HIF; asparaginyl hydroxylase; negative regulator of HIF |
| KDM4A | Lysine-specific histone demethylases 4A, also known as JmjC-KDM2A; |
| histone demethylation/regulate DEPTOR | |
| Signaling pathway regulator | |
| DEPTOR | DEP domain-containing mTOR-interacting protein; negative regulator of the mTOR pathway mediated by KDM4A |
| Molecules of anti-oxidative pathways | |
| GSH | Glutathione (reduced form); antioxidants, against ROS and maintains redox homeostasis |
| GSSG | Glutathione disulfide (oxidized form); GSSG can be reduced to GSH by glutathione reductase |
| DNA repair pathways | |
| HR | Homologous recombination; manage DNA double-strand breaks (DSBs) |
| NHEJ | Non-homologous end-joining; DNA double-strand breaks (DSBs) |
| BER | Base excision repair; manage DNA base methylation |
| Chemotherapy agents | |
| TMZ | Temozolomide; DNA alkylating agent for gliomas treatment; result in DNA methylation |
| PCV | procarbazine-cisplatin-vincristine; multi-drug chemotherapy for gliomas |
| Compound | Drug Name | Route | Target | Clinical Trials and Preclinical Studies | References |
|---|---|---|---|---|---|
| AGI-5198 | Oral | IDH1mut R132H | Phase 3 clinical trial (NCT03173248) in IDH mutated AML | ||
| Delays growth and promotes differentiation in IDH1 mutated glioma cells | [117,135] | ||||
| AG-120 | Ivosidenib | Oral | IDH1mut R132H and R132C | Phase 1 clinical trial (NCT03343197) in patients with recurrent, non-enhancing IDH1 mutated low grade glioma | [137] |
| (FDA-approved) | Phase 1 clinical trial (NCT02073994) in IDH1 mutated advanced solid tumors, including glioma | ||||
| AG-221 | Enasidenib | Oral | IDH2mut R140Q and R172K | Phase 1/2 clinical trial (NCT02273739) in adults with IDH2 mutated advanced solid tumors, including glioma | |
| (FDA-approved) | |||||
| DS-1001b | Oral | IDH1mutations | Phase 1 clinical trial (NCT03030066) in patients with gene IDH1 mutated gliomas | [134] | |
| Phase 2 clinical trial (NCT04458272) in patients with chemo- and radiotherapy-naive IDH1 mutated WHO grade II glioma | |||||
| BAY1436032 | Oral | pan IDH1 mutations | Phase 1 clinical trial (NCT02746081) in IDH1 mutated advanced solid tumors, including glioma | [140] | |
| (R132H, R132C, R132G, R132L, and R132S) | |||||
| AG-881 | Vorasidenib | Oral | IDH1/2 mutation | Phase 1 clinical trial (NCT02481154) in patients with IDH1 or IDH2 mutated advanced solid tumors, including gliomas | [143] |
| (IDH1mut R132H, R132C, R132G, R132L, and R132S) | Phase 1 clinical trial (NCT03343197) in patients with recurrent, non-enhancing IDH1 mutated low grade glioma | ||||
| (IDH2mut R140Q and R172K) | Phase 3 clinical trial (NCT04164901) in patients with residual or recurrent grade 2 IDH1 or IDH2 mutated glioma | [144] | |||
| AGI-6780 | IDH2 mut R140Q | Reverses IDH2 R140Q induced histone hypermethylation expression in IDH2 mutated AML cell model | [145] | ||
| MRK-A | IDH1mut R132H and R132C | Show survival benefit in IDH1 mutated patient-derived cells xenograft model | [146] | ||
| FT-2102 | Olutasidenib | Oral | Phase 1 dose escalation study in patients with IDH1 mutated AML or MDS | [147] | |
| HMS-101 | IDH1mut R132C | In vitro and in vivo in IDH1 mutated AML | [148] | ||
| IDH305 | Oral | IDH1mut R132H and R132C | Phase 1 clinical trial (NCT02381886) in patients with IDH1R132 mutated advanced malignancies, including glioma | [149] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, F.-J.; Liu, Y.; Lang, F.; Yang, C. D-2-Hydroxyglutarate in Glioma Biology. Cells 2021, 10, 2345. https://doi.org/10.3390/cells10092345
Chou F-J, Liu Y, Lang F, Yang C. D-2-Hydroxyglutarate in Glioma Biology. Cells. 2021; 10(9):2345. https://doi.org/10.3390/cells10092345
Chicago/Turabian StyleChou, Fu-Ju, Yang Liu, Fengchao Lang, and Chunzhang Yang. 2021. "D-2-Hydroxyglutarate in Glioma Biology" Cells 10, no. 9: 2345. https://doi.org/10.3390/cells10092345
APA StyleChou, F.-J., Liu, Y., Lang, F., & Yang, C. (2021). D-2-Hydroxyglutarate in Glioma Biology. Cells, 10(9), 2345. https://doi.org/10.3390/cells10092345