
Applications of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme



Abstract
:1. Introduction
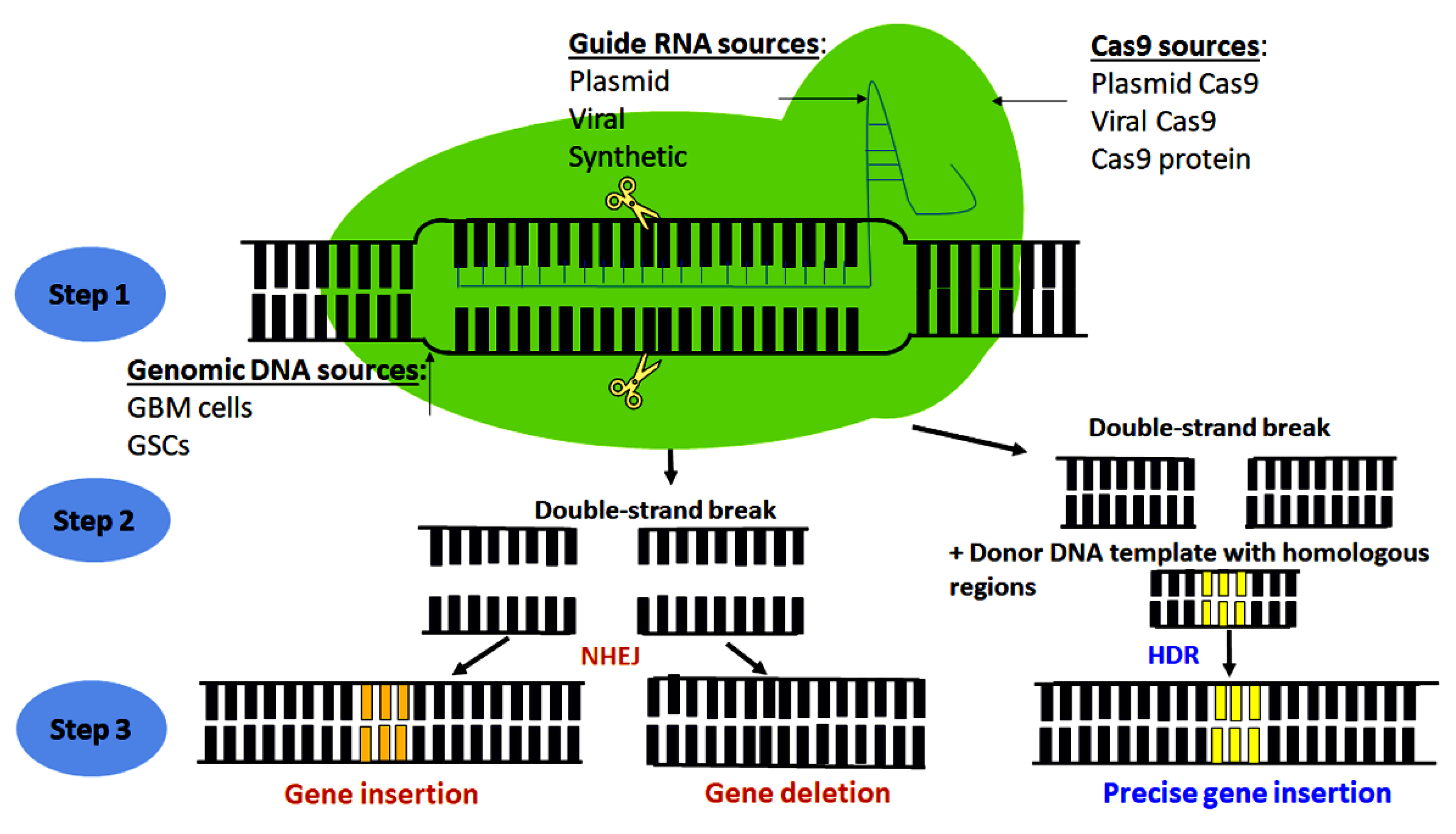
2. Principle of CRISPR-Cas9 Genome Editing Technology
3. Genome-Wide CRISPR-Cas9 Screens in GBM Research

{kind=link}
{kind=link}
| Tumor Model | Type of Screen | References |
|---|---|---|
| SNB19 | Genome-scale CRISPR knockout screen | [43] |
| U138MG | Large-scale CRISPR-Cas9 mediated loss of function screen | [46] |
| GSCs | Whole-genome CRISPR screening | [49] |
| U87MG | CRISPR interference (CRISPRi) screen | [51] |
| GBM3565, GSC23 | CRISPRi screen | [52] |
| Patient-derived GSCs | Genome-wide CRISPR-Cas9 screens | [48] |
| Mice | In vivo CRISPR screen | [45] |
| Patient-derived GSCs and human NSCs | Genome-wide CRISPR-Cas9 screen | [50] |
| U87MG and U87MG-EGFRvIII cells | Pooled genome wide CRISPR screening | [44] |
4. Application of CRISPR-Cas9 to Identifying Genetic Regulators of Autophagy in GBM
5. Application of CRISPR-Cas9 to Identifying genetic Regulators of Apoptosis in GBM
| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| ERN1 IGFBP3 IGFBP5 | Knockout | U251 | [63] |
| FAT1 | Knockout | U251MG | [59] |
| CHAF1A | Knockout | U251MG, U87MG | [65] |
| GLI1 | Knock down | GBM28 | [61] |
| TRIM45 | Knockout | U87MG LN229 | [62] |
| RGS4 | Knockout | GSC20 | [64] |
| ATM | Knockouts | LN18, LN229 | [58] |
| Podoplanin (PDPN) | Knockout | GBMF2, GBMF3, LN308, LN319 | [60] |
| ATG5 ATG7 | Knockout | MZ-54 GBM cells | [57] |
| ATG5 | knockout | TGS01 or TGS04 | [55] |
| C14-IP-3 | CRISPR-induced activation | LN229 | [35] |
6. Use of CRISPR-Cas9 Editing for Identifying Genetic Regulators of Angiogenesis in GBM
7. CRISPR-Cas9 Editing of the Genes for Down Regulation of Cell Invasion and Migration in GBM
| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| QKI | Knockout | GSCs | [81] |
| PAX6 | Knockout | U251 | [77] |
| NRP1 and NRP2 | Knockout | U87MG | [42] |
| ATRX | Knockdown | U251, LN229 | [75] |
| TP53 exon 4 | Homologous recombination to disrupt the TP53 | Cerebral organoids of human embryonic stem cell line (H9) | [82] |
| C14-IP-3 | CRISPR-induced activation | LN229 | [35] |
| AhR | Knockdown | Patient-derived 15-037 cells | [39] |
| KDM6B | CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) | U87MG and U251 | [79] |
| TEAD1 or TEAD4 | Knockout | Patient-derived GBM cells | [80] |
| PDPN | Knockdown | GBMF3 | [60] |
| DCX | Overexpression or knockdown | Rat C6 and human U251 cell lines | [74] |
| ZEB1 | Knockdown | Bevacizumab-resistant xenograft models | [76] |
| Dazl | Knockdown | A172, U251, and LN229 cell lines | [22] |
| DDX39B | Knockdown | GBM34, GBM44, GSCs | [70] |
| RGS4 | Knockout | GSC20 and GSC28 | [64] |
| Nanos3 | Knockdown | GBM | [41] |
| Caveolin-1 and cavin | Knockout | U251 | [78] |
| ATM | Knockout | LN18 LN229 | [58] |
8. CRISPR-Cas9 Editing of the Inflammatory and Immune Response Genes in GBM
9. CRISPR-Cas9 Editing of the Genes That Provide Proliferative Signals in GBM
| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| Laminin-411 | Knockout | U87MG and LN229 and patient derived GBM cell lines TS543 and TS576 | [94] |
| KIF3A and IFT88 | Knockout | L0 | [95] |
| KDM6B | CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) | U87MG and U251MG | [79] |
| AIM2 | Knockout | U251MG | [83] |
| H3K27M | Knockout | HGG lines | [98] |
| PDPN | Knockout | GBMF2, GBMF3, and human GBM cell lines LN308 and LN319 | [60] |
| SRSF3 | Knockout | GSCs | [99] |
| ATM | Knockout | LN18 and LN229 | [58] |
| DCX | High DCX expression or knockdown | Rat C6 and human U251MG | [74] |
| TERT promoter | Correction of mutated TERT promoter | U87, U251 | [86] |
| tRNAiMet | Knockdown | U251 | [89] |
| HOTAIRM1 | Knockdown | U251MG | [87] |
| CXCR7 CXCL16 or CX3CL1 | Knockouts | LN229 | [97] |
| RGS4 | Knockouts | GSC20 and GSC28 | [64] |
| Nanos3 | knockdown | A172, U251, and LN229 | [41] |
| NRF2 | Knockdown | U87MG neurospheres | [90] |
| Dazl | Knockdown | A172, U251, and LN229 | [22] |
| CDK7 | knockout | U87MG and U251MG | [92] |
| PCM1 | Deletion | L0 and SN186 GBM cell lines | [88] |
| ID1 | Deletion | GBM cell line | [85] |
| Enhancer between Ki67 and MGMT genes | Deletion of enhancer between Ki67 and MGMT genes | SKMG3 | [23] |
| LXRβ | Deletion | GBM cell line | [91] |
| PAX6 | Knockout | U251MG | [77] |
| STAT3 | Knockout | MT330 GBM | [93] |
| ERβ | knockout | U87MG | [96] |
10. CRISPR-Cas9 Editing of the Genes That Regulate Self-Renewal Capacity in GBM
11. Conclusions, Limitations, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Eulate-Beramendi, S.A.; Piña-Batista, K.M.; Rodrigo, V.; Torres-Rivas, H.E.; Rial-Basalo, J.C. Multicentric spinal cord and brain glioblastoma without previous craniotomy. Surg. Neurol. Int. 2016, 7 (Suppl. 17), S492–S494. [Google Scholar] [CrossRef] [Green Version]
- Huse, J.T.; Holland, E.; De Angelis, L.M. Glioblastoma: Molecular analysis and clinical implications. Annu. Rev. Med. 2013, 64, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Jacob, G.; Dinca, E.B. Current data and strategy in glioblastoma multiforme. J. Med. Life 2009, 2, 386–393. [Google Scholar]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Hengel, S.K.; Balvers, R.K.; Dautzenberg, I.J.; van den Wollenberg, D.J.; Kloezeman, J.J.; Lamfers, M.L.; Sillivis-Smit, P.A.; Hoeben, R.C. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer Gene Ther. Sep. 2013, 20, 507–513. [Google Scholar] [CrossRef]
- Haar, C.P.; Hebbar, P.; Wallace, G.C., 4th; Das, A.; Vandergrift, W.A., 3rd; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug resistance in glioblastoma: A mini review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef] [Green Version]
- Ortensi, B.; Setti, M.; Osti, D.; Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res. Ther. 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic signaling pathways in glioblastoma and therapeutic implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune evasion strategies of glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Banik, N.L.; Ray, S.K. Combination of hTERT knockdown and IFN-gamma treatment inhibited angiogenesis and tumor progression in glioblastoma. Clin. Cancer Res. 2009, 15, 7186–7195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, J.D.; Aghi, M.K.; Taylor, J.W. Anti-angiogenic therapies in the management of glioblastoma. Chin. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Heuser, V.D.; Kiviniemi, A.; Lehtinen, L.; Munthe, S.; Kristensen, B.W.; Posti, J.P.; Sipilä, J.; Vuorinen, V.; Carpén, O.; Gardberg, M. Multiple formin proteins participate in glioblastoma migration. BMC Cancer 2020, 20, 710. [Google Scholar] [CrossRef]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.S.; et al. Effect of CRISPR/Cas9-mediated PD-1-disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, R.; Zhang, H.; Liu, C.; Lu, Y. Suppressing Dazl modulates tumorigenicity and stemness in human glioblastoma cells. BMC Cancer 2020, 20, 673. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.; He, L.J.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xue, W.; Anderson, D.G. CRISPR-Cas: A tool for cancer research and therapeutics. Nat. Rev. Clin. Oncol. 2019, 16, 281–295. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. Mar. 2010, 11, 181–190. [Google Scholar] [CrossRef]
- Horvath, P.; Romero, D.A.; Coûté-Monvoisin, A.C.; Richards, M.; Deveau, H.; Moineau, S.; Boyaval, P.; Fremaux, C.; Barrangou, R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1401–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Godde, J.S.; Bickerton, A. The repetitive DNA elements called CRISPRs and their associated genes: Evidence of horizontal transfer among prokaryotes. J. Mol. Evol. 2006, 62, 718–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating, and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef] [Green Version]
- Yelton, C.J.; Ray, S.K. Histone deacetylase enzymes and selective histone deacetylase inhibitors for antitumor effects and enhancement of antitumor immunity in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.; Aich, M.; Kumar, A.; Sengupta, S.; Bajad, P.; Dhapola, P.; Paul, D.; Narta, K.; Purkrait, S.; Mehani, B.; et al. Novel internal regulators and candidate miRNAs within miR-379/miR-656 miRNA cluster can alter cellular phenotype of human glioblastoma. Sci. Rep. 2018, 8, 7673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150496. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Dudás, A.; Chovanec, M. DNA double-strand break repair by homologous recombination. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Karki, K.; Cheng, Y.; Michelhaugh, S.K.; Mittal, S.; Safe, S. The aryl hydrocarbon receptor is a tumor suppressor-like gene in glioblastoma. J. Biol. Chem. 2019, 294, 11342–11353. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, R.; Liu, C.; Zhang, H.; Lu, Y. Nanos3, a cancer-germline gene, promotes cell proliferation, migration, chemoresistance, and invasion of human glioblastoma. Cancer Cell Int. 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Smolkin, T.; Nir-Zvi, I.; Duvshani, N.; Mumblat, Y.; Kessler, O.; Neufeld, G. Complexes of plexin-A4 and plexin-D1 convey semaphorin-3C signals to induce cytoskeletal collapse in the absence of neuropilins. J. Cell Sci. 2018, 131, jcs208298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awah, C.U.; Chen, L.; Bansal, M.; Mahajan, A.; Winter, J.; Lad, M.; Warnke, L.; Gonzalez-Buendia, E.; Park, C.; Zhang, D.; et al. Ribosomal protein S11 influences glioma response to TOP2 poisons. Oncogene 2020, 39, 5068–5081. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, X.; Li, Y.; Wang, Q.; Zhou, J.; Wang, Y.; Dong, F.; Yang, C.; Sun, Z.; Fang, C.; et al. Genome-wide CRISPR-Cas9 screening identifies NF-κB/E2F6 responsible for EGFRvIII-associated temozolomide resistance in glioblastoma. Adv. Sci. 2019, 6, 1900782. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prolo, L.M.; Li, A.; Owen, S.F.; Parker, J.J.; Foshay, K.; Nitta, R.T.; Morgens, D.W.; Bolin, S.; Wilson, C.M.; Vega, L.J.; et al. Targeted genomic CRISPR-Cas9 screen identifies MAP4K4 as essential for glioblastoma invasion. Sci. Rep. 2019, 9, 14020. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Park, J.J.; Dong, M.B.; Yang, Q.; Chow, R.D.; Peng, L.; Du, Y.; Guo, J.; Dai, X.; Wang, G.; et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat. Biotechnol. 2019, 37, 1302–1313. [Google Scholar] [CrossRef]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.; et al. Genome-wide CRISPR-Cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef] [PubMed]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 screens reveal loss of redundancy between PKMYT1 and WEE1 in glioblastoma stem-like cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21, 83. [Google Scholar] [CrossRef]
- Morton, A.R.; Dogan-Artun, N.; Faber, Z.J.; MacLeod, G.; Bartels, C.F.; Piazza, M.S.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 2019, 179, 1330–1341.e13. [Google Scholar] [CrossRef]
- Hönscheid, P.; Datta, K.; Muders, M.H. Autophagy: Detection, regulation and its role in cancer and therapy response. Int. J. Radiat. Biol. 2014, 90, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Hsueh, W.T.; Chuang, J.Y.; Chang, K.Y. Role of autophagy in therapeutic resistance of glioblastoma. J. Cancer Metastasis Treat. 2019, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Vu, H.T.; Kobayashi, M.; Hegazy, A.M.; Tadokoro, Y.; Ueno, M.; Kasahara, A.; Takase, Y.; Nomura, N.; Peng, H.; Ito, C.; et al. Autophagy inhibition synergizes with calcium mobilization to achieve efficient therapy of malignant gliomas. Cancer Sci. 2018, 109, 2497–2508. [Google Scholar] [CrossRef]
- Fettweis, G.; Di Valentin, E.; L’homme, L.; Lassence, C.; Dequiedt, F.; Fillet, M.; Coupienne, I.; Piette, J. RIP3 antagonizes a TSC2-mediated pro-survival pathway in glioblastoma cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Zielke, S.; Meyer, N.; Mari, M.; Abou-El-Ardat, K.; Reggiori, F.; van Wijk, S.; Kögel, D.; Fulda, S. Loperamide, pimozide, and STF-62247 trigger autophagy-dependent cell death in glioblastoma cells. Cell Death Dis. 2018, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Alabdullah, M.; Miligy, I.; Normatova, M.; Babaei-Jadidi, R.; Nateri, A.S.; Rakha, E.A.; Madhusudan, S. ATM regulated PTEN degradation is XIAP E3 ubiquitin ligase mediated in p85α Deficient cancer cells and influence platinum sensitivity. Cells 2019, 8, 1271. [Google Scholar] [CrossRef] [Green Version]
- Kranz, D.; Boutros, M. A synthetic lethal screen identifies FAT1 as an antagonist of caspase-8 in extrinsic apoptosis. EMBO J. 2014, 33, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Eisemann, T.; Costa, B.; Harter, P.N.; Wick, W.; Mittelbronn, M.; Angel, P.; Peterziel, H. Podoplanin expression is a prognostic biomarker but may be dispensable for the malignancy of glioblastoma. Neuro Oncol. 2019, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8, 32960–32976. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, C.; Cui, J.; Ou, J.; Han, J.; Qin, Y.; Zhi, F.; Wang, R.F. TRIM45 functions as a tumor suppressor in the brain via its E3 ligase activity by stabilizing p53 through K63-linked ubiquitination. Cell Death Dis. 2017, 8, e2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodvold, J.J.; Xian, S.; Nussbacher, J.; Tsui, B.; Cameron Waller, T.; Searles, S.C.; Lew, A.; Jiang, P.; Babic, I.; Nomura, N.; et al. IRE1α and IGF signaling predict resistance to an endoplasmic reticulum stress-inducing drug in glioblastoma cells. Sci. Rep. 2020, 10, 8348. [Google Scholar] [CrossRef] [PubMed]
- Guda, M.R.; Velpula, K.K.; Asuthkar, S.; Cain, C.P.; Tsung, A.J. Targeting RGS4 ablates glioblastoma proliferation. Int. J. Mol. Sci. 2020, 21, 3300. [Google Scholar] [CrossRef]
- Peng, H.; Du, B.; Jiang, H.; Gao, J. Over-expression of CHAF1A promotes cell proliferation and apoptosis resistance in glioblastoma cells via AKT/FOXO3a/Bim pathway. Biochem. Biophys. Res. Commun. 2016, 469, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Banik, N.L.; Ray, S.K. Combination of taxol and Bcl-2 siRNA induces apoptosis in human glioblastoma cells and inhibits invasion, angiogenesis and tumour growth. J. Cell Mol. Med. 2009, 13, 4205–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Szymura, S.J.; Bernal, G.M.; Wu, L.; Zhang, Z.; Crawley, C.D.; Voce, D.J.; Campbell, P.A.; Ranoa, D.E.; Weichselbaum, R.R.; Yamini, B. DDX39B interacts with the pattern recognition receptor pathway to inhibit NF-κB and sensitize to alkylating chemotherapy. BMC Biol. 2020, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Mikhailova, V.; Gulaia, V.; Tiasto, V.; Rybtsov, S.; Yatsunskaya, M.; Kagansky, A. Towards an advanced cell based. AIMS Genet. 2018, 5, 91–112. [Google Scholar] [CrossRef]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The extracellular matrix and biocompatible materials in glioblastoma treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Ayanlaja, A.A.; Ji, G.; Wang, J.; Gao, Y.; Cheng, B.; Kanwore, K.; Zhang, L.; Xiong, Y.; Kambey, P.A.; Gao, D. Doublecortin undergo nucleocytoplasmic transport via the RanGTPase signaling to promote glioma progression. Cell Commun. Signal. 2020, 18, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Cai, J.; Gao, W.; Meng, X.; Gao, F.; Wu, P.; Duan, C.; Wang, R.; Dinislam, M.; Lin, L.; et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018, 419, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Jahangiri, A.; Chen, W.; Nguyen, A.T.; Yagnik, G.; Pereira, M.P.; Jain, S.; Garcia, J.H.; Shah, S.S.; Wadhwa, H.; et al. Clonal ZEB1-driven mesenchymal transition promotes targetable oncologic antiangiogenic therapy resistance. Cancer Res. 2020, 80, 1498–1511. [Google Scholar] [CrossRef]
- Hegge, B.; Sjøttem, E.; Mikkola, I. Generation of a PAX6 knockout glioblastoma cell line with changes in cell cycle distribution and sensitivity to oxidative stress. BMC Cancer. 2018, 18, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, W.; Qiu, J.; Nassar, Z.D.; Shaw, P.N.; McMahon, K.A.; Ferguson, C.; Parton, R.G.; Riggins, G.J.; Harris, J.M.; Parat, M.O. A role for caveola-forming proteins caveolin-1 and CAVIN1 in the pro-invasive response of glioblastoma to osmotic and hydrostatic pressure. J. Cell Mol. Med. 2020, 24, 3724–3738. [Google Scholar] [CrossRef]
- Sui, A.; Xu, Y.; Yang, J.; Pan, B.; Wu, J.; Guo, T.; Shen, Y.; Guo, X. The histone H3 Lys 27 demethylase KDM6B promotes migration and invasion of glioma cells partly by regulating the expression of SNAI1. Neurochem. Int. 2019, 124, 123–129. [Google Scholar] [CrossRef]
- Tome-Garcia, J.; Erfani, P.; Nudelman, G.; Tsankov, A.M.; Katsyv, I.; Tejero, R.; Zhang, B.; Walsh, M.; Friedel, R.H.; Zaslavsky, E.; et al. Analysis of chromatin accessibility uncovers TEAD1 as a regulator of migration in human glioblastoma. Nat. Commun. 2018, 9, 4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Wang, R.; Chen, Y.; Meng, X.; Wu, P.; Li, Z.; Duan, C.; Li, Q.; Li, Y.; Zhao, S.; et al. QKI deficiency maintains glioma stem cell stemness by activating the SHH/GLI1 signaling pathway. Cell Oncol. 2019, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma model using human cerebral organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.A.; Shrivastava, G.; Balcom, E.F.; McKenzie, B.A.; Fernandes, J.; Branton, W.G.; Wheatley, B.M.; Petruk, K.; van Landeghem, F.; Power, C. Absent in melanoma 2 regulates tumor cell proliferation in glioblastoma multiforme. J. Neurooncol. 2019, 144, 265–273. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.; et al. ID1 is critical for tumorigenesis and regulates chemoresistance in glioblastoma. Cancer Res. 2019, 79, 4057–4071. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Qian, X.; Wang, B.; Xia, Y.; Zheng, Y.; Du, L.; Xu, D.; Xing, D.; DePinho, R.A.; Lu, Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22, 282–288. [Google Scholar] [CrossRef]
- Shi, T.; Guo, D.; Xu, H.; Su, G.; Chen, J.; Zhao, Z.; Shi, J.; Wedemeyer, M.; Attenello, F.; Zhang, L.; et al. HOTAIRM1, an enhancer lncRNA, promotes glioma proliferation by regulating long-range chromatin interactions within HOXA cluster genes. Mol. Biol. Rep. 2020, 47, 2723–2733. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Nakamura, N.S.; Parker, A.K.; Martuscello, R.T.; Reynolds, B.A.; Sarkisian, M.R. PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl. Oncol. 2016, 9, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Smith, D.K.; Ni, H.; Wu, K.; Huang, D.; Pan, S.; Sathe, A.A.; Tang, Y.; Liu, M.L.; Xing, C.; et al. SOX4-mediated repression of specific tRNAs inhibits proliferation of human glioblastoma cells. Proc. Natl. Acad. Sci. USA 2020, 117, 5782–5790. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.R.D.V.; Pour Khavari, A.; Rizzo, M.; Sakamoto-Hojo, E.T.; Haghdoost, S. Targeting NRF2, regulator of antioxidant system, to sensitize glioblastoma neurosphere cells to radiation-induced oxidative stress. Oxidative Med. Cell Longev. 2020, 2020, 2534643. [Google Scholar] [CrossRef]
- Patel, D.; Ahmad, F.; Kambach, D.M.; Sun, Q.; Halim, A.S.; Kramp, T.; Camphausen, K.A.; Stommel, J.M. LXRβ controls glioblastoma cell growth, lipid balance, and immune modulation independently of ABCA1. Sci. Rep. 2019, 9, 15458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, W.; Wang, J.; Wang, B.; Liu, F.; Li, M.; Zhao, Y.; Zhang, C.; Li, Q.; Chen, J.; Zhang, L.; et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo. Cancer Manag. Res. 2018, 10, 5747–5758. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018, 9, 22095–22112. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Patil, R.; Galstyan, A.; Klymyshyn, D.; Ding, H.; Chesnokova, A.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Shatalova, E.S.; et al. Blockade of a Laminin-411-Notch axis with CRISPR/Cas9 or a nanobioconjugate inhibits glioblastoma growth through tumor-microenvironment cross-talk. Cancer Res. 2019, 79, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Dutra-Clarke, M.; Breunig, J.J.; Sarkisian, M.R. Glioma cell proliferation is enhanced in the presence of tumor-derived cilia vesicles. Cilia 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sareddy, G.R.; Zhou, M.; Viswanadhapalli, S.; Li, X.; Lai, Z.; Tekmal, R.R.; Brenner, A.; Vadlamudi, R.K. Differential effects of estrogen receptor β isoforms on glioblastoma progression. Cancer Res. 2018, 78, 3176–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 39, 4421–4435. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De, J.N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.; Banik, N.L.; Ray, S.K. Synergistic anti-cancer mechanisms of curcumin and paclitaxel for growth inhibition of human brain tumor stem cells and LN18 and U138MG cells. Neurochem. Int. 2012, 61, 1102–1113. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.; Vazquez, N.; Lopez, A.; Fanniel, V.; Sanchez, L.; Marks, R.; Hinojosa, L.; Cuello, V.; Cuevas, M.; Rodriguez, A.; et al. The PI3K pathway impacts stem gene expression in a set of glioblastoma cell lines. J. Cancer Res. Clin. Oncol. 2020, 146, 593–604. [Google Scholar] [CrossRef]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Wu, Y.; Mayer, K.; von Rosenstiel, C.; Schecker, J.; Baur, S.; Würstle, S.; Liesche-Starnecker, F.; Gempt, J.; Schlegel, J. Lipid peroxidation plays an important role in chemotherapeutic effects of temozolomide and the development of therapy resistance in human glioblastoma. Transl. Oncol. 2020, 13, 100748. [Google Scholar] [CrossRef]
- Koga, T.; Chen, C.C.; Furnari, F.B. Genome engineering evolves brain tumor modeling. Neurol. Med. Chir. 2020, 60, 329–336. [Google Scholar] [CrossRef]

| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| DDX39B | Knockdown | U87MG | [70] |
| PDPN | Knockout | GBMF2, GBMF3, LN308, LN319 | [60] |
| Notch1 | Knockdown | U87MG, U251 | [33] |
| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| OPN | Knockout | GSCs | [40] |
| DDX39B | Knockout | U87MG | [70] |
| AIM2 | Knockdown | U251 | [83] |
| Target Gene | Type of CRISPR-Cas9 Mediated Genome Editing | GBM Model | References |
|---|---|---|---|
| FOXO3 | Knockdown | U87MG | [101] |
| FOXG1 | Deletion | GSCs | [102] |
| RGS4 | Deletion | GSC20 and GSC28 | [64] |
| ALDH1A3 | Knockdown | LN229, U87MG, T98G, GSC-like cells T84 and X01 | [103] |
| Dazl | Knockdown | A172, U251, and LN229 | [22] |
| Nrf2 | Knockdown | U87MG | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Sammarraie, N.; Ray, S.K. Applications of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme. Cells 2021, 10, 2342. https://doi.org/10.3390/cells10092342
Al-Sammarraie N, Ray SK. Applications of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme. Cells. 2021; 10(9):2342. https://doi.org/10.3390/cells10092342
Chicago/Turabian StyleAl-Sammarraie, Nadia, and Swapan K. Ray. 2021. "Applications of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme" Cells 10, no. 9: 2342. https://doi.org/10.3390/cells10092342
APA StyleAl-Sammarraie, N., & Ray, S. K. (2021). Applications of CRISPR-Cas9 Technology to Genome Editing in Glioblastoma Multiforme. Cells, 10(9), 2342. https://doi.org/10.3390/cells10092342