
The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters



{kind=link}
Abstract
1. Introduction
1.1. Myeloproliferative Neoplasms
1.2. MPN Microenvironment: Inflammation, Immunity and Beyond
1.3. MPN Microenvironment: The Power of Extracellular Vesicles
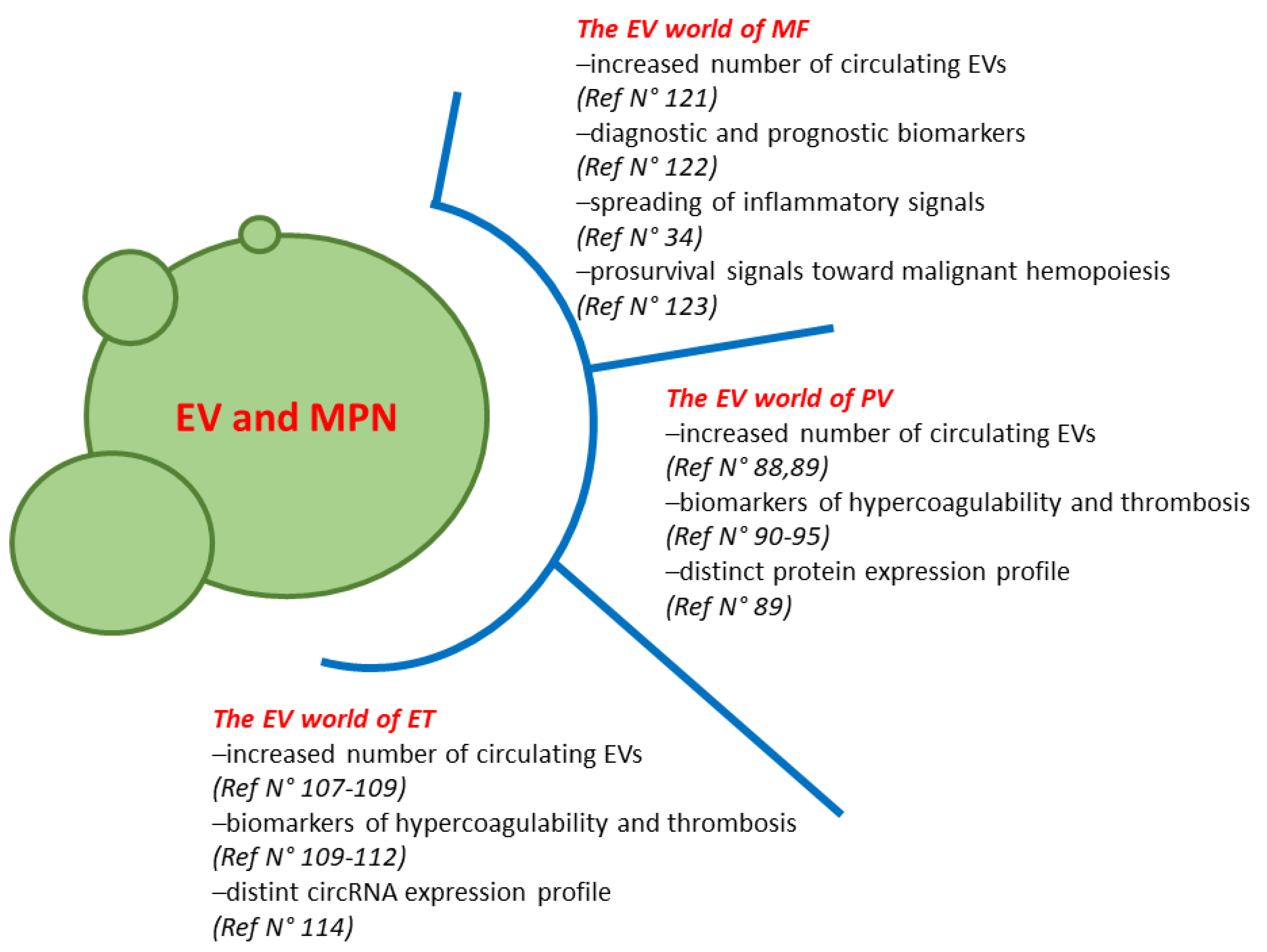
2. The EV World of PV
3. The EV World of ET
4. The EV World of MF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guy, A.; Poisson, J.; James, C. Pathogenesis of cardiovascular events in BCR-ABL1-negative myeloproliferative neoplasms. Leukemia 2021, 35, 935–955. [Google Scholar] [CrossRef]
- Reeves, B.N.; Beckman, J.D. Novel Pathophysiological Mechanisms of Thrombosis in Myeloproliferative Neoplasms. Curr. Hematol. Malig. Rep. 2021, 6, 2992–2993. [Google Scholar] [CrossRef]
- Schafer, A.I. Thrombotic, Vascular, and Bleeding Complications of the Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 305–324. [Google Scholar] [CrossRef]
- Brabrand, M.; Frederiksen, H. Risks of Solid and Lymphoid Malignancies in Patients with Myeloproliferative Neoplasms: Clinical Implications. Cancers 2020, 12, 3061. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, S.N.; Vainchenker, W.; Levy, G.; Papadopoulos, N. Functional Consequences of Mutations in Myeloproliferative Neoplasms. Hemasphere 2021, 5, e578. [Google Scholar] [CrossRef]
- Shallis, R.M.; Zeidan, A.M.; Wang, R.; Podoltsev, N.A. Epidemiology of the Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Mora, B.; Gangat, N.; Catani, L. Is there a gender effect in polycythemia vera? Ann. Hematol. 2021, 100, 11–25. [Google Scholar] [CrossRef]
- Karantanos, T.; Jain, T.; Moliterno, A.R.; Jones, R.J.; DeZern, A.E. Sex-Related Differences in Chronic Myeloid Neoplasms: From the Clinical Observation to the Underlying Biology. Int. J. Mol. Sci. 2021, 22, 2595. [Google Scholar] [CrossRef] [PubMed]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Guglielmelli, P.; Vannucchi, A.M. Impact of Mutational Profile on the Management of Myeloproliferative Neoplasms: A Short Review of the Emerging Data. Oncotargets Ther. 2020, 13, 12367–12382. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Hernandez, A.; Vizmanos, J.L. A Broad Overview of Signaling in pH-Negative Classic Myeloproliferative Neoplasms. Cancers 2021, 13, 984. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, G.; Fleischman, A.G. The Microenvironment in Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. [Google Scholar] [CrossRef] [PubMed]
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef]
- Zhan, H.; Kaushansky, K. The Hematopoietic Microenvironment in Myeloproliferative Neoplasms: The Interplay Between Nature (Stem Cells) and Nurture (the Niche). Adv. Exp. Med. Biol. 2020, 1273, 135–145. [Google Scholar] [CrossRef]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 3401. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.S.; How, J.; Allen, M.J.; Oh, S.T. Young versus old age at diagnosis confers distinct genomic profiles in patients with polycythemia vera. Leukemia 2019, 33, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Begna, K.; Finke, C.; Lasho, T.; Tefferi, A. Circulating levels of MCP-1, sIL-2R, IL-15, and IL-8 predict anemia response to pomalidomide therapy in myelofibrosis. Am. J. Hematol. 2011, 86, 343–345. [Google Scholar] [CrossRef]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Cacemiro, M.D.C.; Cominal, J.G.; Tognon, R.; Nunes, N.S.; Simoes, B.P.; Figueiredo-Pontes, L.L.; Catto, L.F.B.; Traina, F.; Souto, E.X.; Zambuzi, F.A.; et al. Philadelphia-negative myeloproliferative neoplasms as disorders marked by cytokine modulation. Hematol. Transfus. Cell Ther. 2018, 40, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.N.; Oh, S.T. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFkappaB signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: WheTher. to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediat. Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef]
- Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. [Google Scholar] [CrossRef]
- Barosi, G. An immune dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef]
- Sanchez, C.; Baier, C.; Colle, J.G.; Chelbi, R.; Rihet, P.; Le Treut, T.; Imbert, J.; Sebahoun, G.; Venton, G.; Costello, R.T. Natural killer cells in patients with polycythemia vera. Hum. Immunol. 2015, 76, 644–650. [Google Scholar] [CrossRef]
- Gersuk, G.M.; Carmel, R.; Pattamakom, S.; Challita, P.M.; Rabinowitz, A.P.; Pattengale, P.K. Quantitative and functional studies of impaired natural killer (NK) cells in patients with myelofibrosis, essential thrombocythemia, and polycythemia vera. I. A potential role for platelet-derived growth factor in defective NK cytotoxicity. Nat. Immun. 1993, 12, 136–151. [Google Scholar]
- Briard, D.; Brouty-Boye, D.; Giron-Michel, J.; Azzarone, B.; Jasmin, C.; Le Bousse-Kerdiles, C. Impaired NK cell differentiation of blood-derived CD34+ progenitors from patients with myeloid metaplasia with myelofibrosis. Clin. Immunol. 2003, 106, 201–212. [Google Scholar] [CrossRef]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-alpha in myeloproliferative neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef]
- Spanoudakis, E.; Papoutselis, M.; Bazdiara, I.; Lamprianidi, E.; Kordella, X.; Tilkeridis, C.; Tsatalas, C.; Kotsianidis, I. The JAK2V617F Point Mutation Increases the Osteoclast Forming Ability of Monocytes in Patients with Chronic Myeloproliferative Neoplasms and Makes their Osteoclasts more Susceptible to JAK2 Inhibition. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018058. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; Catani, L.; Ricci, F.; Romano, M.; Forte, D.; Auteri, G.; Bartoletti, D.; Ottaviani, E.; Tazzari, P.L.; Vianelli, N.; et al. The role of circulating monocytes and JAK inhibition in the infectious- driven inflammatory response of myelofibrosis. Oncoimmunology 2020, 9, 1782575. [Google Scholar] [CrossRef] [PubMed]
- Obro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-alpha and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. Hemasphere 2020, 4, e371. [Google Scholar] [CrossRef]
- Elliott, M.A.; Verstovsek, S.; Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Barraco, D.; Cerquozzi, S.; Gangat, N.; Patnaik, M.M.; Lasho, T.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Monocytosis in polycythemia vera: Clinical and molecular correlates. Am. J. Hematol. 2017, 92, 640–645. [Google Scholar] [CrossRef]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology 2017, 6, e1345402. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Holmstrom, M.O.; Riley, C.H.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia 2016, 30, 2413–2416. [Google Scholar] [CrossRef]
- Holmstrom, M.O.; Hjortso, M.D.; Ahmad, S.M.; Met, O.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F mutation is a target for specific T cells in the JAK2V617F-positive myeloproliferative neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, O.; Friese, C.; Kjaer, L.; Riley, C.H.; Thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef]
- Bozkus, C.C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef]
- Riley, C.H.; Jensen, M.K.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M. Increase in circulating CD4(+)CD25(+)Foxp3(+) T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-alpha. Blood 2011, 118, 2170–2173. [Google Scholar] [CrossRef][Green Version]
- Massa, M.; Campanelli, R.; Fois, G.; Villani, L.; Bonetti, E.; Catarsi, P.; Poletto, V.; Viarengo, G.; De Amici, M.; Rosti, V.; et al. Reduced frequency of circulating CD4+CD25brightCD127lowFOXP3+ regulatory T cells in primary myelofibrosis. Blood 2016, 128, 1660–1662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Korn, C.; Mendez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Gleitz, H.F.E.; Dugourd, A.J.F.; Leimkuhler, N.B.; Snoeren, I.A.M.; Fuchs, S.N.R.; Menzel, S.; Ziegler, S.; Kroger, N.; Triviai, I.; Busche, G.; et al. Increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood 2020, 136, 2051–2064. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.S.; Lai, D.M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Tai, Y.L.; Chu, P.Y.; Lee, B.H.; Chen, K.C.; Yang, C.Y.; Kuo, W.H.; Shen, T.L. Basics and applications of tumor-derived extracellular vesicles. J. Biomed. Sci. 2019, 26, 35. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Ratajczak, J. Extracellular microvesicles/exosomes: Discovery, disbelief, acceptance, and the future? Leukemia 2020, 34, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Matsumoto, K.; Akiyoshi, T.; Kubo, M.; Yamanaka, N.; Tasaki, A.; Nakashima, H.; Nakamura, M.; Kuroki, S.; Tanaka, M.; et al. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 2005, 25, 3703–3707. [Google Scholar]
- Flaumenhaft, R.; Mairuhu, A.T.; Italiano, J.E. Platelet- and megakaryocyte-derived microparticles. Semin. Thromb. Hemost. 2010, 36, 881–887. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrugger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell Vesicles 2019, 8, 1684862. [Google Scholar] [CrossRef]
- Pezzicoli, G.; Tucci, M.; Lovero, D.; Silvestris, F.; Porta, C.; Mannavola, F. Large Extracellular Vesicles-A New Frontier of Liquid Biopsy in Oncology. Int. J. Mol. Sci. 2020, 21, 6543. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L.; Yu, H.; McKenzie, A.J.; Franklin, J.L.; Patton, J.G.; Liu, Q.; Weaver, A.M. Quantitative Proteomic Analysis of Small and Large Extracellular Vesicles (EVs) Reveals Enrichment of Adhesion Proteins in Small EVs. J. Proteome Res. 2019, 18, 947–959. [Google Scholar] [CrossRef]
- Ciardiello, C.; Migliorino, R.; Leone, A.; Budillon, A. Large extracellular vesicles: Size matters in tumor progression. Cytokine Growth Factor Rev. 2020, 51, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Clancy, J.W.; Schmidtmann, M.; D’Souza-Schorey, C. The ins and outs of microvesicles. FASEB Bioadv. 2021, 3, 399–406. [Google Scholar] [CrossRef]
- Latifkar, A.; Hur, Y.H.; Sanchez, J.C.; Cerione, R.A.; Antonyak, M.A. New insights into extracellular vesicle biogenesis and function. J. Cell Sci. 2019, 132, 2406. [Google Scholar] [CrossRef]
- Donoso-Quezada, J.; Ayala-Mar, S.; Gonzalez-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 92, 1672. [Google Scholar] [CrossRef]
- Corbeil, D.; Santos, M.F.; Karbanova, J.; Kurth, T.; Rappa, G.; Lorico, A. Uptake and Fate of Extracellular Membrane Vesicles: Nucleoplasmic Reticulum-Associated Late Endosomes as a New Gate to Intercellular Communication. Cells 2020, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D. Isolation and molecular characterization of extracellular vesicles. Methods 2015, 87, 1–2. [Google Scholar] [CrossRef]
- Kreimer, S.; Belov, A.M.; Ghiran, I.; Murthy, S.K.; Frank, D.A.; Ivanov, A.R. Mass-spectrometry-based molecular characterization of extracellular vesicles: Lipidomics and proteomics. J. Proteome Res. 2015, 14, 2367–2384. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Tanasi, I.; Adamo, A.; Kamga, P.T.; Bazzoni, R.; Krampera, M. High-throughput analysis and functional interpretation of extracellular vesicle content in hematological malignancies. Comput Struct Biotechnol J. 2020, 18, 2670–2677. [Google Scholar] [CrossRef]
- Alzhrani, G.N.; Alanazi, S.T.; Alsharif, S.Y.; Albalawi, A.M.; Alsharif, A.A.; Abdel-Maksoud, M.S.; Elsherbiny, N. Exosomes: Isolation, characterization, and biomedical applications. Cell Biol. Int. 2021, 45, 1807–1831. [Google Scholar] [CrossRef]
- Forte, D.; Barone, M.; Palandri, F.; Catani, L. The “Vesicular Intelligence” Strategy of Blood Cancers. Genes 2021, 12, 416. [Google Scholar] [CrossRef]
- Laurenzana, I.; Lamorte, D.; Trino, S.; De Luca, L.; Ambrosino, C.; Zoppoli, P.; Ruggieri, V.; Del Vecchio, L.; Musto, P.; Caivano, A.; et al. Extracellular Vesicles: A New Prospective in Crosstalk between Microenvironment and Stem Cells in Hematological Malignancies. Stem Cells Int. 2018, 2018, 9863194. [Google Scholar] [CrossRef] [PubMed]
- Niazi, V.; Parseh, B.; Ahani, M.; Karami, F.; Gilanchi, S.; Atarodi, K.; Soufi, M.; Soleimani, M.; Ghafouri-Fard, S.; Taheri, M.; et al. Communication between stromal and hematopoietic stem cell by exosomes in normal and malignant bone marrow niche. Biomed. Pharmacother. 2020, 132, 110854. [Google Scholar] [CrossRef]
- Lombardi, M.; Gabrielli, M.; Adinolfi, E.; Verderio, C. Role of ATP in Extracellular Vesicle Biogenesis and Dynamics. Front. Pharmacol. 2021, 12, 654023. [Google Scholar] [CrossRef]
- Ciardiello, C.; Leone, A.; Lanuti, P.; Roca, M.S.; Moccia, T.; Minciacchi, V.R.; Minopoli, M.; Gigantino, V.; De Cecio, R.; Rippa, M.; et al. Large oncosomes overexpressing integrin alpha-V promote prostate cancer adhesion and invasion via AKT activation. J. Exp. Clin. Cancer Res. 2019, 38, 317. [Google Scholar] [CrossRef]
- Garnier, D. Reprogramming of GBM microenvironment by large oncosomes: ‘Traveling’ V-ATPases are doing more than acidification. EBioMedicine 2019, 41, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Spinelli, C.; Reis-Sobreiro, M.; Cavallini, L.; You, S.; Zandian, M.; Li, X.; Mishra, R.; Chiarugi, P.; Adam, R.M.; et al. MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res. 2017, 77, 2306–2317. [Google Scholar] [CrossRef]
- Morello, M.; Minciacchi, V.R.; de Candia, P.; Yang, J.; Posadas, E.; Kim, H.; Griffiths, D.; Bhowmick, N.; Chung, L.W.; Gandellini, P.; et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 2013, 12, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Garnier, D.; Lee, T.H.; D’Asti, E.; Montermini, L.; Meehan, B.; Rak, J. PML-RARa modulates the vascular signature of extracellular vesicles released by acute promyelocytic leukemia cells. Angiogenesis 2016, 19, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.T.; Abdelhamed, S.; Kurre, P. Extracellular vesicles in the hematopoietic microenvironment. Haematologica 2018, 103, 382–394. [Google Scholar] [CrossRef]
- Trino, S.; Lamorte, D.; Caivano, A.; De Luca, L.; Sgambato, A.; Laurenzana, I. Clinical relevance of extracellular vesicles in hematological neoplasms: From liquid biopsy to cell biopsy. Leukemia 2020, 27, 2776–2777. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef]
- Sankar, K.; Stein, B.L.; Rampal, R.K. Thrombosis in the Philadelphia Chromosome-Negative Myeloproliferative Neoplasms. Cancer Treat. Res. 2019, 179, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Cerquozzi, S.; Barraco, D.; Lasho, T.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; Tefferi, A. Risk factors for arterial versus venous thrombosis in polycythemia vera: A single center experience in 587 patients. Blood Cancer J. 2017, 7, 662. [Google Scholar] [CrossRef] [PubMed]
- Putter, J.S.; Seghatchian, J. Polycythaemia vera: Molecular genetics, diagnostics and therapeutics. Vox Sang. 2021, 6, 69. [Google Scholar] [CrossRef]
- Ahadon, M.; Abdul Aziz, S.; Wong, C.L.; Leong, C.F. Plasma-derived microparticles in polycythaemia vera. Malays. J. Pathol. 2018, 40, 41–48. [Google Scholar]
- Fel, A.; Lewandowska, A.E.; Petrides, P.E.; Wisniewski, J.R. Comparison of Proteome Composition of Serum Enriched in Extracellular Vesicles Isolated from Polycythemia Vera Patients and Healthy Controls. Proteomes 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Duchemin, J.; Ugo, V.; Ianotto, J.C.; Lecucq, L.; Mercier, B.; Abgrall, J.F. Increased circulating procoagulant activity and thrombin generation in patients with myeloproliferative neoplasms. Thromb. Res. 2010, 126, 238–242. [Google Scholar] [CrossRef]
- Tan, X.; Shi, J.; Fu, Y.; Gao, C.; Yang, X.; Li, J.; Wang, W.; Hou, J.; Li, H.; Zhou, J. Role of erythrocytes and platelets in the hypercoagulable status in polycythemia vera through phosphatidylserine exposure and microparticle generation. Thromb. Haemost. 2013, 109, 1025–1032. [Google Scholar] [CrossRef]
- Kissova, J.; Ovesna, P.; Bulikova, A.; Zavrelova, J.; Penka, M. Increasing procoagulant activity of circulating microparticles in patients with Philadelphia-negative myeloproliferative neoplasms: A single-centre experience. Blood Coagul. Fibrinolysis 2015, 26, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qi, J.; Zhao, S.; Shen, W.; Dai, L.; Han, W.; Huang, M.; Wang, Z.; Ruan, C.; Wu, D.; et al. Clinical significance of circulating microparticles in pH(-) myeloproliferative neoplasms. Oncol. Lett. 2017, 14, 2531–2536. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tanaka, H.; Luis, E.J.; Sakai, K.; Kumode, T.; Sano, K.; Serizawa, K.; Rai, S.; Morita, Y.; Hanamoto, H.; et al. Elevated plasma levels of procoagulant microparticles are a novel risk factor for thrombosis in patients with myeloproliferative neoplasms. Int. J. Hematol. 2017, 106, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Tanguy, M.; Davy, H.; Camara, F.; El Mdawar, M.B.; Kheloufi, M.; Dagher, T.; Devue, C.; Lasselin, J.; Plessier, A.; et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Investig. 2020, 130, 2630–2643. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and oTher. myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Alimam, S.; Villiers, W.; Dillon, R.; Simpson, M.; Runglall, M.; Smith, A.; Chatzikyriakou, P.; Lavender, P.; Kanda, A.; Mills, K.; et al. Patients with triple-negative, JAK2V617F- and CALR-mutated essential thrombocythemia share a unique gene expression signature. Blood Adv. 2021, 5, 1059–1068. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Godfrey, A.L. Low-Risk Essential Thrombocythemia: A Comprehensive Review. Hemasphere 2021, 5, e521. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, D.; Kosiorek, H.E.; Dueck, A.C.; Hoffman, R. Evaluation of Therapeutic Strategies to Reduce the Number of Thrombotic Events in Patients With Polycythemia Vera and Essential Thrombocythemia. Front. Oncol. 2020, 10, 636675. [Google Scholar] [CrossRef]
- Kishtagari, A.; Gerds, A.T. Unmet Need in Essential Thrombocythemia and Polycythemia Vera. Hematol. Oncol. Clin. N. Am. 2021, 35, 295–303. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A. Essential Thrombocythemia. N. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Connor, D.E.; Ma, D.D.; Joseph, J.E. Flow cytometry demonstrates differences in platelet reactivity and microparticle formation in subjects with thrombocytopenia or thrombocytosis due to primary haematological disorders. Thromb. Res. 2013, 132, 572–577. [Google Scholar] [CrossRef]
- Moles-Moreau, M.P.; Ternisien, C.; Tanguy-Schmidt, A.; Boyer, F.; Gardembas, M.; Dib, M.; Ponthieux, A.; Guardiola, P.; Ifrah, N.; Hunault-Berger, M. Flow cytometry-evaluated platelet CD36 expression, reticulated platelets and platelet microparticles in essential thrombocythaemia and secondary thrombocytosis. Thromb. Res. 2010, 126, e394–e396. [Google Scholar] [CrossRef]
- Trappenburg, M.C.; van Schilfgaarde, M.; Marchetti, M.; Spronk, H.M.; ten Cate, H.; Leyte, A.; Terpstra, W.E.; Falanga, A. Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia. Haematologica 2009, 94, 911–918. [Google Scholar] [CrossRef]
- Marchetti, M.; Tartari, C.J.; Russo, L.; Panova-Noeva, M.; Leuzzi, A.; Rambaldi, A.; Finazzi, G.; Woodhams, B.; Falanga, A. Phospholipid-dependent procoagulant activity is highly expressed by circulating microparticles in patients with essential thrombocythemia. Am. J. Hematol. 2014, 89, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, A.; Lebreton, A.; Rauch, A.; Bauters, A.; Trillot, N.; Nibourel, O.; Tintillier, V.; Wemeau, M.; Demory, J.L.; Preudhomme, C.; et al. Microparticle phenotypes are associated with driver mutations and distinct thrombotic risks in essential thrombocythemia. Haematologica 2016, 101, e365–e368. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Steurer, M.; Feistritzer, C.; Murphy, C.; Eakins, E.; Van Schilfgaarde, M.; Corvetta, D.; Di Pierro, A.M.; Pusceddu, I.; Marcheselli, L.; et al. Observational retrospective study of vascular modulator changes during treatment in essential thrombocythemia. Transl. Res. 2017, 184, 21–34. [Google Scholar] [CrossRef]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in human diseases and potential use as biomarkers. Cell Death Dis. 2021, 12, 468. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, G.; He, H.; Zheng, Z.; Li, X.; Lin, R.; Xu, D. Differential expression of circular RNAs in bone marrow-derived exosomes from essential thrombocythemia patients. Cell Biol. Int. 2021, 45, 869–881. [Google Scholar] [CrossRef]
- Garmezy, B.; Schaefer, J.K.; Mercer, J.; Talpaz, M. A provider’s guide to primary myelofibrosis: Pathophysiology, diagnosis, and management. Blood Rev. 2021, 45, 100691. [Google Scholar] [CrossRef]
- Gangat, N.; Tefferi, A. Myelofibrosis biology and contemporary management. Br. J. Haematol. 2020, 191, 152–170. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Melo-Cardenas, J.; Migliaccio, A.R.; Crispino, J.D. The Role of Megakaryocytes in Myelofibrosis. Hematol. Oncol. Clin. N. Am. 2021, 35, 191–203. [Google Scholar] [CrossRef]
- Rumi, E.; Trotti, C.; Vanni, D.; Casetti, I.C.; Pietra, D.; Sant’Antonio, E. The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance. Int. J. Mol. Sci. 2020, 21, 8885. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Mascarenhas, J. Current Clinical Investigations in Myelofibrosis. Hematol. Oncol. Clin. N. Am. 2021, 35, 353–373. [Google Scholar] [CrossRef]
- Caivano, A.; Laurenzana, I.; De Luca, L.; La Rocca, F.; Simeon, V.; Trino, S.; D’Auria, F.; Traficante, A.; Maietti, M.; Izzo, T.; et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol. 2015, 36, 9739–9752. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; Ricci, F.; Sollazzo, D.; Ottaviani, E.; Romano, M.; Auteri, G.; Bartoletti, D.; Reggiani, M.L.B.; Vianelli, N.; Tazzari, P.L.; et al. Circulating megakaryocyte and platelet microvesicles correlate with response to ruxolitinib and distinct disease severity in patients with myelofibrosis. Br. J. Haematol. 2019, 185, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Forte, D.; Barone, M.; Morsiani, C.; Simonetti, G.; Fabbri, F.; Bruno, S.; Bandini, E.; Sollazzo, D.; Collura, S.; Deregibus, M.C.; et al. Distinct profile of CD34(+) cells and plasma-derived extracellular vesicles from triple-negative patients with Myelofibrosis reveals potential markers of aggressive disease. J. Exp. Clin. Cancer Res. 2021, 40, 49. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catani, L.; Cavo, M.; Palandri, F. The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters. Cells 2021, 10, 2316. https://doi.org/10.3390/cells10092316
Catani L, Cavo M, Palandri F. The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters. Cells. 2021; 10(9):2316. https://doi.org/10.3390/cells10092316
Chicago/Turabian StyleCatani, Lucia, Michele Cavo, and Francesca Palandri. 2021. "The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters" Cells 10, no. 9: 2316. https://doi.org/10.3390/cells10092316
APA StyleCatani, L., Cavo, M., & Palandri, F. (2021). The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters. Cells, 10(9), 2316. https://doi.org/10.3390/cells10092316