
Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal



Abstract
1. Introduction
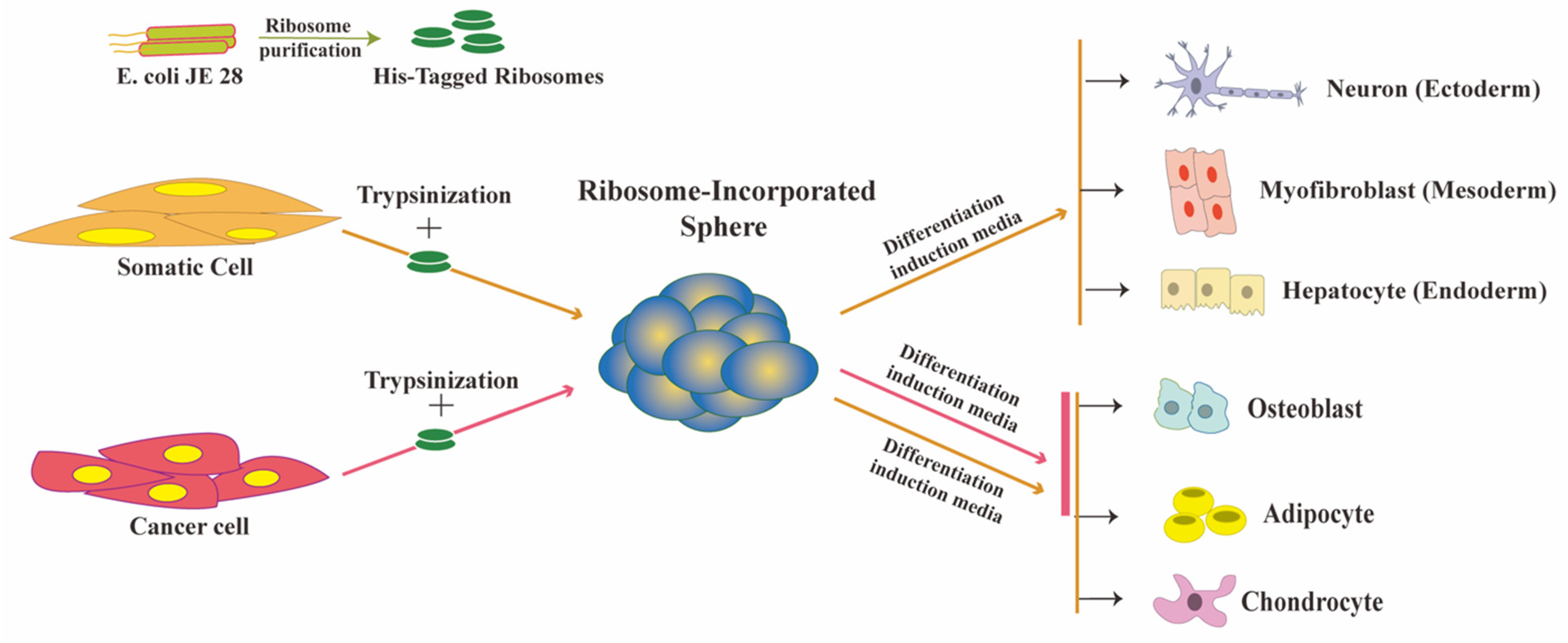
2. Ribosome Incorporation for the Generation of Multipotent Cells from Somatic Cells
2.1. Ribosomes Are the Bacteria-Derived Factors Inducing Cluster Formation and Reprogramming
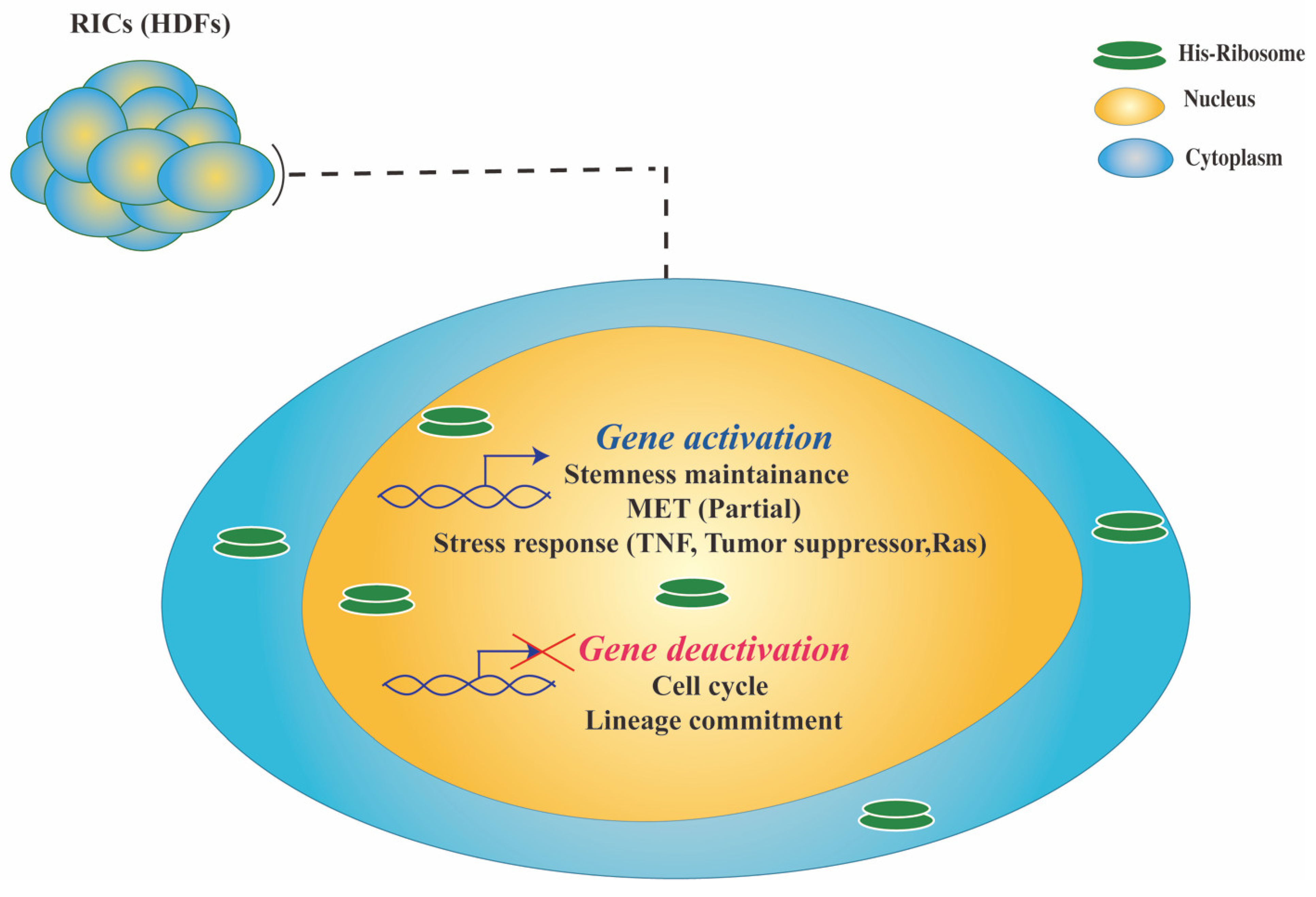
2.2. Downstream Events Underlying Cell Reprogramming Triggered by Ribosomes
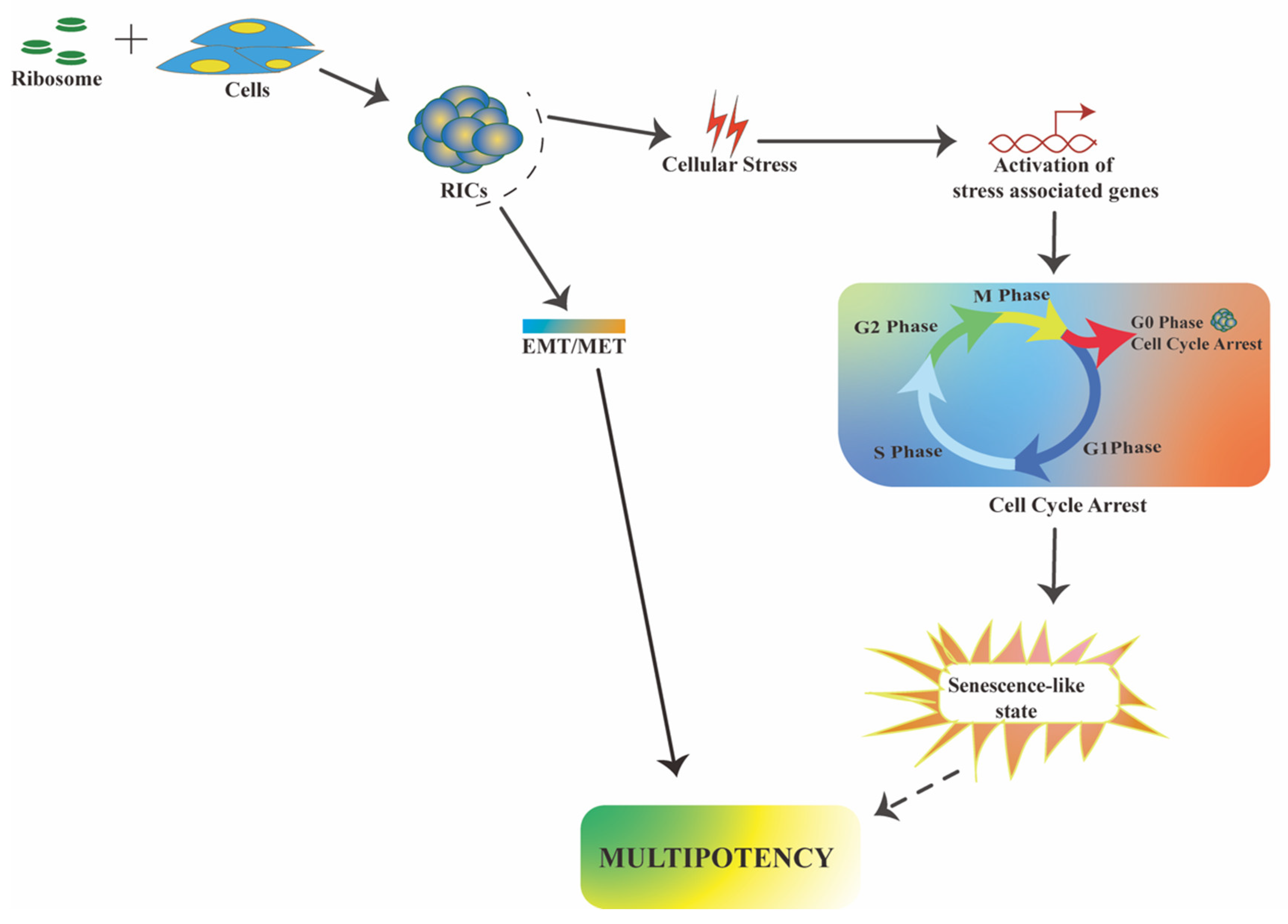
2.2.1. RICs Multipotency Is Coupled with Cellular Stress
2.2.2. RICs Multipotency Induction Involves a Partial MET Process
3. Ribosome Incorporation Modulates Cancer Cell Fate
3.1. Ribosomes Induce Cellular Stress in Cancer Cells
3.2. Ribosomes Induce Partial EMT in Cancer Cells
4. Mechanistic Insights in Ribosome-Induced Multipotency
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahla, R.S. Stem Cells Applications in Regenerative Medicine and Disease Therapeutics. Int. J. Cell Biol. 2016, 2016, 6940283. [Google Scholar] [CrossRef]
- Singh, V.K.; Saini, A.; Kalsan, M.; Kumar, N.; Chandra, R. Describing the Stem Cell Potency: The Various Methods of Functional Assessment and In Silico Diagnostics. Front. Cell Dev. Biol. 2016, 4, 134. [Google Scholar] [CrossRef] [PubMed]
- Romito, A.; Cobellis, G. Pluripotent Stem Cells: Current Understanding and Future Directions. Stem Cells Int. 2016, 2016, 9451492. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, A.; Khanlarkhani, N.; Baazm, M.; Mohammadzadeh, F.; Najafi, A.; Mehdinejadiani, S.; Sargolzaei Aval, F. Multipotent Stem Cell and Current Application. Acta Med. Iran. 2017, 55, 6–23. [Google Scholar]
- Estrov, Z. Stem Cells and Somatic Cells: Reprogramming and Plasticity. Clin. Lymphoma Myeloma 2009, 9, S319–S328. [Google Scholar] [CrossRef] [PubMed]
- Merrell, A.J.; Stanger, B.Z. Adult Cell Plasticity in Vivo: De-Differentiation and Transdifferentiation Are Back in Style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Galliot, B. Hydra, a Fruitful Model System for 270 Years. Int. J. Dev. Biol. 2012, 56, 411–423. [Google Scholar] [CrossRef]
- Baguñà, J. The Planarian Neoblast: The Rambling History of Its Origin and Some Current Black Boxes. Int. J. Dev. Biol. 2012, 56, 19–37. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Stem Cell Plasticity. Plasticity of Epithelial Stem Cells in Tissue Regeneration. Science 2014, 344, 1242281. [Google Scholar] [CrossRef]
- Gurdon, J.B. The Developmental Capacity of Nuclei Taken from Intestinal Epithelium Cells of Feeding Tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar]
- Matoba, S.; Zhang, Y. Somatic Cell Nuclear Transfer Reprogramming: Mechanisms and Applications. Cell Stem Cell 2018, 23, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, A.R.; Fisher, A.G. Reprogramming of Somatic Cells Towards Pluripotency by Cell Fusion. Methods Mol. Biol. 2016, 1480, 289–299. [Google Scholar] [CrossRef]
- Xie, X.; Fu, Y.; Liu, J. Chemical Reprogramming and Transdifferentiation. Curr. Opin. Genet. Dev. 2017, 46, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct Cell Reprogramming: Approaches, Mechanisms and Progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar] [CrossRef]
- Ito, N.; Katoh, K.; Kushige, H.; Saito, Y.; Umemoto, T.; Matsuzaki, Y.; Kiyonari, H.; Kobayashi, D.; Soga, M.; Era, T.; et al. Ribosome Incorporation into Somatic Cells Promotes Lineage Transdifferentiation towards Multipotency. Sci. Rep. 2018, 8, 1634. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Seo, B.J.; Hong, Y.J.; Do, J.T. Cellular Reprogramming Using Protein and Cell-Penetrating Peptides. Int. J. Mol. Sci. 2017, 18, 552. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. A Decade of Transcription Factor-Mediated Reprogramming to Pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- Warner, J.R.; McIntosh, K.B. How Common Are Extra-Ribosomal Functions of Ribosomal Proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liao, W.-J.; Liao, J.-M.; Liao, P.; Lu, H. Ribosomal Proteins: Functions beyond the Ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef]
- Ito, N.; Anam, M.B.; Ahmad, S.A.I.; Ohta, K. Transdifferentiation of Human Somatic Cells by Ribosome. Dev. Growth Differ. 2018, 60, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Anam, M.B.; Istiaq, A.; Kariya, R.; Kudo, M.; Ishtiyaq Ahmad, S.A.; Ito, N.; Okada, S.; Ohta, K. Ribosome Induces Transdifferentiation of A549 and H-111-TC Cancer Cell Lines. Biochem. Biophys. Rep. 2021, 26, 100946. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Anam, M.B.; Istiaq, A.; Ahmad, S.A.I.; Ito, N.; Ohta, K. Ribosome Incorporation Induces EMT-like Phenomenon with Cell Cycle Arrest in Human Breast Cancer Cell. Cells Tissues Organs 2021, 1–10. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Tumor Cells and the Onset of Cancer. In Molecular Cell Biology, 4th ed.; Macmillan: New York, NY, USA, 2000. [Google Scholar]
- Interplay between the Cancer Genome and Epigenome: Cell. Available online: https://www.cell.com/fulltext/S0092-8674(13)00296-1 (accessed on 23 March 2021).
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. Lynn Margulis and the Origin of the Eukaryotes. J. Theor. Biol. 2017, 434, 1. [Google Scholar] [CrossRef]
- Ohta, K.; Kawano, R.; Ito, N. Lactic Acid Bacteria Convert Human Fibroblasts to Multipotent Cells. PLoS ONE 2012, 7, e51866. [Google Scholar] [CrossRef]
- Ito, N.; Ohta, K. Reprogramming of Human Somatic Cells by Bacteria. Dev. Growth Differ. 2015, 57, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes Wide Open: A Critical Review of Sphere-Formation as an Assay for Stem Cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of Neurons and Astrocytes from Isolated Cells of the Adult Mammalian Central Nervous System. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef]
- Ederth, J.; Mandava, C.S.; Dasgupta, S.; Sanyal, S. A Single-Step Method for Purification of Active His-Tagged Ribosomes from a Genetically Engineered Escherichia Coli. Nucleic Acids Res. 2009, 37, e15. [Google Scholar] [CrossRef]
- Lecompte, O.; Ripp, R.; Thierry, J.; Moras, D.; Poch, O. Comparative Analysis of Ribosomal Proteins in Complete Genomes: An Example of Reductive Evolution at the Domain Scale. Nucleic Acids Res. 2002, 30, 5382–5390. [Google Scholar] [CrossRef]
- Nierhaus, K.H. Mg2+, K+, and the Ribosome. J. Bacteriol. 2014, 196, 3817–3819. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, T.; Honma, S.; Nomura, T.; Dabbs, E.R.; Hachimori, A. Translation Elongation by a Hybrid Ribosome in Which Proteins at the GTPase Center of the Escherichia Coli Ribosome Are Replaced with Rat Counterparts. J. Biol. Chem. 2002, 277, 3857–3862. [Google Scholar] [CrossRef] [PubMed]
- García-Marcos, A.; Morreale, A.; Guarinos, E.; Briones, E.; Remacha, M.; Ortiz, A.R.; Ballesta, J.P.G. In Vivo Assembling of Bacterial Ribosomal Protein L11 into Yeast Ribosomes Makes the Particles Sensitive to the Prokaryotic Specific Antibiotic Thiostrepton. Nucleic Acids Res. 2007, 35, 7109–7117. [Google Scholar] [CrossRef][Green Version]
- Ni, J.-Q.; Liu, L.-P.; Hess, D.; Rietdorf, J.; Sun, F.-L. Drosophila Ribosomal Proteins Are Associated with Linker Histone H1 and Suppress Gene Transcription. Genes Dev. 2006, 20, 1959–1973. [Google Scholar] [CrossRef]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal Protein S3: A KH Domain Subunit in NF-KappaB Complexes That Mediates Selective Gene Regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef]
- Mazumder, B.; Sampath, P.; Seshadri, V.; Maitra, R.K.; DiCorleto, P.E.; Fox, P.L. Regulated Release of L13a from the 60S Ribosomal Subunit as a Mechanism of Transcript-Specific Translational Control. Cell 2003, 115, 187–198. [Google Scholar] [CrossRef]
- Imafuku, I.; Masaki, T.; Waragai, M.; Takeuchi, S.; Kawabata, M.; Hirai, S.; Ohno, S.; Nee, L.E.; Lippa, C.F.; Kanazawa, I.; et al. Presenilin 1 Suppresses the Function of C-Jun Homodimers via Interaction with QM/Jif-1. J. Cell Biol. 1999, 147, 121–134. [Google Scholar] [CrossRef]
- Sengupta, J.; Nilsson, J.; Gursky, R.; Spahn, C.M.T.; Nissen, P.; Frank, J. Identification of the Versatile Scaffold Protein RACK1 on the Eukaryotic Ribosome by Cryo-EM. Nat. Struct. Mol. Biol. 2004, 11, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct Analysis of Protein Complexes Using Mass Spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-Mediated Specificity in Hox MRNA Translation and Vertebrate Tissue Patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef]
- Duncan, K.A.; Jimenez, P.; Carruth, L.L. The Selective Estrogen Receptor-Alpha Coactivator, RPL7, and Sexual Differentiation of the Songbird Brain. Psychoneuroendocrinology 2009, 34 (Suppl. 1), S30–S38. [Google Scholar] [CrossRef]
- Anderson, S.J.; Lauritsen, J.P.H.; Hartman, M.G.; Foushee, A.M.D.; Lefebvre, J.M.; Shinton, S.A.; Gerhardt, B.; Hardy, R.R.; Oravecz, T.; Wiest, D.L. Ablation of Ribosomal Protein L22 Selectively Impairs Alphabeta T Cell Development by Activation of a P53-Dependent Checkpoint. Immunity 2007, 26, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Provost, E.; Wehner, K.A.; Zhong, X.; Ashar, F.; Nguyen, E.; Green, R.; Parsons, M.J.; Leach, S.D. Ribosomal Biogenesis Genes Play an Essential and P53-Independent Role in Zebrafish Pancreas Development. Development 2012, 139, 3232–3241. [Google Scholar] [CrossRef]
- Iizumi, Y.; Oishi, M.; Taniguchi, T.; Goi, W.; Sowa, Y.; Sakai, T. The Flavonoid Apigenin Downregulates CDK1 by Directly Targeting Ribosomal Protein S9. PLoS ONE 2013, 8, e73219. [Google Scholar] [CrossRef]
- Kuroda, K.; Takenoyama, M.; Baba, T.; Shigematsu, Y.; Shiota, H.; Ichiki, Y.; Yasuda, M.; Uramoto, H.; Hanagiri, T.; Yasumoto, K. Identification of Ribosomal Protein L19 as a Novel Tumor Antigen Recognized by Autologous Cytotoxic T Lymphocytes in Lung Adenocarcinoma. Cancer Sci. 2010, 101, 46–53. [Google Scholar] [CrossRef]
- Teshigawara, R.; Cho, J.; Kameda, M.; Tada, T. Mechanism of Human Somatic Reprogramming to IPS Cell. Lab. Investig. 2017, 97, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Gil, J. Induced Pluripotent Stem Cells and Senescence: Learning the Biology to Improve the Technology. EMBO Rep. 2010, 11, 353–359. [Google Scholar] [CrossRef]
- Xiao, T.; Xu, Z.; Zhang, H.; Geng, J.; Qiao, Y.; Liang, Y.; Yu, Y.; Dong, Q.; Suo, G. TP53I11 Suppresses Epithelial-Mesenchymal Transition and Metastasis of Breast Cancer Cells. BMB Rep. 2019, 52, 379–384. [Google Scholar] [CrossRef]
- Xiong, X.-F.; Li, H.; Cao, E.-H. PIG11 Protein Binds to DNA in Sequence-Independent Manner in Vitro. Biochem. Biophys. Res. Commun. 2007, 358, 29–34. [Google Scholar] [CrossRef]
- Tong, Y.; Liu, H. P15—A New Tumor Suppressor Gene. Chin. Sci. Bull. 1999, 44, 1157–1163. [Google Scholar] [CrossRef]
- Bergholz, J.; Xiao, Z.-X. Role of P63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron. 2012, 5, 311–322. [Google Scholar] [CrossRef]
- Seibold, S.; Rudroff, C.; Weber, M.; Galle, J.; Wanner, C.; Marx, M. Identification of a New Tumor Suppressor Gene Located at Chromosome 8p21.3–22. FASEB J. 2003, 17, 1180–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yu, W.; Liang, G.; Jia, Z.; Chen, Z.; Zhao, L.; Yuan, Y.; Zhou, X.; Li, D.; Shen, S.; et al. Tumor Suppressor Candidate 1 Suppresses Cell Growth and Predicts Better Survival in Glioblastoma. Cell Mol. Neurobiol. 2017, 37, 37–42. [Google Scholar] [CrossRef]
- Li, Q.; Wang, L.; Tan, W.; Peng, Z.; Luo, Y.; Zhang, Y.; Zhang, G.; Na, D.; Jin, P.; Shi, T.; et al. Identification of C1qTNF-Related Protein 4 as a Potential Cytokine That Stimulates the STAT3 and NF-ΚB Pathways and Promotes Cell Survival in Human Cancer Cells. Cancer Lett. 2011, 308, 203–214. [Google Scholar] [CrossRef]
- Das, T.; Chen, Z.; Hendriks, R.W.; Kool, M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Guo, F.; Yuan, Y. Tumor Necrosis Factor Alpha-Induced Proteins in Malignant Tumors: Progress and Prospects. Onco. Targets Ther. 2020, 13, 3303–3318. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Ashkenazi, A. Apo2L/TRAIL: Apoptosis Signaling, Biology, and Potential for Cancer Therapy. Cytokine Growth Factor Rev. 2003, 14, 337–348. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Cismowski, M.J.; Wang, G.; Vincent, T.S.; Brown, K.D.; Lanier, S.M. The Ras-Related Protein AGS1/RASD1 Suppresses Cell Growth. Oncogene 2004, 23, 5858–5863. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xie, M.; Zhang, L.; Zhou, X.; Xie, H.; Zhou, L.; Zheng, S.; Wang, W. Ras-Related Associated with Diabetes Gene Acts as a Suppressor and Inhibits Warburg Effect in Hepatocellular Carcinoma. Onco. Targets Ther. 2016, 9, 3925–3937. [Google Scholar] [CrossRef][Green Version]
- Leszczynska, K.; Kaur, S.; Wilson, E.; Bicknell, R.; Heath, V.L. The Role of RhoJ in Endothelial Cell Biology and Angiogenesis. Biochem. Soc. Trans. 2011, 39, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Velez, A.M.A.; Howard, M.S. Tumor-Suppressor Genes, Cell Cycle Regulatory Checkpoints, and the Skin. N. Am. J. Med. Sci. 2015, 7, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Manning, A.M. NF-ΚB as a Primary Regulator of the Stress Response. Oncogene 1999, 18, 6163–6171. [Google Scholar] [CrossRef]
- Goodsell, D.S. The Molecular Perspective: The Ras Oncogene. Oncologist 1999, 4, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Pei, D. The Function and Regulation of Mesenchymal-to-Epithelial Transition in Somatic Cell Reprogramming. Curr. Opin. Genet. Dev. 2014, 28, 32–37. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the Mesenchymal–Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 Is a Nuclear Protein Required for Cell Cycle Progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of P53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-Induced Cell Cycle Arrest Compromised by P21 Deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Trocoli, A.; Djavaheri-Mergny, M. The Complex Interplay between Autophagy and NF-ΚB Signaling Pathways in Cancer Cells. Am. J. Cancer Res. 2011, 1, 629–649. [Google Scholar] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Vogelmann, R.; Nguyen-Tat, M.-D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-Induced Downregulation of E-Cadherin-Based Cell-Cell Adhesion Depends on PI3-Kinase and PTEN. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-Mesenchymal Transition (EMT): A Biological Process in the Development, Stem Cell Differentiation and Tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.-J.; Yoo, J. Snail Is Required for Transforming Growth Factor-Beta-Induced Epithelial-Mesenchymal Transition by Activating PI3 Kinase/Akt Signal Pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef]
- Tubiana, M. Tumor Cell Proliferation Kinetics and Tumor Growth Rate. Acta Oncol. 1989, 28, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gaviraghi, M.; Vivori, C.; Tonon, G. How Cancer Exploits Ribosomal RNA Biogenesis: A Journey beyond the Boundaries of RRNA Transcription. Cells 2019, 8, 1098. [Google Scholar] [CrossRef]
- Himanen, S.V.; Sistonen, L. New Insights into Transcriptional Reprogramming during Cellular Stress. J. Cell Sci. 2019, 132, jcs238402. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming in Vivo. Science 2016, 354. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene | Gene Upregulation in RICs (Fold Change) | Stress-Related Functions |
|---|---|---|---|
| Tumor suppressor/growth suppressor | TP53I11, tumor protein p53 inducible protein 11 | 2.58 | Metastasis and EMT inhibition [52], Apoptosis [53] |
| CDKN2B, cyclin-dependent kinase inhibitor 2B (p15) | 1.27 | Inhibits CDK4, growth suppression [54] | |
| TP63, tumor protein 63 | 3.0 | Growth suppression, senescence, survival [55] | |
| MTUS1, microtubule-associated tumor suppressor | 1.47 | Growth suppression and senescence [56] | |
| TUSC1, tumor suppressor candidate 1 | 1.47 | Growth suppression [57] | |
| Tumor necrosis factor | C1QTNF4, C1q, and tumor necrosis factor-related protein 4 | 3.04 | Activates the NF-kappaB pathway, survival [58] |
| TNFAIP3, tumor necrosis factor, alpha-induced protein 3 | 1.68 | Regulates the NF-kappaB pathway, apoptosis [59] | |
| TNFAIP6, tumor necrosis factor, alpha-induced protein 6 | 2.59 | Activates the NF-kappaB pathway [60] | |
| TNFSF10, tumor necrosis factor (ligand) superfamily, member 10 | 4.04 | Apoptosis [61] | |
| Ras-associated genes | RASD1, Ras dexamethasone-induced 1 | 2.47 | Stress response, suppression of aberrant cell growth, proliferation [62] |
| RRAGD, Ras-related GTP binding D | 2.57 | Stress response, regulation of mTORC1 signaling [63] | |
| RRAD, Ras-related associated with diabetes | 1.47 | Regulation of cell cycle, apoptosis [64] | |
| RHOJ, Ras homolog gene family, member J | 3.98 | Proliferation [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istiaq, A.; Ohta, K. Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal. Cells 2021, 10, 2276. https://doi.org/10.3390/cells10092276
Istiaq A, Ohta K. Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal. Cells. 2021; 10(9):2276. https://doi.org/10.3390/cells10092276
Chicago/Turabian StyleIstiaq, Arif, and Kunimasa Ohta. 2021. "Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal" Cells 10, no. 9: 2276. https://doi.org/10.3390/cells10092276
APA StyleIstiaq, A., & Ohta, K. (2021). Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal. Cells, 10(9), 2276. https://doi.org/10.3390/cells10092276