
A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
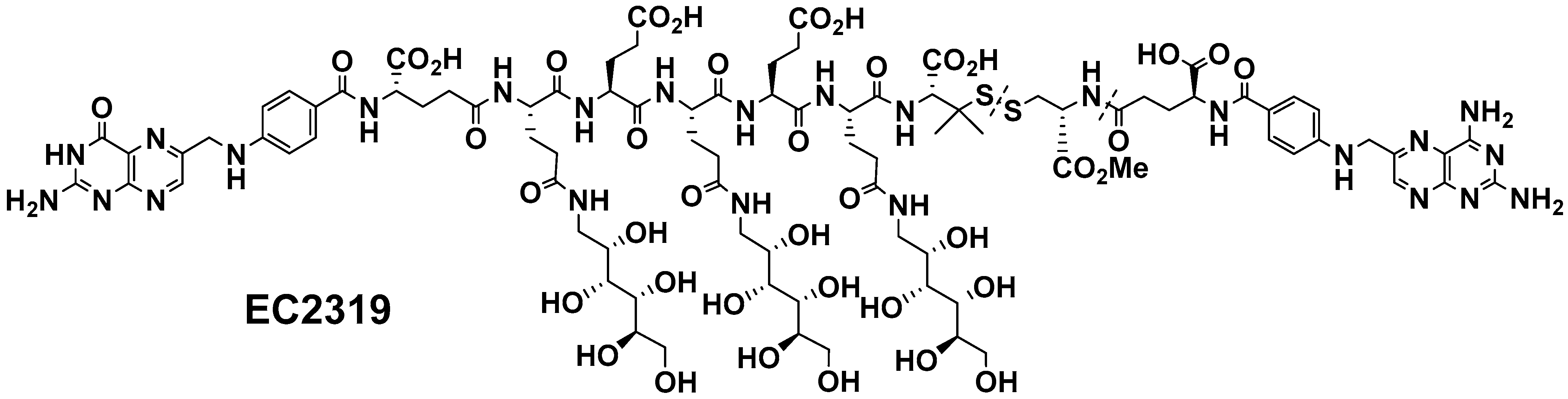
2.1. EC2319 Modular Design
2.2. Induction, Treatment, and Analysis of Accelerated Anti-GBM GN
2.3. mRNA Expression of Chemokines and Cytokines
2.4. Morphological Analysis, Immunohistochemical Phenotyping, and Quantitation of Leukocytes
2.5. Immunohistochemistry of Collagen
2.6. Immunohistochemistry of FRβ
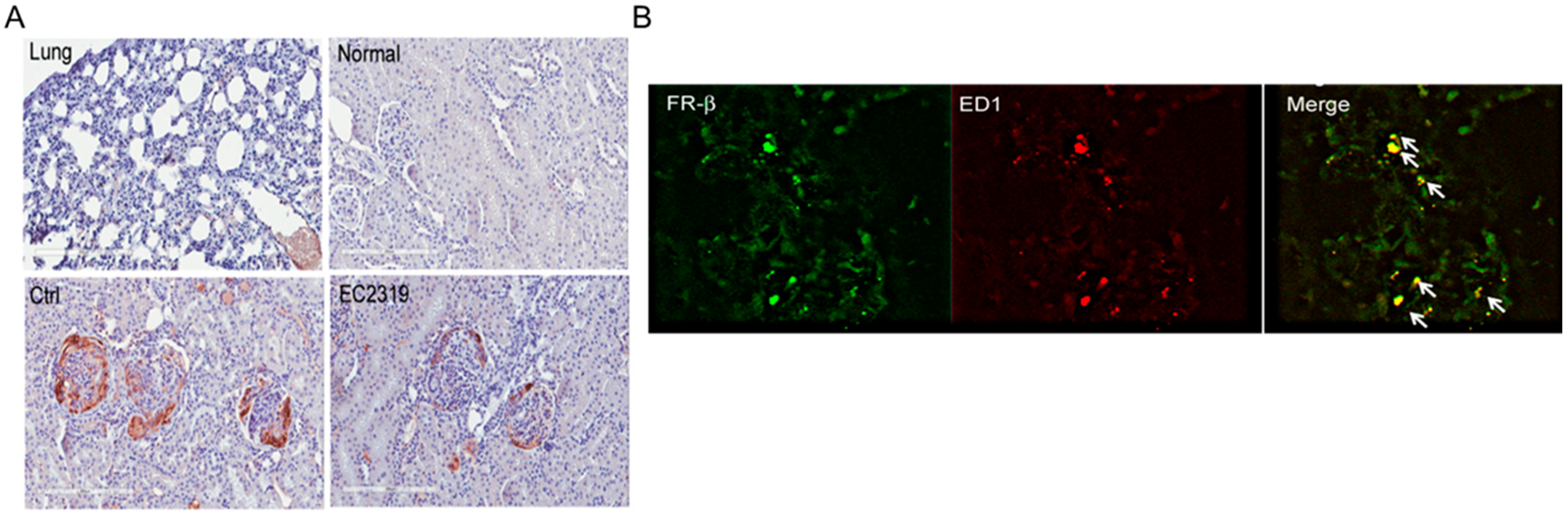
2.7. Immunofluorescence Staining of FRβ and Macrophages
2.8. Circulating Ab and Glomerular IgG Deposition
2.9. Statistics
3. Results
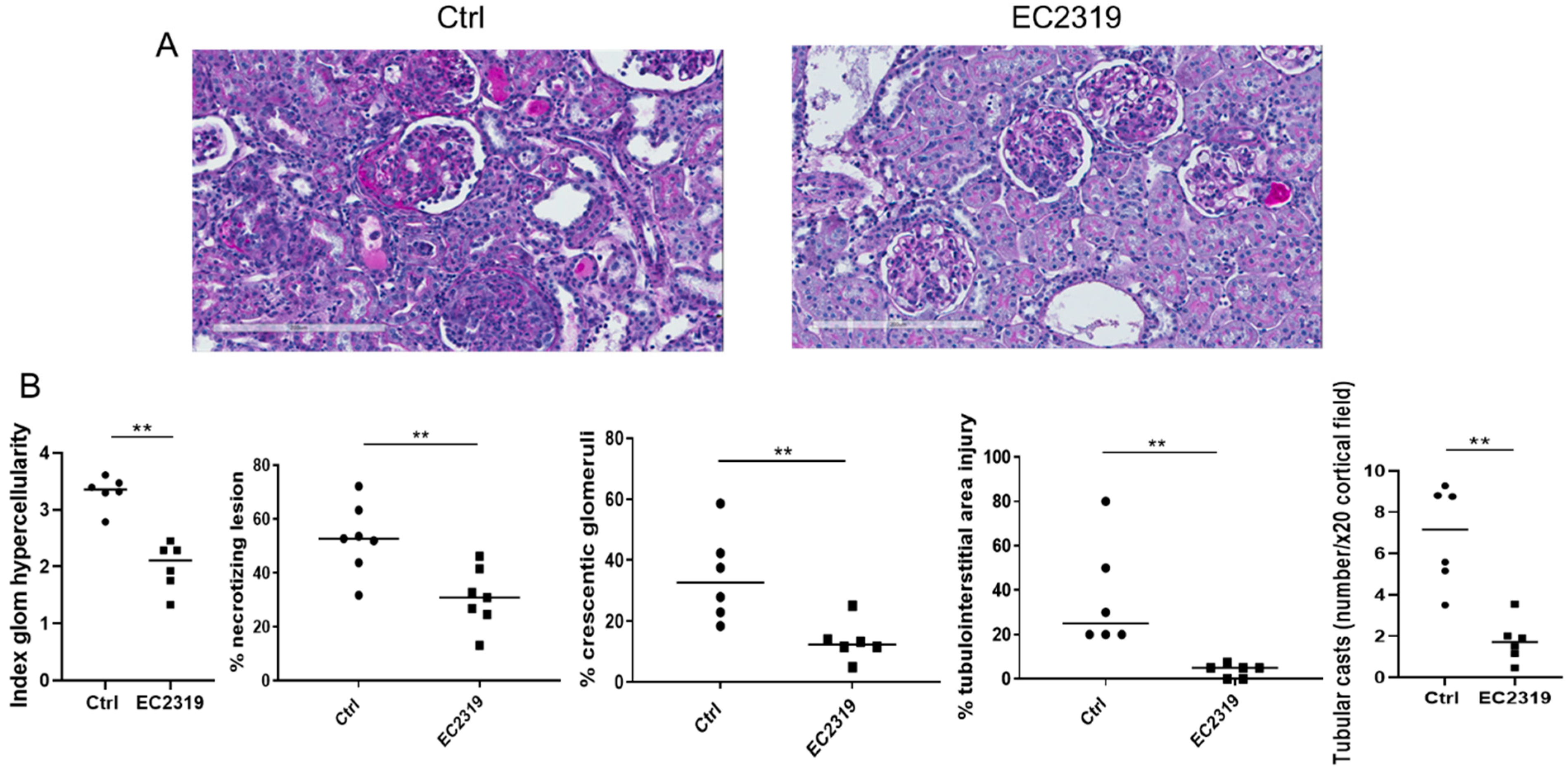
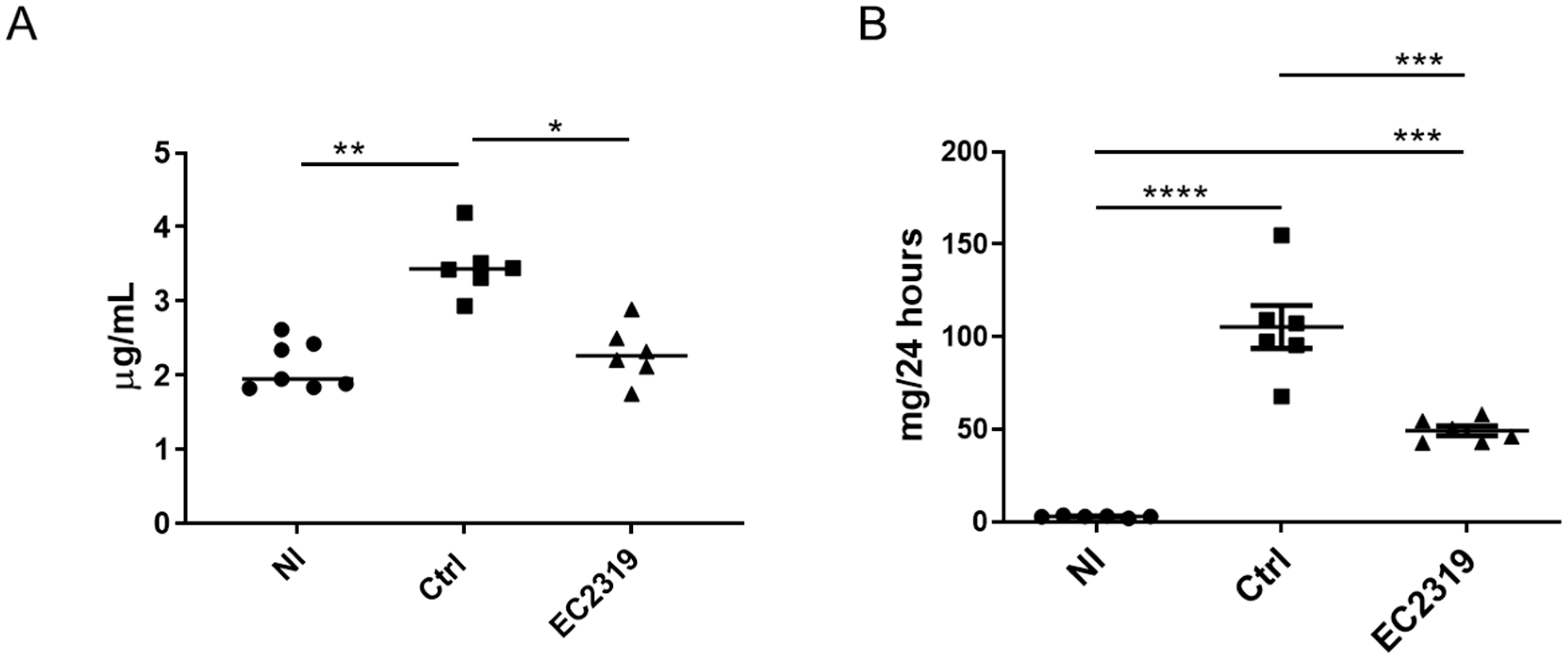
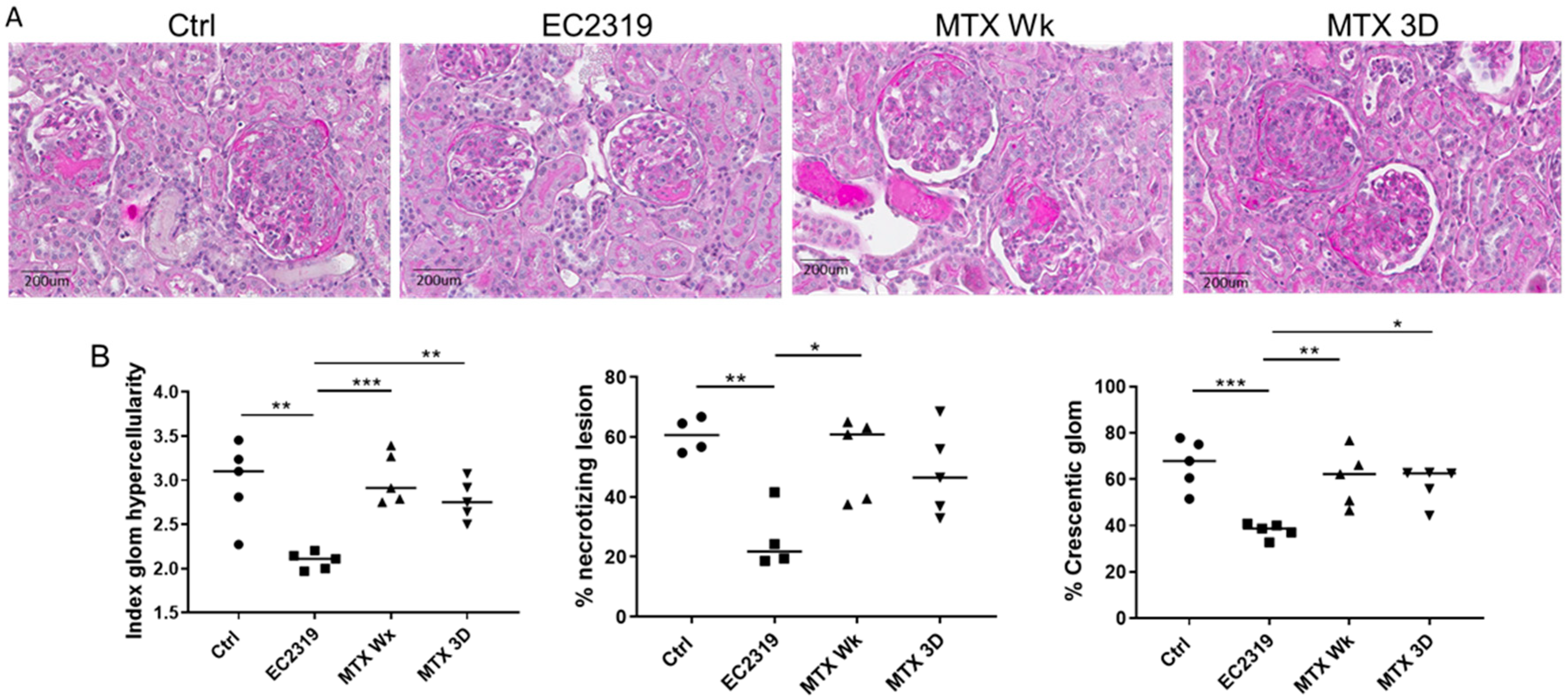
3.1. EC2319 Protects from Kidney Injury in Anti-GBM GN
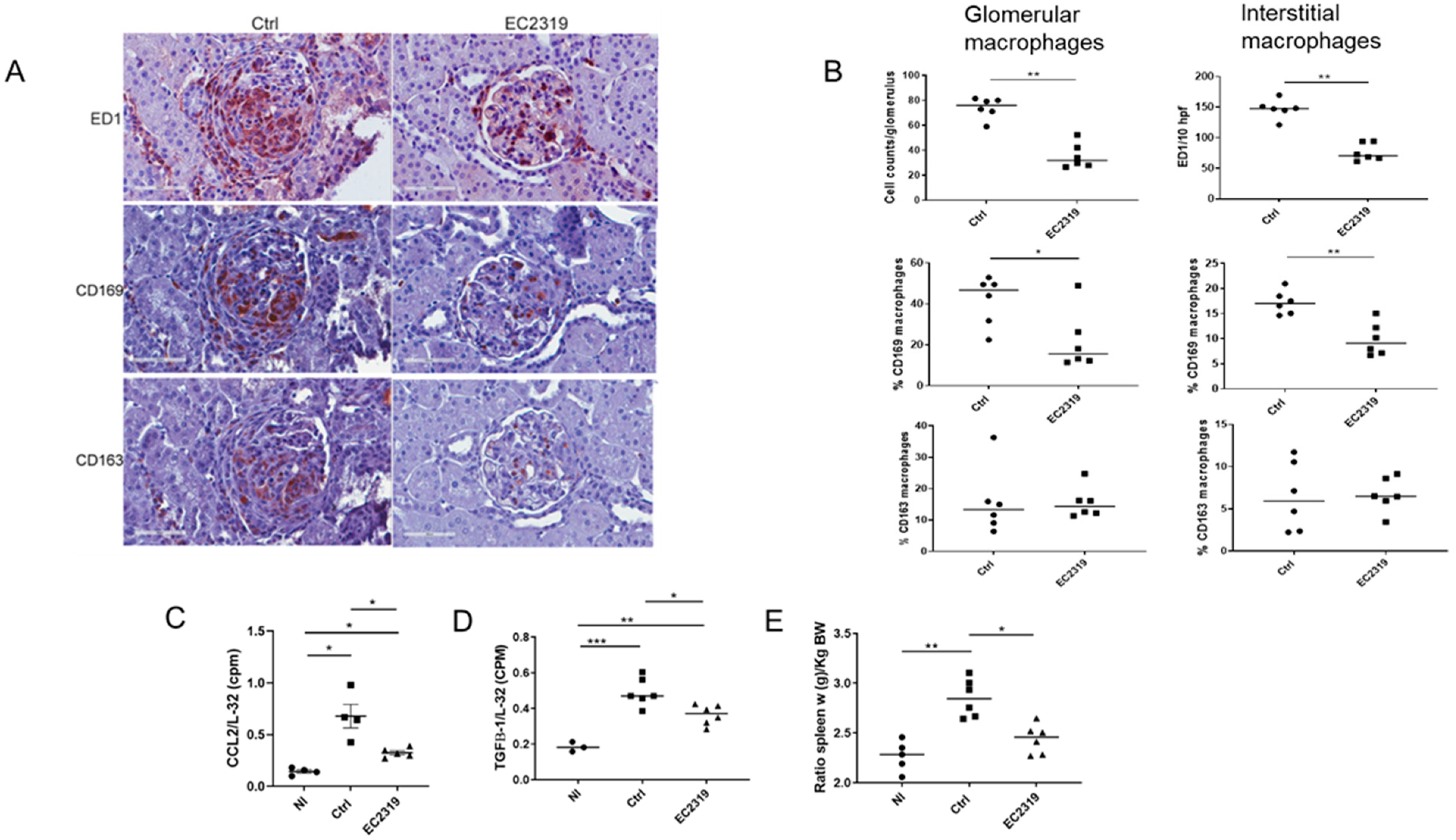
3.1.1. Kidney Protection in GN by EC2319 Is Associated with Reduced Macrophage Infiltration
3.1.2. EC2319 Modulates the Expression of Chemokines and Cytokines
3.2. EC2319 Decreases Collagen Deposition
3.3. FRβ Is Expressed in Nephritic Glomeruli
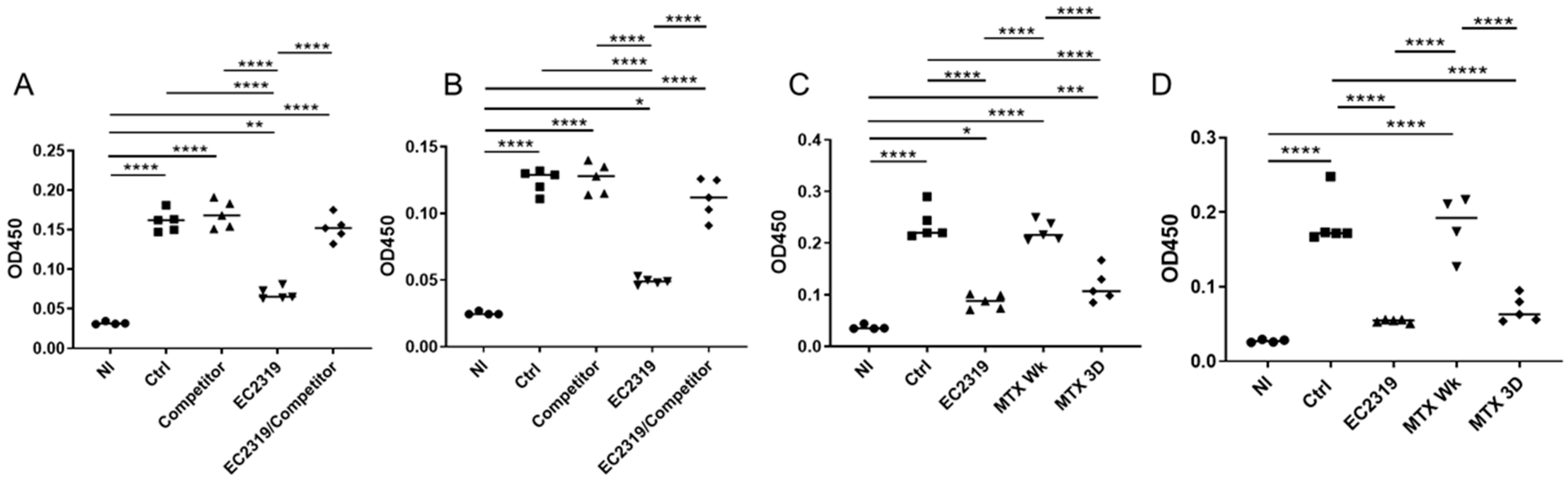
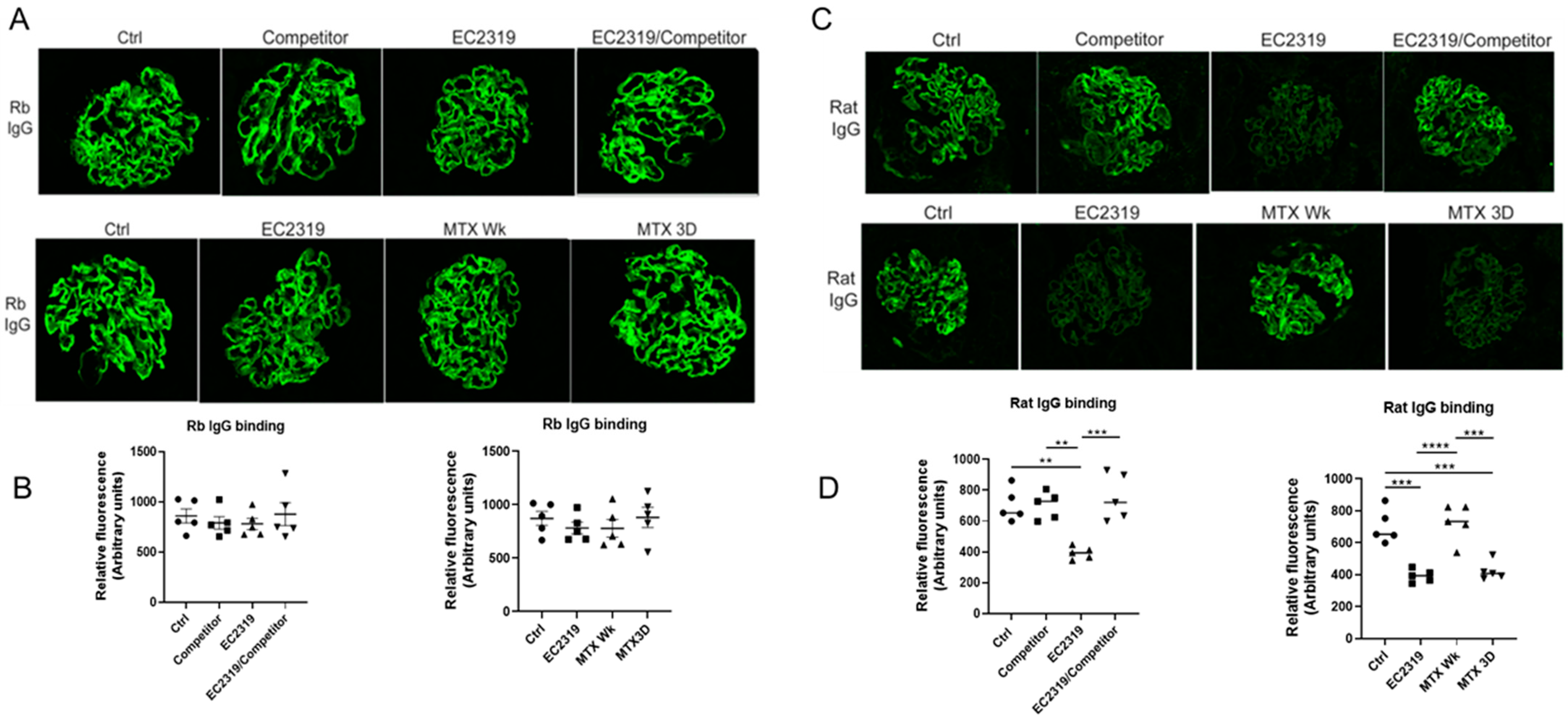
3.4. Blocking EC2319 Binding to Folate Receptor Prevents Its Protective Effect in GN
3.5. MTX Is Unable to Protect from Kidney Injury in Anti-GBM GN
3.6. EC2319 and MTX Decrease Antigen-Specific Humoral Immune Response
3.7. EC2319 and MTX Reduce Glomerular Rat Igg Deposition
4. Discussion
Limitations of Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duffield, J.S. Macrophages and Immunologic Inflammation of the Kidney. Semin. Nephrol. 2010, 30, 234–254. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Moll, S.; Angeletti, A.; Scapozza, L.; Cavalli, A.; Ghiggeri, G.; Prunotto, M. Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis. Cells 2021, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Tipping, P.G.; Kipari, T.; Cailhier, J.-F.; Clay, S.; Lang, R.; Bonventre, J.V.; Hughes, J. Conditional Ablation of Macrophages Halts Progression of Crescentic Glomerulonephritis. Am. J. Pathol. 2005, 167, 1207–1219. [Google Scholar] [CrossRef]
- Holdsworth, S.R.; Neale, T.J.; Wilson, C.B. Abrogation of Macrophage-dependent Injury in Experimental Glomerulonephritis in the Rabbit. J. Clin. Investig. 1981, 68, 686–698. [Google Scholar] [CrossRef]
- Chen, S.; Bacon, K.B.; Li, L.; Garcia, G.E.; Xia, Y.; Lo, D.; Thompson, D.A.; Siani, M.A.; Yamamoto, T.; Harrison, J.K.; et al. In Vivo Inhibition of CC and CX3C Chemokine–induced Leukocyte Infiltration and Attenuation of Glomerulonephritis in Wistar-Kyoto (WKY) Rats by vMIP-II. J. Exp. Med. 1998, 188, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Fujinaka, H.; Yamamoto, T.; Takeya, M.; Feng, L.; Kawasaki, K.; Yaoita, E.; Kondo, D.; Wilson, C.B.; Uchiyama, M.; Kihara, I. Suppression of anti-glomerular basement membrane nephritis by administration of anti-monocyte chemoattractant protein-1 antibody in WKY rats. J. Am. Soc. Nephrol. 1997, 8, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.E.; Truong, L.D.; Li, P.; Zhang, P.; Du, J.; Chen, J.-F.; Feng, L. Adenosine A 2A receptor activation and macrophage-mediated experimental glomerulonephritis. FASEB J. 2007, 22, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.; Minto, A.W.; Dorf, M.E.; Proudfoot, A.; Wells, T.; Salant, D.; Gutierrez-Ramos, J.-C. RANTES and Monocyte Chemoattractant Protein-1 (MCP-1) Play an Important Role in the Inflammatory Phase of Crescentic Nephritis, but Only MCP-1 Is Involved in Crescent Formation and Interstitial Fibrosis. J. Exp. Med. 1997, 185, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Ikezumi, Y.; Hurst, L.A.; Masaki, T.; Atkins, R.C.; Nikolic-Paterson, D.J. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003, 63, 83–95. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Wada, T.; Furuichi, K.; Sakai, N.; Iwata, Y.; Yokoyama, H. CD68 and MCP-1/CCR2 Expression of Initial Biopsies Reflect the Outcomes of Membranous Nephropathy. Nephron Clin. Pract. 2004, 98, c25–c34. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.P.; Lin, S.-L.; Surowy, T.; Nowlin, B.T.; Turlapati, S.A.; Patel, T.; Singh, A.; Li, S.; Lupher, M.L.; Duffield, J.S. Serum Amyloid P Inhibits Fibrosis Through Fc R-Dependent Monocyte-Macrophage Regulation in Vivo. Sci. Transl. Med. 2009, 1, 5ra13. [Google Scholar] [CrossRef]
- Tsou, C.-L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Machacek, C.; Supper, V.; Leksa, V.; Mitulovic, G.; Spittler, A.; Drbal, K.; Suchanek, M.; Ohradanova-Repic, A.; Stockinger, H. Folate Receptor β Regulates Integrin CD11b/CD18 Adhesion of a Macrophage Subset to Collagen. J. Immunol. 2016, 197, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, I.R.; You, F.; Santhapuram, H.K.R.; Wang, Y.; Vaughn, J.F.; Hahn, S.J.; Kleindl, P.J.; Fan, M.; Leamon, C.P. Design and regioselective synthesis of a new generation of targeted therapeutics. Part 3: Folate conjugates of aminopterin hydrazide for the treatment of inflammation. Bioorg. Med. Chem. Lett. 2011, 21, 1202–1205. [Google Scholar] [CrossRef]
- Xia, W.; Hilgenbrink, A.R.; Matteson, E.L.; Lockwood, M.B.; Cheng, J.-X.; Low, P.S. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood 2009, 113, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wollak, K.N.; Cross, V.A.; Westrick, E.; Wheeler, L.W.; Stinnette, T.W.; Vaughn, J.F.; Hahn, S.J.; Xu, L.-C.; Vlahov, I.R.; et al. Folate receptor-targeted aminopterin therapy is highly effective and specific in experimental models of autoimmune uveitis and autoimmune encephalomyelitis. Clin. Immunol. 2014, 150, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Thrash, B.; Cherian, C.; Matherly, L.H.; Wang, L.; Gangjee, A.; Morgan, J.R.; Maeda, D.; Schuler, A.D.; Kahn, S.J.; et al. Intestinal Transport of Aminopterin Enantiomers in Dogs and Humans with Psoriasis Is Stereoselective: Evidence for a Mechanism Involving the Proton-Coupled Folate Transporter. J. Pharmacol. Exp. Ther. 2012, 342, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Stinnette, T.W.; Westrick, E.; Klein, P.J.A.; Gehrke, M.A.; Cross, V.; Vlahov, I.R.; Low, P.S.; Leamon, C.P. Treatment of experimental adjuvant arthritis with a novel folate receptor-targeted folic acid-aminopterin conjugate. Arthritis Res. Ther. 2011, 13, R56. [Google Scholar] [CrossRef]
- Lu, Y.J.; Wheeler, L.W.; Chu, H.; Kleindl, P.J.; Michael, P.; You, F.; Rao, S.; Garcia, G.; Wu, H.Y.; de Cunha, A.P.; et al. Targeting Folate-Receptor-Beta on Monocytes/Macrophages Renders Rapid Inflammation Resolution Independent of Root Causes. Cell Rep. Med. Accepted.
- Leamon, C.P.; Reddy, J.A.; Dorton, R.; Bloomfield, A.; Emsweller, K.; Parker, N.; Westrick, E. Impact of High and Low Folate Diets on Tissue Folate Receptor Levels and Antitumor Responses Toward Folate-Drug Conjugates. J. Pharmacol. Exp. Ther. 2008, 327, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, S.; Garcia, G.E.; Xia, Y.; Siani, M.A.; Botti, P.; Wilson, C.B.; Harrison, J.K.; Bacon, K.B. Prevention of crescentic glomerulonephritis by immunoneutralization of the fractalkine receptor CX3CR1: Rapid Communication. Kidney Int. 1999, 56, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.E.; Xia, Y.; Ku, G.; Johnson, R.J.; Wilson, C.B.; Feng, L. IL-18 translational inhibition restricts IFN-γ expression in crescentic glomerulonephritis. Kidney Int. 2003, 64, 160–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kanellis, J.; Garcia, G.E.; Li, P.; Parra, G.; Wilson, C.B.; Rao, Y.; Han, S.; Smith, C.W.; Johnson, R.J.; Wu, J.Y.; et al. Modulation of Inflammation by Slit Protein In Vivo in Experimental Crescentic Glomerulonephritis. Am. J. Pathol. 2004, 165, 341–352. [Google Scholar] [CrossRef]
- Unanue, E.R.; Dixon, F.J. Experimental Glomerulonephritis. J. Exp. Med. 1965, 121, 697–714. [Google Scholar] [CrossRef]
- Kawasaki, K.; Yaoita, E.; Yamamoto, T.; Kihara, I. Depletion of CD8 positive cells in nephrotoxic serum nephritis of WKY rats. Kidney Int. 1992, 41, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Boysen, G.; Li, F.; Li, Y.; Swenberg, J. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int. 2007, 71, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Goolsby, M.J. NKF-K/DOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. J. Am. Acad. Nurse Pract. 2002, 14, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Troxell, M.L.; Houghton, D.C. Atypical anti-glomerular basement membrane disease. Clin. Kidney J. 2016, 9, 211–221. [Google Scholar] [CrossRef]
- Braun, J.; Rau, R. An update on methotrexate. Curr. Opin. Rheumatol. 2009, 21, 216–223. [Google Scholar] [CrossRef]
- Bujor, A.M.; Janjua, S.; LaValley, M.P.; Duran, J.; Braun, J.; Felson, D.T. Comparison of oral versus parenteral methotrexate in the treatment of rheumatoid arthritis: A meta-analysis. PLoS ONE 2019, 14, e0221823. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.E.; Truong, L.D.; Chen, J.-F.; Johnson, R.J.; Feng, L. Adenosine A2A receptor activation prevents progressive kidney fibrosis in a model of immune-associated chronic inflammation. Kidney Int. 2011, 80, 378–388. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Garcia, G.E.; Xia, Y.; Harrison, J.; Wilson, C.B.; Johnson, R.J.; Bacon, K.B.; Feng, L. Mononuclear Cell-Infiltrate Inhibition by Blocking Macrophage-Derived Chemokine Results in Attenuation of Developing Crescentic Glomerulonephritis. Am. J. Pathol. 2003, 162, 1061–1073. [Google Scholar] [CrossRef]
- Feng, L.; Xia, Y.; Yoshimura, T.; Wilson, C.B. Modulation of neutrophil influx in glomerulonephritis in the rat with anti-macrophage inflammatory protein-2 (MIP-2) antibody. J. Clin. Investig. 1995, 95, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.E.; Xia, Y.; Chen, S.; Wang, Y.; Ye, R.D.; Harrison, J.K.; Bacon, K.B.; Zerwes, H.-G.; Feng, L. NF-κB-dependent fractalkine induction in rat aortic endothelial cells stimulated by IL-1β, TNF-α, and LPS. J. Leukoc. Biol. 2000, 67, 577–584. [Google Scholar] [CrossRef]
- Xia, Y.; Pauza, M.E.; Feng, L.; Lo, D. RelB regulation of chemokine expression modulates local inflammation. Am. J. Pathol. 1997, 151, 375–387. [Google Scholar]
- Feng, L.; Garcia, G.E.; Yang, Y.; Xia, Y.; Gabbai, F.B.; Peterson, O.W.; Abraham, J.A.; Blantz, R.C.; Wilson, C.B. Heparin-binding EGF-like growth factor contributes to reduced glomerular filtration rate during glomerulonephritis in rats. J. Clin. Investig. 2000, 105, 341–350. [Google Scholar] [CrossRef]
- Xia, Y.; Feng, L.; Yoshimura, T.; Wilson, C.B. LPS-induced MCP-1, IL-1 beta, and TNF-alpha mRNA expression in isolated erythrocyte-perfused rat kidney. Am. J. Physiol. Physiol. 1993, 264, F774–F780. [Google Scholar] [CrossRef]
- Nagai, T.; Kyo, A.; Hasui, K.; Takao, S.; Matsuyama, T. Efficacy of an immunotoxin to folate receptor beta in the intra-articular treatment of antigen-induced arthritis. Arthritis Res. Ther. 2012, 14, R106. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: A foe or ally? Cell. Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.A.; Robinson, E.; Tanaka, S.; Appella, E.; Leonard, E.J. Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. J. Immunol. 1989, 142, 1956–1962. [Google Scholar] [PubMed]
- Hellmark, T.; Segelmark, M. Diagnosis and classification of Goodpasture’s disease (anti-GBM). J. Autoimmun. 2014, 48, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Carvelli, J.; Biard, L.; Faguer, S.; Provôt, F.; Matignon, M.; Boffa, J.-J.; Plaisier, E.; Hertig, A.; Touzot, M.; et al. Prognostic Factors in Anti-glomerular Basement Membrane Disease: A Multicenter Study of 119 Patients. Front. Immunol. 2019, 10, 1665. [Google Scholar] [CrossRef] [PubMed]
- Van Daalen, E.E.; Jennette, J.C.; McAdoo, S.P.; Pusey, C.D.; Alba, M.A.; Poulton, C.J.; Wolterbeek, R.; Nguyen, T.Q.; Goldschmeding, R.; Alchi, B.; et al. Predicting Outcome in Patients with Anti-GBM Glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2017, 13, 63–72. [Google Scholar] [CrossRef]
- Atkins, R.; Holdsworth, S.; Glasgow, E.; Matthews, F. The Macrophage in Human Rapidly Progressive Glomerulonephritis. Lancet 1976, 307, 830–832. [Google Scholar] [CrossRef]
- Shen, J.; Hilgenbrink, A.R.; Xia, W.; Feng, Y.; Dimitrov, D.S.; Lockwood, M.B.; Amato, R.J.; Low, P.S. Folate receptor-β constitutes a marker for human proinflammatory monocytes. J. Leukoc. Biol. 2014, 96, 563–570. [Google Scholar] [CrossRef]
- Kovalenko, P.; Fujinaka, H.; Yoshida, Y.; Kawamura, H.; Qu, Z.; El-Shemi, A.; Li, H.; Matsuki, A.; Bilim, V.; Yaoita, E.; et al. Fc receptor-mediated accumulation of macrophages in crescentic glomerulonephritis induced by anti-glomerular basement membrane antibody administration in WKY rats. Int. Immunol. 2004, 16, 625–634. [Google Scholar] [CrossRef]
- Park, S.Y.; Ueda, S.; Ohno, H.; Hamano, Y.; Tanaka, M.; Shiratori, T.; Yamazaki, T.; Arase, H.; Arase, N.; Karasawa, A.; et al. Resistance of Fc receptor- deficient mice to fatal glomerulonephritis. J. Clin. Investig. 1998, 102, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, D.B.; Curi, P.R. Treatment of rat nephrotoxic nephritis. Use of 5-fluorouracil or methotrexate-5-fluorouracil association. Clin. Exp. Immunol. 1984, 57, 591–599. [Google Scholar]
- Nagamatsu, T.; Kojima, N.; Kondo, N.; Hattori, T.; Kojima, R.; Ito, M.; Suzuki, Y. Suppression by Cyclosporin A of Anti-GBM Nephritis in Rats. Jpn. J. Pharmacol. 1992, 58, 27–36. [Google Scholar] [CrossRef]
- Unanue, E.R.; Dixon, F.J. Experimental Glomerulonephritis. J. Exp. Med. 1965, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, G.E.; Lu, Y.J.; Truong, L.D.; Roncal-Jiménez, C.A.; Miyazaki, M.; Miyazaki-Anzai, S.; Cara-Fuentes, G.; Andres-Hernando, A.; Lanaspa, M.; Johnson, R.J.; et al. A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats. Cells 2021, 10, 2113. https://doi.org/10.3390/cells10082113
Garcia GE, Lu YJ, Truong LD, Roncal-Jiménez CA, Miyazaki M, Miyazaki-Anzai S, Cara-Fuentes G, Andres-Hernando A, Lanaspa M, Johnson RJ, et al. A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats. Cells. 2021; 10(8):2113. https://doi.org/10.3390/cells10082113
Chicago/Turabian StyleGarcia, Gabriela E., Yingjuan J. Lu, Luan D. Truong, Carlos A. Roncal-Jiménez, Makoto Miyazaki, Shinobu Miyazaki-Anzai, Gabriel Cara-Fuentes, Ana Andres-Hernando, Miguel Lanaspa, Richard J. Johnson, and et al. 2021. "A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats" Cells 10, no. 8: 2113. https://doi.org/10.3390/cells10082113
APA StyleGarcia, G. E., Lu, Y. J., Truong, L. D., Roncal-Jiménez, C. A., Miyazaki, M., Miyazaki-Anzai, S., Cara-Fuentes, G., Andres-Hernando, A., Lanaspa, M., Johnson, R. J., & Leamon, C. P. (2021). A Novel Treatment for Glomerular Disease: Targeting the Activated Macrophage Folate Receptor with a Trojan Horse Therapy in Rats. Cells, 10(8), 2113. https://doi.org/10.3390/cells10082113