
Autophagy and Mitophagy-Related Pathways at the Crossroads of Genetic Pathways Involved in Familial Sarcoidosis and Host-Pathogen Interactions Induced by Coronaviruses

 ,
, 

Abstract
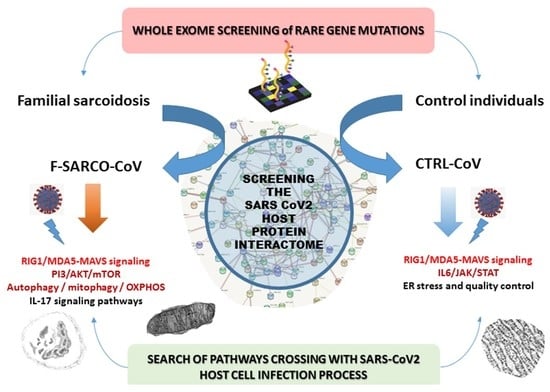
1. Introduction
2. Materials and Methods
2.1. Selection of Sarcoidosis Patients and Families
2.2. Genetic Analysis by Whole Exome Sequencing
2.3. Bioinformatics
2.4. Selection of Pathogenic Gene Variants and Gene Set Enrichment Analysis
2.5. Crossbreeding of the Genetic Data of Sarcoidosis and Control Individuals with the Genes Identified in the SARS-CoV2 Interactome
3. Results
3.1. Gene Variants Identified by the Selection Procedure
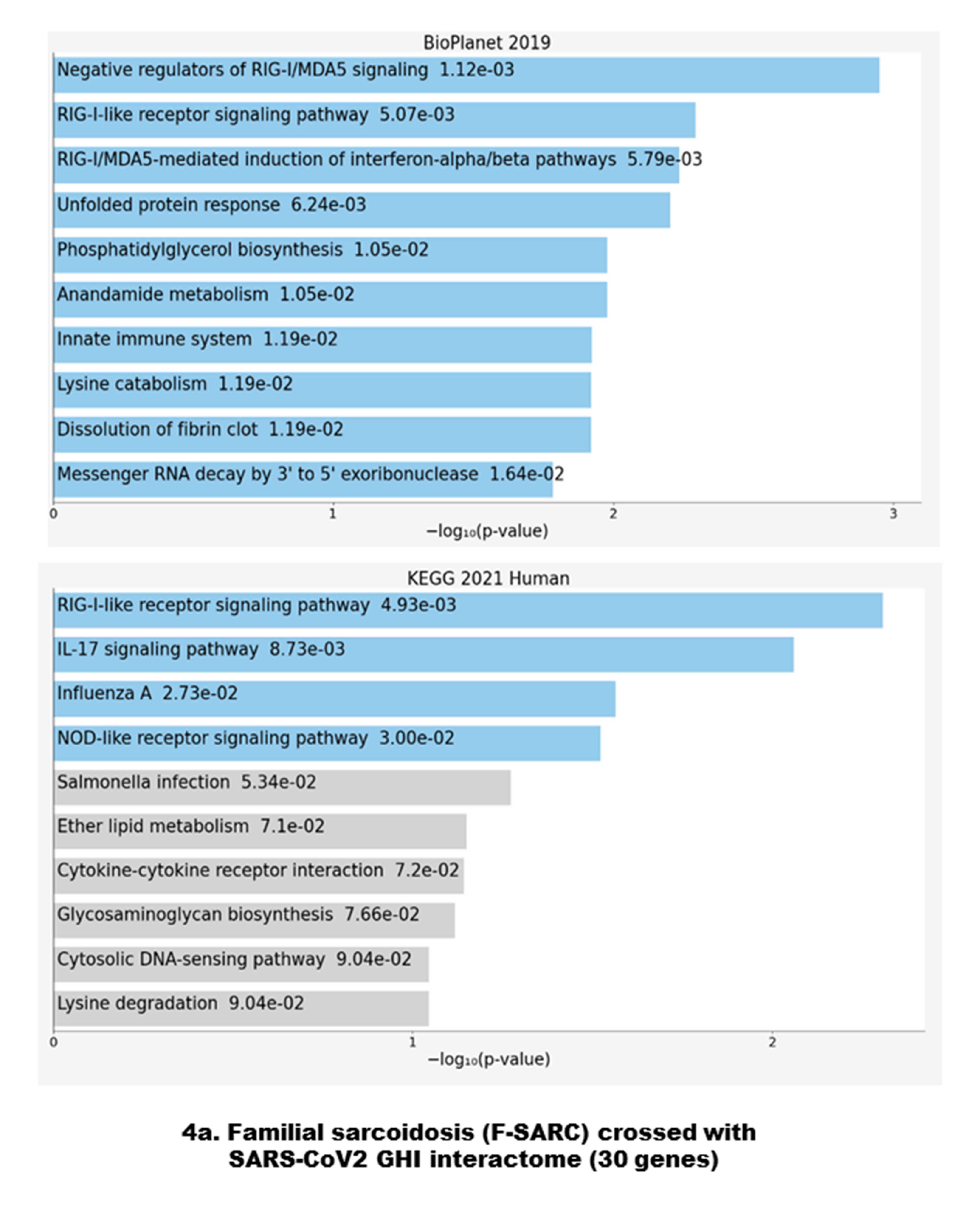
3.2. Data Crossing with the SARS-CoV2 Gordon Hoffmann Interactome (GHI)
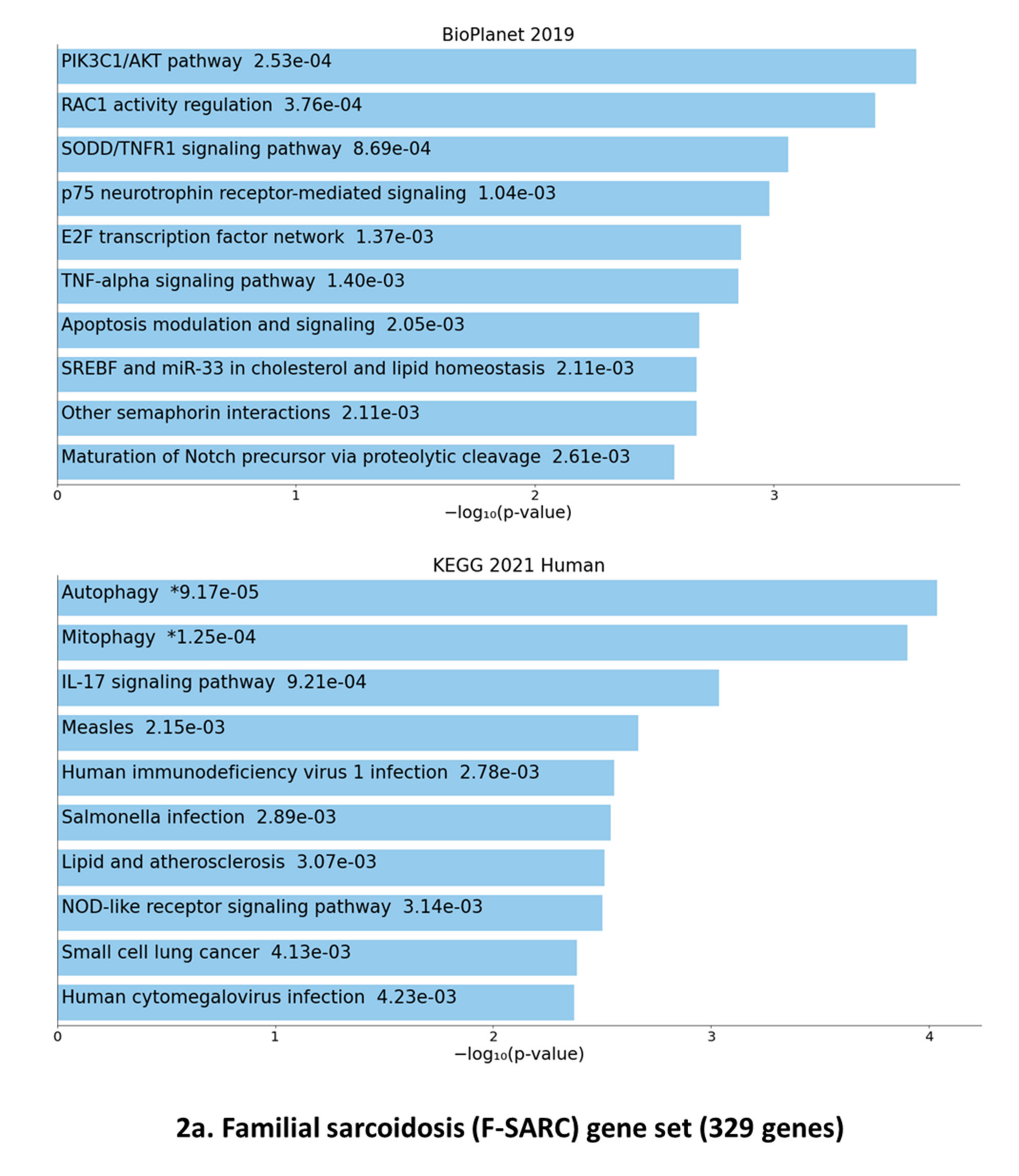
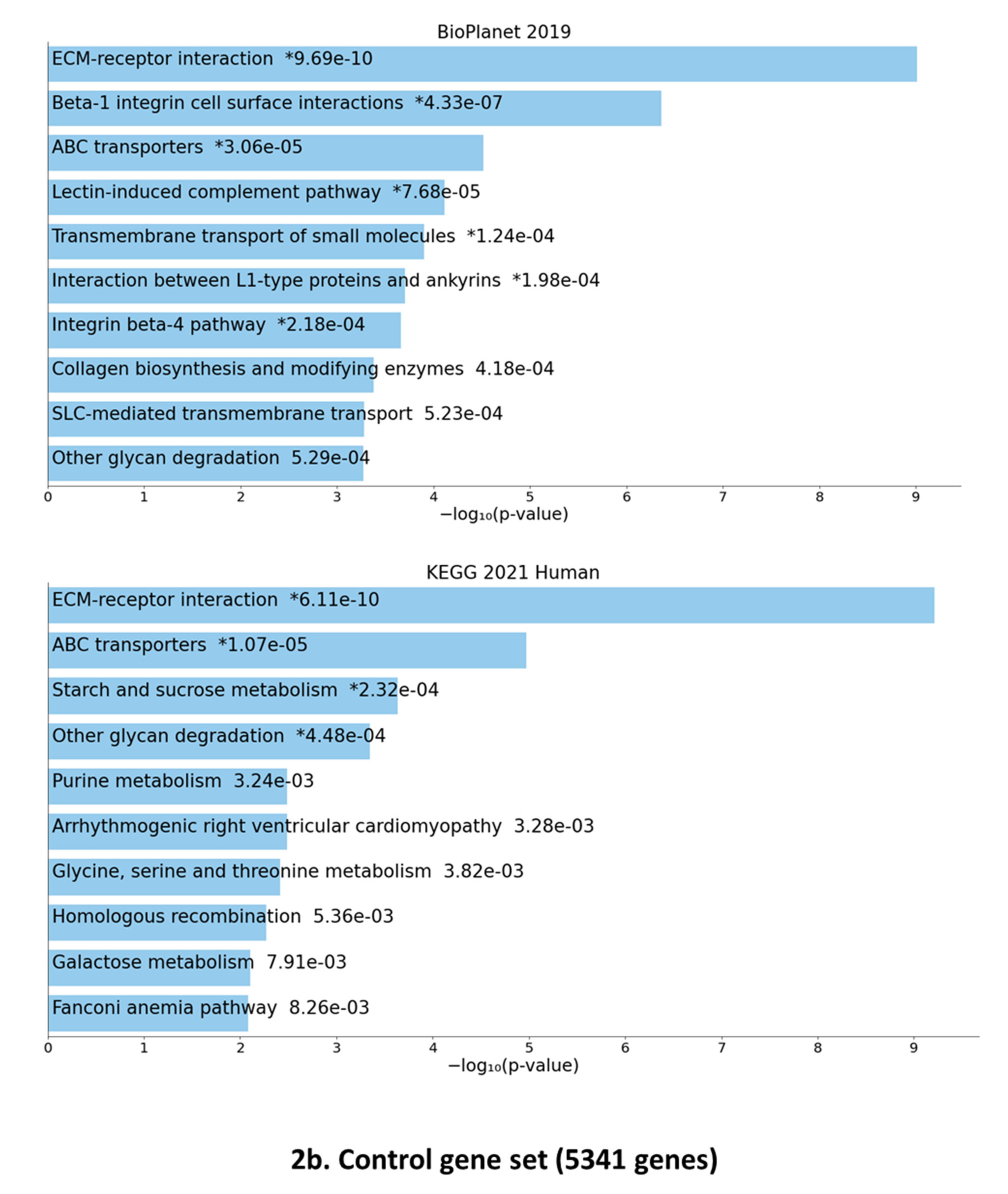
3.3. Gene Set Enrichment of F-SARC-CoV and Control-CoV Gene Panels
4. Discussion
4.1. Antiviral Immune Signaling and Autophagy
4.2. Mitochondrial Dysfunction
4.3. Rho GTPases Related Pathways
4.4. Interleukin-17-Related Pathways
4.5. Endoplasmic Reticulum Stress-Related Pathways
4.6. F-SARCO-CoV Genes Related to Virus Entry
4.7. Impact of Genetic Data on Sarcoidosis and SARS-CoV2 Infection
4.8. Pharmacological Impact of Identified Pathways
4.9. Limitations
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grunewald, J.; Grutters, J.C.; Arkema, E.V.; Saketkoo, L.A.; Moller, D.R.; Müller-Quernheim, J. Sarcoidosis. Nat. Rev. Dis. Primers 2019, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Rybicki, B.A. Granuloma genes in sarcoidosis: What is new? Curr. Opin. Pulm. Med. 2015, 21, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Petersen, B.S.; Nutsua, M.; Müller-Quernheim, J.; Franke, A.; Fischer, A.; Schreiber, S.; Petrek, M. Whole-exome sequencing identifies rare genetic variations in German families with pulmonary sarcoidosis. Hum. Genet. 2018, 137, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Calender, A.; Lim, C.X.; Weichhart, T.; Buisson, A.; Besnard, V.; Rollat-Farnier, P.A.; Bardel, C.; Roy, P.; Cottin, V.; Devouassoux, G.; et al. Exome sequencing and pathogenicity-network analysis of five French families implicate mTOR signaling and autophagy in familial sarcoidosis. Eur. Respir. J. 2019, 54, 1900430. [Google Scholar] [CrossRef] [PubMed]
- Calender, A.; Weichhart, T.; Valeyre, D.; Pacheco, Y. Current Insights in Genetics of Sarcoidosis: Functional and Clinical Impacts. J. Clin. Med. 2020, 9, 2633. [Google Scholar] [CrossRef] [PubMed]
- Culver, D.A. Sarcoidosis. Immunol. Allergy Clin. N. Am. 2012, 32, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.P.; Moral, R.A. Global short-term forecasting of COVID-19 cases. Sci. Rep. 2021, 11, 7555. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Lower, E.E.; Buchanan, M.; Rottoli, P.; Drent, M.; Sellares, J.; Terwiel, M.; Elfferich, M.; Francesqui, J.; Barriuso Cabrerizo, M.R.; et al. Risk and outcome of COVID-19 infection in sarcoidosis patients: Results of a self-reporting questionnaire. Sarcoidosis Vasc. Diffus. Lung Dis. 2020, 37, e2020009. [Google Scholar] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; O’Meara, M.J.; Guo, J.Z.; Swaney, D.L.; Tummino, T.A.; Hüttenhain, R.; et al. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, H.H.; Sánchez-Rivera, F.J.; Schneider, W.M.; Luna, J.M.; Soto-Feliciano, Y.M.; Ashbrook, A.W.; Le Pen, J.; Leal, A.A.; Ricardo-Lax, I.; Michailidis, E.; et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 2021, 29, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, Y.; Calender, A.; Israël-Biet, D.; Roy, P.; Lebecque, S.; Cottin, V.; Bouvry, D.; Nunes, H.; Sève, P.; Pérard, L.; et al. Familial vs. sporadic sarcoidosis: BTNL2 polymorphisms, clinical presentations, and outcomes in a French cohort. Orphanet J. Rare Dis. 2016, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, Y.; Lim, C.X.; Weichhart, T.; Valeyre, D.; Bentaher, A.; Calender, A. Sarcoidosis and the mTOR, Rac1, and Autophagy Triad. Trends Immunol. 2020, 41, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Tana, C.; Schiavone, C.; Cipollone, F.; Giamberardino, M.A. Management Issues of Sarcoidosis in the Time of Coronavirus Disease 2019. Chest 2021, 159, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Aveyard, P.; Gao, M.; Lindson, N.; Hartmann-Boyce, J.; Watkinson, P.; Young, D.; Coupland, C.A.C.; Tan, P.S.; Clift, A.K.; Harrison, D.; et al. Association between pre-existing respiratory disease and its treatment, and severe COVID-19: A population cohort study. Lancet Respir. Med. 2021, 9, 909–923. [Google Scholar] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Calender, A.; Israel-Biet, D.; Valeyre, D.; Pacheco, Y. Modeling Potential Autophagy Pathways in COVID-19 and Sarcoidosis. Trends Immunol. 2020, 41, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Poduri, R.; Joshi, G.; Jagadeesh, G. Drugs targeting various stages of the SARS-CoV-2 life cycle: Exploring promising drugs for the treatment of Covid-19. Cell Signal. 2020, 74, 109721. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between endoplasmic reticulum stress and anti-viral activities: A novel therapeutic target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2020, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Khatoon, F.; Rashid, S.; Ali, N.; AlAsmari, A.F.; Ahmed, M.Z.; Alqahtani, A.S.; Alqahtani, M.S.; Kumar, V. Targeting hub genes and pathways of innate immune response in COVID-19: A network biology perspective. Int. J. Biol. Macromol. 2020, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. A STING to inflammation and autoimmunity. J. Leukoc. Biol. 2019, 106, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Wang, S.Y.; Zheng, Z.Q.; Yi, H.; Li, W.W.; Xu, Z.S.; Wang, Y.Y. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhuang, M.W.; Han, L.; Zhang, J.; Nan, M.L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.H. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Pham, H.T.; Katholnig, K.; Schnöller, T.; Miller, A.; Demel, F.; Schütz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Karam, B.S.; Morris, R.S.; Bramante, C.T.; Puskarich, M.; Zolfaghari, E.J.; Lotfi-Emran, S.; Ingraham, N.E.; Charles, A.; Odde, D.J.; Tignanelli, C.J. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J. Med. Virol. 2021, 93, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Cohen Aubart, F.; Lhote, R.; Amoura, A.; Valeyre, D.; Haroche, J.; Amoura, Z.; Lebrun-Vignes, B. Drug-induced sarcoidosis: An overview of the WHO pharmacovigilance database. J. Intern. Med. 2020, 288, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Miedema, J.; Nunes, H. Drug-induced sarcoidosis-like reactions. Curr. Opin. Pulm. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2021, 39, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M.H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.F.; Masuyer, G.; Pinho, C.M.; Branca, R.M.M.; Kmiec, B.; Wallin, C.; Wärmländer, S.K.T.S.; Berntsson, R.P.; Ankarcrona, M.; Gräslund, A.; et al. Mechanism of Peptide Binding and Cleavage by the Human Mitochondrial Peptidase Neurolysin. J. Mol. Biol. 2018, 430, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2020, 1–18. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; He, C.; Carter, A.B. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020, 33, 101426. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J. Macrophage Polarization in Sarcoidosis: An Unexpected Accomplice? Am. J. Respir. Cell Mol. Biol. 2019, 60, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Mayr, H.K.; Weichhart, T. Metabolic Programming of Macrophages: Implications in the Pathogenesis of Granulomatous Disease. Front. Immunol. 2019, 10, 2265. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.M.; Brewer, S.M.; Thurston, T.; Massis, L.M.; Honeycutt, J.; Lugo, K.; Jacobson, A.R.; Vilches-Moure, J.G.; Hamblin, M.; Helaine, S.; et al. Salmonella-Driven Polarization of Granuloma Macrophages Antagonizes TNF-Mediated Pathogen Restriction during Persistent Infection. Cell Host Microbe 2020, 27, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.; Yu, K.L.; Teuling, E.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. The ALS8 protein VAPB interacts with the ER-Golgi recycling protein YIF1A and regulates membrane delivery into dendrites. EMBO J. 2013, 32, 2056–2072. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.; Wang, M.; Chang, Y.; Zhang, F.; Ban, Z.; Tang, R.; Gan, Q.; Wu, S.; Guo, Y.; et al. The lysine catabolite saccharopine impairs development by disrupting mitochondrial homeostasis. J. Cell Biol. 2019, 218, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.; Zornoza, M.; Jourdain, A.A.; Delmiro Magdalena, A.; García-Consuegra, I.; Torres Merino, R.; Orduña, A.; Martín, M.A.; Martinou, J.C.; De la Fuente, M.A.; et al. Role of FAST Kinase Domains 3 (FASTKD3) in Post-transcriptional Regulation of Mitochondrial Gene Expression. J. Biol. Chem. 2016, 291, 25877–25887. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Sripada, L.; Lipatova, A.; Roy, M.; Prajapati, P.; Gohel, D.; Bhatelia, K.; Chumakov, P.M.; Singh, R. NLRX1 resides in mitochondrial RNA granules and regulates mitochondrial RNA processing and bioenergetic adaptation. Biochim. Biophys Acta Mol. Cell Res. 2018, 1865, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.R.G.; Lepelley, A.; Seeley, J.J.; Hayden, M.S.; Ghosh, S. An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages. Cell Rep. 2018, 22, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Nagai-Singer, M.A.; Morrison, H.A.; Allen, I.C. NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 2019, 10, 2419. [Google Scholar] [CrossRef]
- Huynen, M.A.; Mühlmeister, M.; Gotthardt, K.; Guerrero-Castillo, S.; Brandt, U. Evolution and structural organization of the mitochondrial contact site (MICOS) complex and the mitochondrial intermembrane space bridging (MIB) complex. Biochim. Biophys. Acta 2016, 1863, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. A Biochemical Perspective of the Nonstructural Proteins (NSPs) and the Spike Protein of SARS CoV-2. Protein J. 2021, 24, 1–36. [Google Scholar] [PubMed]
- Mysior, M.M.; Simpson, J.C. Emerging roles for Rho GTPases operating at the Golgi complex. Small GTPases 2020, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell. Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Shimizu, T.; Ohto, U. Crystal structure of the FYCO1 RUN domain suggests possible interfaces with small GTPases. Acta Crystallogr. F Struct. Biol. Commun. 2020, 76, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [PubMed]
- Verde, I.; Pahlke, G.; Salanova, M.; Zhang, G.; Wang, S.; Coletti, D.; Onuffer, J.; Jin, S.L.; Conti, M. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 2001, 276, 11189–11198. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Guan, Y.; Du, Y.; Zhao, M.; Chen, X.; Zhu, F.; Guo, C.; Jia, Y.; Li, Y.; et al. TNFAIP8L2/TIPE2 impairs autolysosome reformation via modulating the RAC1-MTORC1 axis. Autophagy 2021, 17, 1410–1425. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, Y.; Zhang, Z.; Wang, Q.; Liu, X. The signaling axis of Rac1-TFEB regulates autophagy and tumorigenesis. Anti-Cancer Drugs 2019, 30, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Z.; Song, L.; Meng, L.; Xu, G.; Zhang, H.; Hu, J.; Li, F.; Liu, C. GEFT Inhibits Autophagy and Apoptosis in Rhabdomyosarcoma via Activation of the Rac1/Cdc42-mTOR Signaling Pathway. Front. Oncol. 2021, 11, 656608. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, B.; Paats, M.S.; Bergen, I.M.; van den Blink, B.; Hoogsteden, H.C.; Lambrecht, B.N.; Hendriks, R.W.; Kleinjan, A. Increased IL-17A expression in granulomas and in circulating memory T cells in sarcoidosis. Rheumatology 2012, 51, 37–46. [Google Scholar] [CrossRef]
- Georas, S.N.; Chapman, T.J.; Crouser, E.D. Sarcoidosis and T-Helper Cells. Th1, Th17, or Th17.1? Am. J. Respir. Crit. Care Med. 2016, 193, 1198–1200. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.T.; Pasadhika, S.; Crouser, E.D.; Choi, D.; Harrington, C.A.; Lewis, J.A.; Austin, C.R.; Diebel, T.N.; Vance, E.; Braziel, R.M.; et al. Hypothesis: Sarcoidosis is a STAT1-mediated disease. Clin. Immunol. 2009, 132, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, G.M.; Guevara, C.A.; Hajjar, K.A. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J. Biol. Chem. 1994, 269, 21198–21203. [Google Scholar] [CrossRef] [PubMed]
- Goljan-Geremek, A.; Geremek, M.; Puscinska, E.; Sliwinski, P. Venous thromboembolism and sarcoidosis: Co-incidence or coexistence? Cent. Eur. J. Immunol. 2015, 40, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; You, Q.; Feng, X.; Kovach, A.; Li, H. Structure of the ER membrane complex, a transmembrane-domain insertase. Nature 2020, 584, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Wideman, J.G. The ubiquitous and ancient ER membrane protein complex (EMC): Tether or not? F1000Res. 2015, 4, 624. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Chao, J.T.; Tavassoli, S.; Wong, A.K.; Choudhary, V.; Young, B.P.; Loewen, C.J.; Prinz, W.A. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014, 12, e1001969. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, P.; Sadeghalvad, M.; Rezaei, N. An inflammatory triangle in Sarcoidosis: PPAR-γ, immune microenvironment, and inflammation. Expert Opin. Biol. Ther. 2021, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nackenoff, A.G.; Hohman, T.J.; Neuner, S.M.; Akers, C.S.; Weitzel, N.C.; Shostak, A.; Ferguson, S.M.; Mobley, B.; Bennett, D.A.; Schneider, J.A.; et al. PLD3 is a neuronal lysosomal phospholipase D associated with β-amyloid plaques and cognitive function in Alzheimer’s disease. PLoS Genet. 2021, 17, e1009406. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, C.R.; Maier, L.A.; Griffin, T.J.; Higgins, L.; Najt, C.P.; Perlman, D.M.; Bhargava, M. Application of Proteomics in Sarcoidosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Martin, J.H.; Weis, M.A.; Krejci, P.; Konik, P.; Li, B.; Alanay, Y.; Lietman, C.; Lee, B.; Eyre, D.; et al. A Chaperone Complex Formed by HSP47, FKBP65, and BiP Modulates Telopeptide Lysyl Hydroxylation of Type I Procollagen. J. Bone Miner. Res. 2017, 32, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.W.; Huang, Y.K.; Kuo, T.T.; Liu, J.P.; Sher, Y.P. An Overview of ADAM9: Structure, Activation, and Regulation in Human Diseases. Int. J. Mol. Sci. 2020, 21, 790. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Peterson, G.M.; Patel, R.P.; Pathinayake, P.S.; Dua, K.; Hansbro, N.G.; Hsu, A.C.; Wark, P.A.; Shukla, S.D.; et al. The complex interplay between endoplasmic reticulum stress and the NLRP3 inflammasome: A potential therapeutic target for inflammatory disorders. Clin. Transl. Immunol. 2021, 10, e1247. [Google Scholar] [CrossRef] [PubMed]
- Fasken, M.B.; Morton, D.J.; Kuiper, E.G.; Jones, S.K.; Leung, S.W.; Corbett, A.H. The RNA Exosome and Human Disease. Methods Mol. Biol. 2020, 2062, 3–33. [Google Scholar] [PubMed]
- Wahlund, C.J.E.; Gucluler Akpinar, G.; Steiner, L.; Ibrahim, A.; Bandeira, E.; Lepzien, R.; Lukic, A.; Smed-Sörensen, A.; Kullberg, S.; Eklund, A.; et al. Sarcoidosis exosomes stimulate monocytes to produce pro-inflammatory cytokines and CCL2. Sci. Rep. 2020, 10, 15328. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [PubMed]
- Roig-Arcos, J.; López-Malo, D.; Díaz-Llopis, M.; Romero, F.J. Exosomes derived from stimulated monocytes promote endothelial dysfunction and inflammation in vitro. Ann. Transl. Med. 2017, 5, 258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Yuan, H.X.; Ou, Z.J.; Ou, J.S. Microparticles (Exosomes) and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Chirivino, D.; Del Maestro, L.; Formstecher, E.; Hupé, P.; Raposo, G.; Louvard, D.; Arpin, M. The ERM proteins interact with the HOPS complex to regulate the maturation of endosomes. Mol. Biol. Cell. 2011, 22, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Jung, S.H.; Lee, G.H.; Lee, S.; Park, H.J.; Ko, Y.G.; Kim, Y.N.; Lee, J.S. Sulfated syndecan 1 is critical to preventing cellular senescence by modulating fibroblast growth factor receptor endocytosis. FASEB J. 2020, 34, 10316–10328. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Ebine, K.; Askani, J.C.; Krüger, F.; Gonzalez, Z.A.; Ito, E.; Goh, T.; Schumacher, K.; Nakano, A.; Ueda, T. Distinct sets of tethering complexes, SNARE complexes, and Rab GTPases mediate membrane fusion at the vacuole in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E2457–E2466. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, A.; Bucci, C. Coordination between Rac1 and Rab Proteins: Functional Implications in Health and Disease. Cells 2019, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, S.; Chang, V.S.; Navarro-Gomez, D.; Stanwyck, L.K.; Sevgi, D.D.; Papavasileiou, E.; Ren, A.; Uchiyama, E.; Sullivan, L.; Lobo, A.M.; et al. Association of genetic variants in RAB23 and ANXA11 with uveitis in sarcoidosis. Mol. Vis. 2018, 24, 59–74. [Google Scholar] [PubMed]
- Brooks, R.; Williamson, R.; Bass, M. Syndecan-4 independently regulates multiple small GTPases to promote fibroblast migration during wound healing. Small GTPases 2012, 3, 73–79. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fergie, J.; Srivastava, A. Immunity to SARS-CoV-2: Lessons Learned. Front. Immunol. 2021, 12, 654165. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Choudhari, R.; Nema, V.; Khan, A.A. ACE2 and TMPRSS2 polymorphisms in various diseases with special reference to its impact on COVID-19 disease. Microb. Pathog. 2021, 150, 104621. [Google Scholar] [CrossRef] [PubMed]
- Southern, B.D. Patients with interstitial lung disease and pulmonary sarcoidosis are at high risk for severe illness related to COVID-19. Clevel. Clin. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Rentsch, C.T.; Morton, C.E.; Hulme, W.J.; Schultze, A.; MacKenna, B.; Eggo, R.M.; Bhaskaran, K.; Wong, A.Y.S.; Williamson, E.J.; et al. Ethnic differences in SARS-CoV-2 infection and COVID-19-related hospitalisation, intensive care unit admission, and death in 17 million adults in England: An observational cohort study using the OpenSAFELY platform. Lancet 2021, 397, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Morgenthau, A.S.; Levin, M.A.; Freeman, R.; Reich, D.L.; Klang, E. Moderate or Severe Impairment in Pulmonary Function is Associated with Mortality in Sarcoidosis Patients Infected with SARS-CoV-2. Lung 2020, 198, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Gracia-Tello, B.; Robles, A.; Alguacil, A.; Bonet, M.; De-Escalante, B.; Noblejas-Mosso, A.; Gómez-de-la-Torre, R.; Akasbi, M.; Pérez-de-Lis, M.; et al. Characterization and Outcomes of SARS-CoV-2 Infection in Patients with Sarcoidosis. Viruses 2021, 13, 1000. [Google Scholar] [CrossRef] [PubMed]
- Manansala, M.; Chopra, A.; Baughman, R.P.; Novak, R.; Lower, E.E.; Culver, D.A.; Korsten, P.; Drake, W.P.; Judson, M.A.; Sweiss, N. COVID-19 and Sarcoidosis, Readiness for Vaccination: Challenges and Opportunities. Front. Med. 2021, 8, 672028. [Google Scholar] [CrossRef] [PubMed]
- Jeny, F.; Lhote, R.; Lorillon, G.; Belhomme, N.; Pugnet, G.; Borie, R.; Justet, A.; Jouneau, S.; Freymond, N.; Mekinian, A.; et al. Correspondence on ’Glucocorticoid-induced relapse of COVID-19 in a patient with sarcoidosis’. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Manansala, M.; Ascoli, C.; Alburquerque, A.G.; Perkins, D.; Mirsaedi, M.; Finn, P.; Sweiss, N.J. Case Series: COVID-19 in African American Patients With Sarcoidosis. Front. Med. 2020, 7, 588527. [Google Scholar] [CrossRef] [PubMed]
- Badary, O.A. Pharmacogenomics and COVID-19: Clinical implications of human genome interactions with repurposed drugs. Pharm. J. 2021, 4, 1–10. [Google Scholar] [PubMed]
- Raju, R.; Prajith, V.; Biatris, P.S.J. Therapeutic role of corticosteroids in COVID-19: A systematic review of registered clinical trials. Future J. Pharm. Sci. 2021, 7, 1–18. [Google Scholar]
- Judson, M.A. Corticosteroids in Sarcoidosis. Rheum. Dis. Clin. N. Am. 2016, 42, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Rios, M.; Fernández-Fierro, A.; Covián, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Wasan, H.; Reeta, K.H. Heme oxygenase-1 modulation: A potential therapeutic target for COVID-19 and associated complications. Free Radic. Biol. Med. 2020, 161, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020, 25, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Zahedipour, F.; Hosseini, S.A.; Sathyapalan, T.; Majeed, M.; Jamialahmadi, T.; Al-Rasadi, K.; Banach, M.; Sahebkar, A. Potential effects of curcumin in the treatment of COVID-19 infection. Phytother. Res. 2020, 34, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Ramdani, L.H.; Bachari, K. Potential therapeutic effects of Resveratrol against SARS-CoV-2. Acta Virol. 2020, 64, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Cherian, J.J.; Sharma, A. Exploring links between vitamin D deficiency and COVID-19. PLoS Pathog. 2020, 16, e1008874. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; Young, B.D.; Sloan, B.; Miller, E.J.; Obando, J.A.; King, B. Treatment of Multiorgan Sarcoidosis with Tofacitinib. ACR Open Rheumatol. 2020, 2, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92, e00321-18. [Google Scholar] [CrossRef] [PubMed]
- Bottermann, M.; Foss, S.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; Higginson, K.; Hirst, J.C.; et al. TRIM21 mediates antibody inhibition of adenovirus-based gene delivery and vaccination. Proc. Natl. Acad. Sci. USA 2018, 115, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020, 92, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.; Donnelly, S.C.; Moller, D.R. Tocilizumab in sarcoidosis patients failing steroid sparing therapies and anti-TNF agents. Respir. Med. X 2019, 1, 100004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 2013, 587, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, Y.; Yin, D.Q.; Hong, S.; Son, Y.J.; Kim, J.H.; Cho, J.Y. TBK1 inhibitors: A review of patent literature (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli, R.M.L.; Berrill, M.; Catravas, J.D.; Marik, P.E. Quercetin and Vitamin C: An Experimental, Synergistic Therapy for the Prevention and Treatment of SARS-CoV-2 Related Disease (COVID-19). Front. Immunol. 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Autophagy as a molecular target of quercetin underlying its protective effects in human diseases. Arch. Physiol. Biochem. 2019, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; An, M.H.; Kim, W.J.; Hwang, T.H. Comparative efficacy and safety of pharmacological interventions for the treatment of COVID-19: A systematic review and network meta-analysis. PLoS Med. 2020, 17, e1003501. [Google Scholar] [CrossRef] [PubMed]
- Hatipoglu, D.; Tanski, C.; Hachamovitch, R.; Legha, S.; Moudgil, R. Ixekizumab-Induced Cardiac Sarcoidosis: A Case Report. CJC Open 2020, 3, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, V.M.M. Interleukin-17: A potential therapeutic target in COVID-19. J. Infect. 2020, 81, e136–e138. [Google Scholar] [CrossRef] [PubMed]
- El Jammal, T.; Jaillou, Y.; Gerfaud-Valentin, M.; Valeyre, D.; Sève, P. Refractory Sarcoidosis: A Review. Ther. Clin. Risk Manag. 2020, 16, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Khullar, N.; Reddy, A.P.; Reddy, P.H. Therapeutic Strategies in the Development of Anti-viral Drugs and Vaccines Against SARS-CoV-2 Infection. Mol. Neurobiol. 2020, 57, 4856–4877. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Perfetto, L.; Micarelli, E.; Iannuccelli, M.; Lo Surdo, P.; Giuliani, G.; Latini, S.; Monia Pugliese, M.; Massacci, G.; Vumbaca, S.; Riccio, F.; et al. A Ressource for the Network Representation of Cell Perturbations Caused by SARS-CoV2 infection. Genes 2021, 12, 450. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
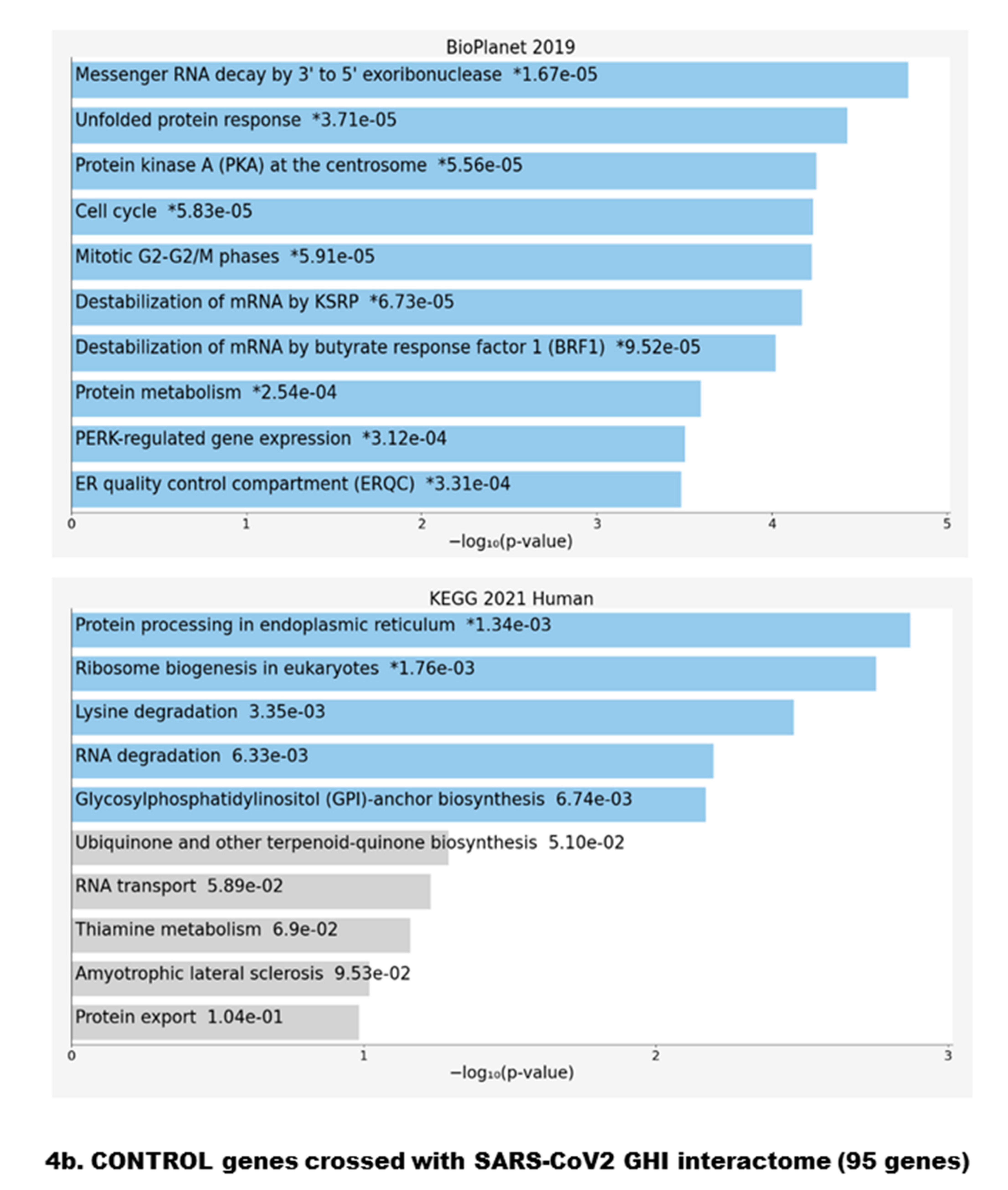{kind=link}
{kind=link}
{kind=link}
| Gene | Denomination | SARS-CoV2 | Functional Key Words | Identified in |
|---|---|---|---|---|
| Symbol | ||||
| Virus—Host cell interacting proteins involved in autophagy, mitophagy and mitochondrial processes | ||||
| YIF1A | Yip1 interacting factor homolog A | M | Mitochondrial factor involved in autophagy | F-SARC + Control |
| PITRM1 | Pitrilysin metallopeptidase 1 | M | Member of the mitochondrial OXPHOS system | F-SARC + Control |
| FASTKD5 | FAST kinase domain 5 | M | Regulation of all mitochondrial-encoded RNA | F-SARC |
| STOM | Stomatin | M | Cellular/mitochondrial ion channel activity | F-SARC |
| AASS | Aminoadipate-Semialdehyde Synthase | M | Saccharopine-induced mitochondrial damage | F-SARC + Control |
| TBK1 | TANK Binding Kinase 1 | Nsp13 | Regulation of IFN-I and mTOR, aggregates with MAVS | F-SARC |
| GOLGA2 | Golgin subfamily A member 2 | Nsp13 | Golgi-derived vesicles traffic regulation, endosomes | F-SARC |
| GOLGA3 | Golgin subfamily A member 3 | Nsp13 | Golgi-derived vesicles traffic regulation, endosomes | F-SARC |
| GOLGA7 | Golgin subfamily A member 7 | SPIKE | Interacts with ZDHHC5, Golgi vesicles traffic regulation | F-SARC |
| GOLGB1 | Golgin subfamily B member 1 | Nsp13 | Golgi-derived vesicles traffic regulation, endosomes | F-SARC |
| TYSDN1 | Trypsin Like Peroxisomal Matrix Peptidase 1 | Nsp12 | Component of peroxisomes, role in pexophagy | F-SARC + Control |
| ECSIT | ECSIT Signaling Integrator | Orf9b | Mitochondrial factor, regulation of viral signaling | F-SARC |
| NLRX1 | NLR Family Member X1 | Orf9b | Mitochondrial factor, interfere in MAVS-RIG1 signaling | F-SARC + Control |
| ACAD9 | Acyl-CoA Dehydrogenase Family Member 9 | Orf9b | Component of the mitochondrial complex I | F-SARC + Control |
| PUSL1 | tRNA pseudouridine synthase-like 1 | Orf8 | Mitochondrial protein for efficient mt-RNA translation | F-SARC + Control |
| DNAJC11 | DnaJ homolog subfamily C member 11 | Nsp4 | Component of the mitochondrial complex I | F-SARC + Control |
| BRD4 | Bromodomain-containing protein 4 | E | Regulation of autophagy, mitophagy through PINK1 | F-SARC + Control |
| Virus—Host cell interacting proteins involved in Rho-GTPases (Rab/Rac1) related pathways | ||||
| VPS11 | Vacuolar protein sorting-associated protein 11 homolog | Orf3A | Connects Rab GTPases to Golgi and autophagosome | F-SARC |
| PDE4DIP | Phosphodiesterase 4D Interacting Protein | Nsp13 | PDE4D-related regulatory role in Golgi microtubules | F-SARC |
| FYCO1 | FYVE And Coiled-Coil Domain Autophagy Adaptor 1 | Nsp13 | Rab7-mediated transport of late endosomes | F-SARC + Control |
| HS2ST1 | Heparan Sulfate 2-O-Sulfotransferase 1 | HS2ST1 | Involved in Rab and Rac1 signaling | F-SARC |
| Virus-host cell interacting proteins involved in ER-associated vesicular traffic, extracellular matrix, and coagulation | ||||
| ZDHHC5 | Zinc finger DHHC-type palmitoyltransferase 5 | SPIKE | Intracellular protein trafficking and S-acylation | F-SARC |
| EMC1 | ER Membrane Protein Complex Subunit 1 | Orf8 | The largest component of ER transmembrane complex | F-SARC |
| PLD3 | Phospholipase D3 | Orf8 | ER transmembrane protein, acts on amyloid secretion | F-SARC + Control |
| GDF15 | Growth Differentiation Factor 15 | Orf8 | Role in IPF, correlation with pulmonary function | F-SARC |
| FKBP10 | FK506-binding protein 10 | Orf8 | Involved in transmembrane signal transducers IRE1 | F-SARC + Control |
| ADAM9 | Disintegrin metalloproteinase domain-containing protein 9. | Orf8 | Involved in transmembrane signal transducers IRE1 | F-SARC + Control |
| IL17RA | Interleukin 17 Receptor A | Orf8 | Innate immune signaling, role in thrombosis | F-SARC + Control |
| PLAT | Plasminogen Activator, Tissue Type | Orf8 | Interaction with annexin A2, role in coagulation | F-SARC + Control |
| EXOSC2 | Exosome Component 2 | Nsp8 | Extracellular vesicles, clearance of viral particles | F-SARC |
| UGGT2 | UDP-Glucose Glycoprotein Glucosyltransferase 2 | Orf8 | Provides quality control for protein folding in the ER | CTRL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco, Y.; Valeyre, D.; El Jammal, T.; Vallee, M.; Chevalier, F.; Lamartine, J.; Sigaudo-Roussel, D.; Verrier, B.; Israel-Biet, D.; Freymond, N.; et al. Autophagy and Mitophagy-Related Pathways at the Crossroads of Genetic Pathways Involved in Familial Sarcoidosis and Host-Pathogen Interactions Induced by Coronaviruses. Cells 2021, 10, 1995. https://doi.org/10.3390/cells10081995
Pacheco Y, Valeyre D, El Jammal T, Vallee M, Chevalier F, Lamartine J, Sigaudo-Roussel D, Verrier B, Israel-Biet D, Freymond N, et al. Autophagy and Mitophagy-Related Pathways at the Crossroads of Genetic Pathways Involved in Familial Sarcoidosis and Host-Pathogen Interactions Induced by Coronaviruses. Cells. 2021; 10(8):1995. https://doi.org/10.3390/cells10081995
Chicago/Turabian StylePacheco, Yves, Dominique Valeyre, Thomas El Jammal, Maxime Vallee, Fabien Chevalier, Jérôme Lamartine, Dominique Sigaudo-Roussel, Bernard Verrier, Dominique Israel-Biet, Nathalie Freymond, and et al. 2021. "Autophagy and Mitophagy-Related Pathways at the Crossroads of Genetic Pathways Involved in Familial Sarcoidosis and Host-Pathogen Interactions Induced by Coronaviruses" Cells 10, no. 8: 1995. https://doi.org/10.3390/cells10081995
APA StylePacheco, Y., Valeyre, D., El Jammal, T., Vallee, M., Chevalier, F., Lamartine, J., Sigaudo-Roussel, D., Verrier, B., Israel-Biet, D., Freymond, N., Cottin, V., & Calender, A. (2021). Autophagy and Mitophagy-Related Pathways at the Crossroads of Genetic Pathways Involved in Familial Sarcoidosis and Host-Pathogen Interactions Induced by Coronaviruses. Cells, 10(8), 1995. https://doi.org/10.3390/cells10081995