
Inhibition of Phosphodiesterase 3A by Cilostazol Dampens Proinflammatory Platelet Functions

 , ,
, ,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Blood Collection
2.2. Mice
2.3. Cell Culture
2.4. Statistical Analysis
3. Results
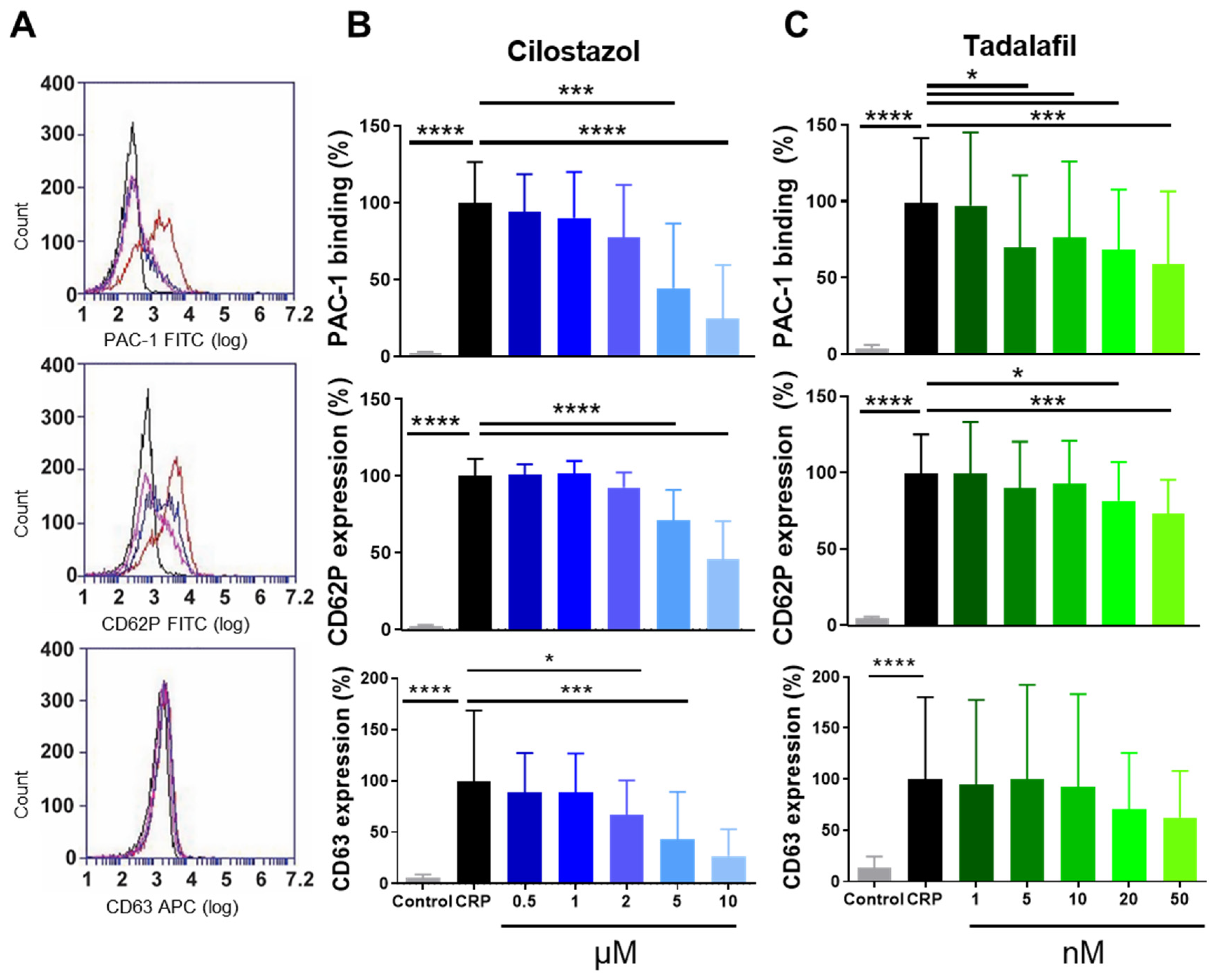
3.1. Function of PDE3A and PDE5 in P-Selectin Expression and αIIbβ3 Activation of Human and Mouse Platelets
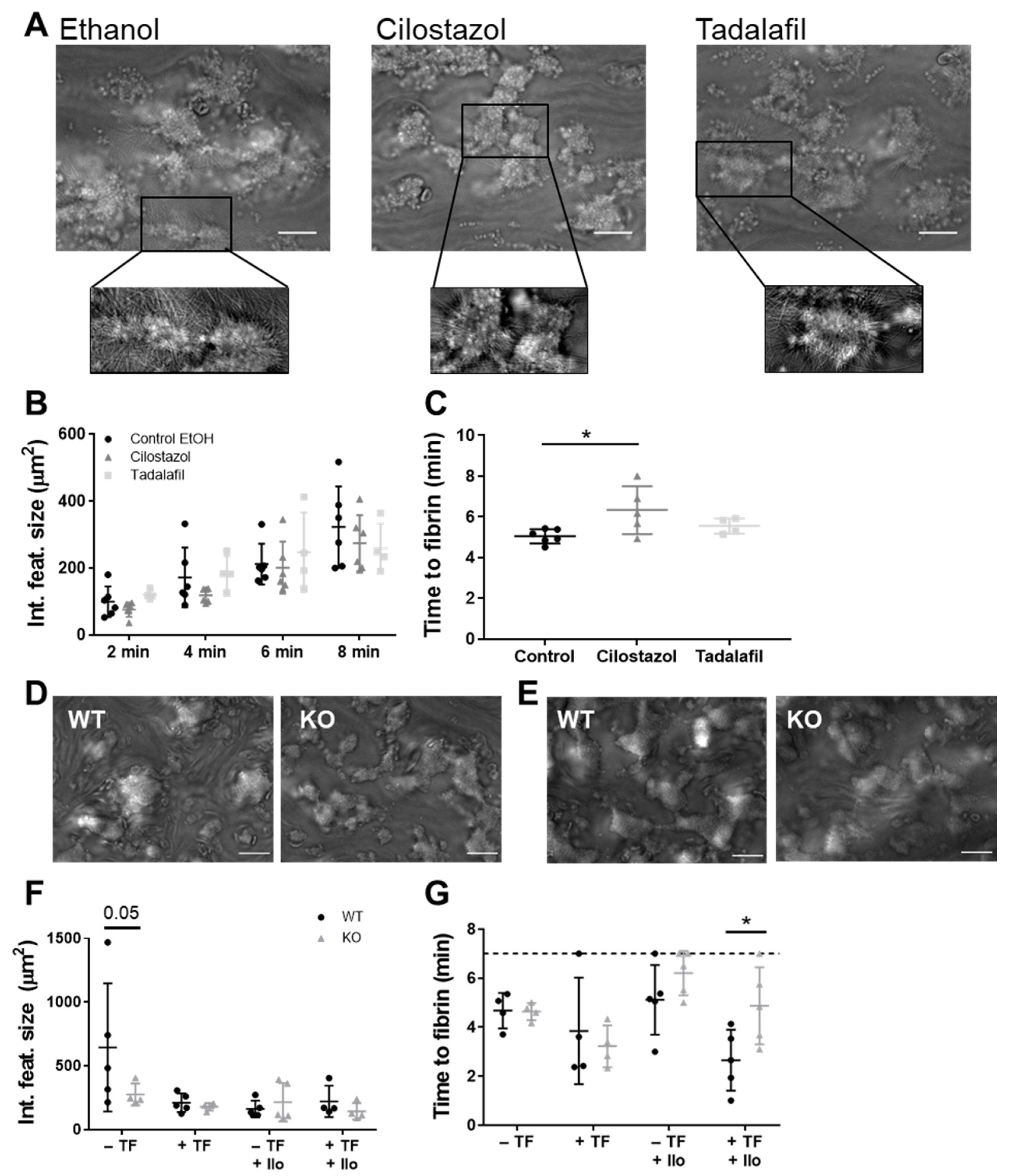
3.2. PDE3A, but not PDE5, Inhibition Delays Platelet-Dependent Coagulation, while Maintaining Initial Haemostatic Thrombus Formation in Human and Mouse Platelets
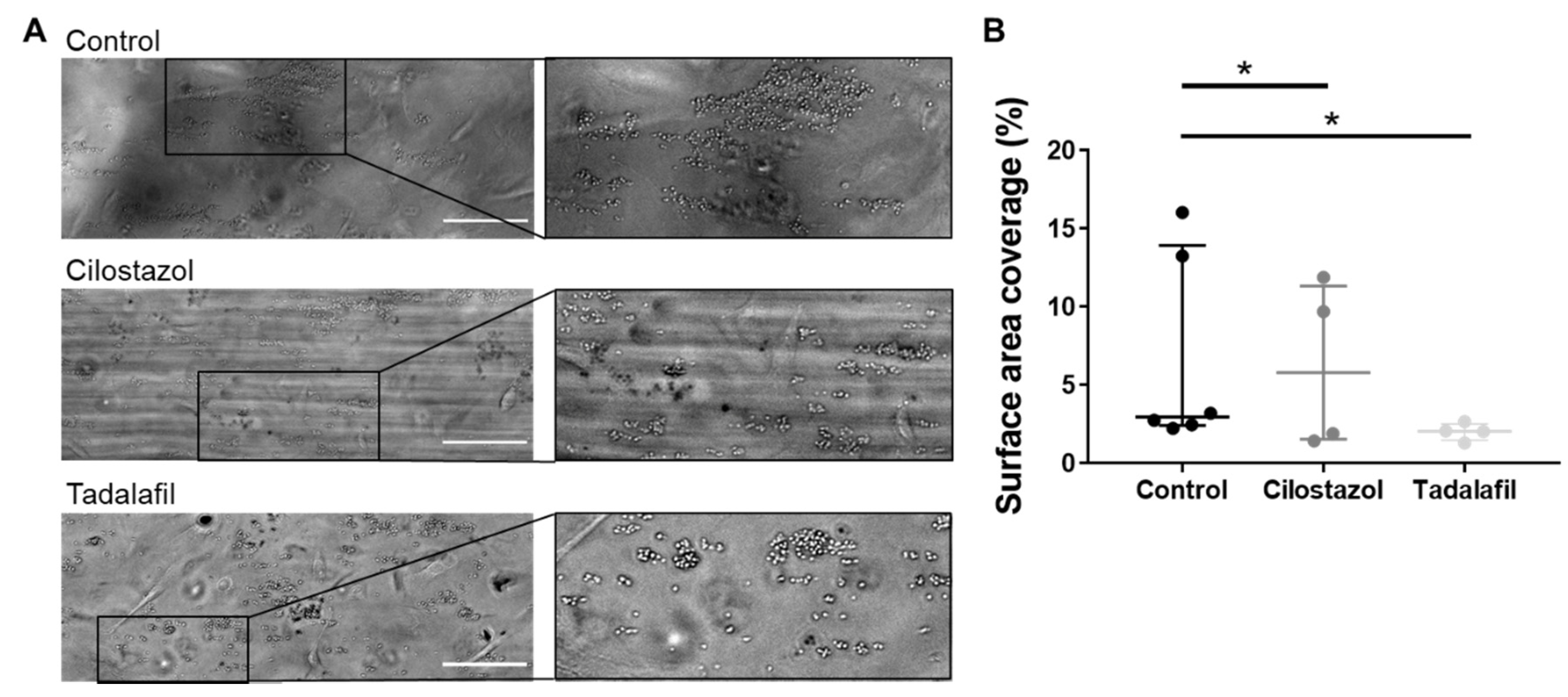
3.3. PDE3A and PDE5 Inhibition Decreases Platelet Adhesion to Inflamed Endothelial Cells
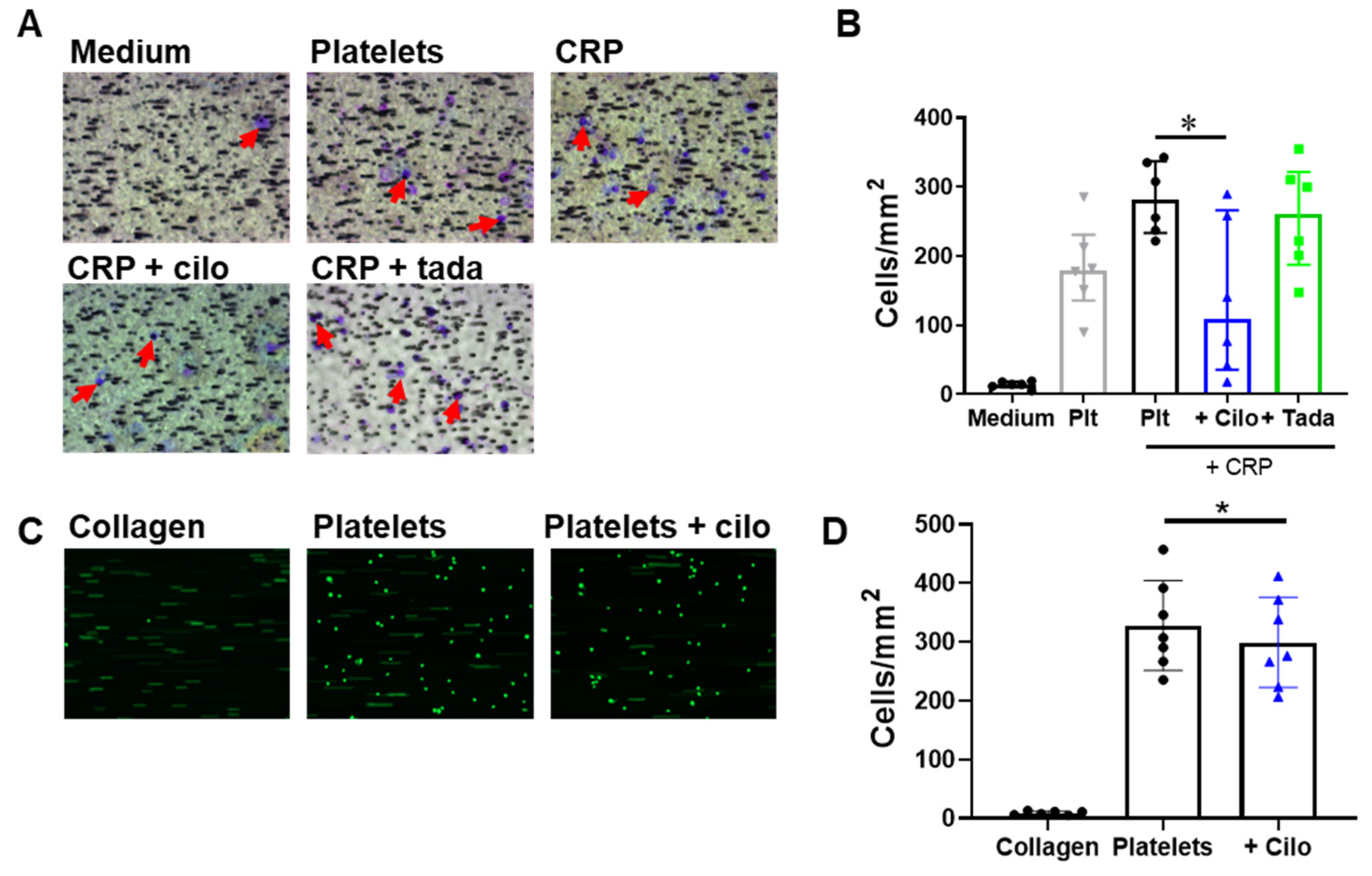
3.4. Monocyte Migration to and Adhesion on Platelets Is Reduced by Inhibition of PDE3A but not of PDE5
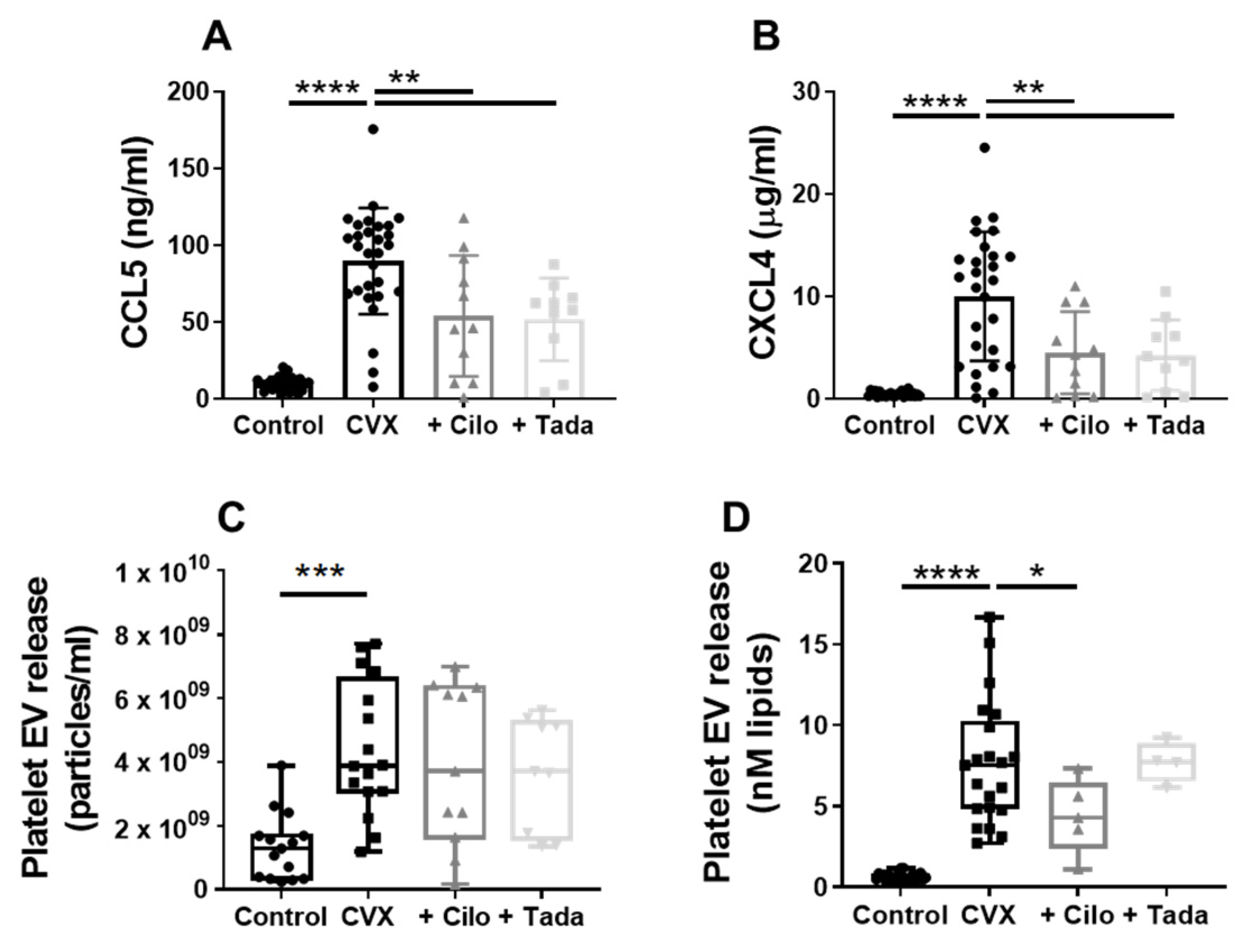
3.5. Platelet Chemokine- and Procoagulant EV Release Are Regulated by PDE3A and Partly by PDE5
4. Discussion
5. Highlights
- We identified novel roles for platelet PDE3A and PDE5 in promoting procoagulant and proinflammatory platelet functions.
- PDE3A inhibition with cilostazol reduced procoagulant extracellular vesicle release.
- Release of the chemokines CCL5 and CXCL4 was decreased by either cilostazol or the PDE5 inhibitor tadalafil.
- Our findings add mechanistic insight substantiating a beneficial effect of cilostazol in thromboinflammatory diseases.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coenen, D.M.; Heinzmann, A.C.A.; Karel, M.F.A.; Cosemans, J.M.E.M.; Koenen, R.R. The multifaceted contribution of platelets in the emergence and aftermath of acute cardiovascular events. Atherosclerosis 2021, 319, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Angiolillo, D.J.; Geisler, T.; Heitmeier, S. Dual pathway inhibition for vascular protection in patients with atherosclerotic disease: Rationale and review of the evidence. Thromb. Haemost. 2020, 120, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the American Heart Association. Circulation 2018, 137, e67–e92. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases (cvds). Available online: https://www.Who.Int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 19 February 2019).
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.R.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: A report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2017, 69, e71–e126. [Google Scholar] [CrossRef]
- Noma, K.; Higashi, Y. Cilostazol for treatment of cerebral infarction. Expert Opin. Pharm. 2018, 19, 1719–1726. [Google Scholar] [CrossRef]
- Toyoda, K.; Uchiyama, S.; Yamaguchi, T.; Easton, J.D.; Kimura, K.; Hoshino, H.; Sakai, N.; Okada, Y.; Tanaka, K.; Origasa, H.; et al. Dual antiplatelet therapy using cilostazol for secondary prevention in patients with high-risk ischaemic stroke in Japan: A multicentre, open-label, randomised controlled trial. Lancet Neurol. 2019, 18, 539–548. [Google Scholar] [CrossRef]
- Tamai, Y.; Takami, H.; Nakahata, R.; Ono, F.; Munakata, A. Comparison of the effects of acetylsalicylic acid, ticlopidine and cilostazol on primary hemostasis using a quantitative bleeding time test apparatus. Haemostasis 1999, 29, 269–276. [Google Scholar] [CrossRef]
- Wilhite, D.B.; Comerota, A.J.; Schmieder, F.A.; Throm, R.C.; Gaughan, J.P.; Rao, A.K. Managing PAD with multiple platelet inhibitors: The effect of combination therapy on bleeding time. J. Vasc. Surg. 2003, 38, 710–713. [Google Scholar] [CrossRef]
- Florescu, C.; Mustafa, E.R.; Târtea, E.A.; Florescu, D.R.; Albu, V.C. Antiplatelet therapy in secondary ischemic stroke prevention—a short review. Rom. J. Morphol. Embryol. 2019, 60, 383–387. [Google Scholar]
- Erdman, M.K.; Leary, M.C. Antiplatelet agents: Mechanisms and their role in stroke prevention. In Primer on Cerebrovascular Diseases; Academic Press: Cambridge, MA, USA, 2017; pp. 874–881. [Google Scholar]
- Bath, P.M.; Woodhouse, L.J.; Appleton, J.P.; Beridze, M.; Christensen, H.; Dineen, R.A.; Duley, L.; England, T.J.; Flaherty, K.; Havard, D.; et al. Antiplatelet therapy with aspirin, clopidogrel, and dipyridamole versus clopidogrel alone or aspirin and dipyridamole in patients with acute cerebral ischaemia (TARDIS): A randomised, open-label, phase 3 superiority trial. Lancet 2018, 391, 850–859. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar]
- Gresele, P.; Zoja, C.; Deckmyn, H.; Arnout, J.; Vermylen, J.; Verstraete, M. Dipyridamole inhibits platelet aggregation in whole blood. Thromb. Haemost. 1983, 50, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.; Bhardwaj, A.; Nair, A. Pharmacotherapy for pulmonary arterial hypertension. J. Thorac. Dis. 2019, 11, S1767–S1781. [Google Scholar] [CrossRef] [PubMed]
- Masciarelli, S.; Horner, K.; Liu, C.; Park, S.H.; Hinckley, M.; Hockman, S.; Nedachi, T.; Jin, C.; Conti, M.; Manganiello, V. Cyclic nucleotide phosphodiesterase 3A-deficient mice as a model of female infertility. J. Clin. Investig. 2004, 114, 196–205. [Google Scholar] [CrossRef]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Takimoto, E.; Hsu, S.; Lee, D.I.; Nagayama, T.; Danner, T.; Koitabashi, N.; Barth, A.S.; Bedja, D.; Gabrielson, K.L.; et al. Myocardial remodeling is controlled by myocyte-targeted gene regulation of phosphodiesterase type 5. J. Am. Coll. Cardiol. 2010, 56, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- de Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.E.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M.E.M. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Bourne, J.H.; Brill, A.; Watson, S.P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res. Pract. Thromb. Haemost. 2020, 4, 23–35. [Google Scholar] [CrossRef]
- Vajen, T.; Mause, S.F.; Koenen, R.R. Microvesicles from platelets: Novel drivers of vascular inflammation. Thromb. Haemost. 2015, 114, 228–236. [Google Scholar] [CrossRef]
- Dickhout, A.; Koenen, R.R. Extracellular vesicles as biomarkers in cardiovascular disease; chances and risks. Front. Cardiovasc. Med. 2018, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Huczek, Z.; Kapłon-Cieślicka, A.; Lacroix, R.; Opolski, G.; et al. Randomized controlled trial protocol to investigate the antiplatelet therapy effect on extracellular vesicles (AFFECT EV) in acute myocardial infarction. Platelets 2020, 31, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R. The prowess of platelets in immunity and inflammation. Thromb. Haemost. 2016, 116, 605–612. [Google Scholar] [CrossRef]
- Yagi, H.; Yamaguchi, N.; Shida, Y.; Hayakawa, M.; Matsumoto, M.; Sugimoto, M.; Wada, H.; Tsubaki, K.; Fujimura, Y. Cilostazol down-regulates the height of mural platelet thrombi formed under a high-shear rate flow in the absence of ADAMTS13 activity. Eur. J. Pharmacol. 2012, 691, 151–155. [Google Scholar] [CrossRef]
- Minami, N.; Suzuki, Y.; Yamamoto, M.; Kihira, H.; Imai, E.; Wada, H.; Kimura, Y.; Ikeda, Y.; Shiku, H.; Nishikawa, M. Inhibition of shear stress-induced platelet aggregation by cilostazol, a specific inhibitor of cGMP-inhibited phosphodiesterase, in vitro and ex vivo. Life Sci. 1997, 61, PL383–PL389. [Google Scholar] [CrossRef]
- Nakamura, T.; Uchiyama, S.; Yamazaki, M.; Iwata, M. Synergistic effect of cilostazol and dipyridamole mediated by adenosine on shear-induced platelet aggregation. Thromb. Res. 2007, 119, 511–516. [Google Scholar] [CrossRef]
- Onoda, K.; Ohashi, K.; Hashimoto, A.; Okuda, M.; Shimono, T.; Nishikawa, M.; Shimpo, H. Inhibition of platelet aggregation by combined therapy with aspirin and cilostazol after off-pump coronary artery bypass surgery. Ann. Thorac. Cardiovasc. Surg. 2008, 14, 230–237. [Google Scholar] [PubMed]
- Ryu, K.H.; Han, H.Y.; Lee, S.Y.; Jeon, S.D.; Im, G.J.; Lee, B.Y.; Kim, K.; Lim, K.M.; Chung, J.H. Ginkgo biloba extract enhances antiplatelet and antithrombotic effects of cilostazol without prolongation of bleeding time. Thromb Res. 2009, 124, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, T.; Nishikawa, M.; Kitai, T.; Ueda, Y.; Okinaka, T.; Makino, K.; Ito, M.; Isaka, N.; Ikeda, Y.; Shiku, H.; et al. Increased platelet aggregability in response to shear stress in acute myocardial infarction and its inhibition by combined therapy with aspirin and cilostazol after coronary intervention. Am. J. Cardiol. 2000, 85, 1054–1059. [Google Scholar] [CrossRef]
- Yamazaki, M.; Uchiyama, S.; Xiong, Y.; Nakano, T.; Nakamura, T.; Iwata, M. Effect of remnant-like particle on shear-induced platelet activation and its inhibition by antiplatelet agents. Thromb. Res. 2005, 115, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Miyakoda, G.; Mori, T. Cilostazol inhibits platelet-leukocyte interaction by suppression of platelet activation. Platelets 2004, 15, 293–301. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kang, M.J.; Cha, J.K. Cilostazol reduces PAC-1 expression on platelets in ischemic stroke. J. Clin. Neurol. 2008, 4, 148–152. [Google Scholar] [CrossRef][Green Version]
- Nomura, S.; Inami, N.; Iwasaka, T.; Liu, Y. Platelet activation markers, microparticles and soluble adhesion molecules are elevated in patients with arteriosclerosis obliterans: Therapeutic effects by cilostazol and potentiation by dipyridamole. Platelets 2004, 15, 167–172. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef]
- Zeiler, M.; Moser, M.; Mann, M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol. Cell Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef]
- Sun, B.; Li, H.; Shakur, Y.; Hensley, J.; Hockman, S.; Kambayashi, J.; Manganiello, V.C.; Liu, Y. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 2007, 19, 1765–1771. [Google Scholar] [CrossRef]
- Hashimoto, A.; Miyakoda, G.; Hirose, Y.; Mori, T. Activation of endothelial nitric oxide synthase by cilostazol via a cAMP/protein kinase A- and phosphatidylinositol 3-kinase/Akt-dependent mechanism. Atherosclerosis 2006, 189, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Hase, Y.; Okamoto, Y.; Fujita, Y.; Kitamura, A.; Nakabayashi, H.; Ito, H.; Maki, T.; Washida, K.; Takahashi, R.; Ihara, M. Cilostazol, a phosphodiesterase inhibitor, prevents no-reflow and hemorrhage in mice with focal cerebral ischemia. Exp. Neurol. 2012, 233, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, T.; Hayashi, T.; Hirayama, M.; Maruyama, H.; Tanahashi, N. Cilostazol inhibits platelet-endothelial cell interaction in murine microvessels after transient bilateral common carotid artery occlusion. J. Stroke Cerebrovasc. Dis. 2014, 23, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Sim, D.S.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B.; Flaumenhaft, R. Initial accumulation of platelets during arterial thrombus formation in vivo is inhibited by elevation of basal cAMP levels. Blood 2004, 103, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Projahn, D.; Koenen, R.R. Platelets: Key players in vascular inflammation. J. Leukoc. Biol. 2012, 92, 1167–1175. [Google Scholar] [CrossRef]
- van Gils, J.M.; Zwaginga, J.J.; Hordijk, P.L. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc Biol. 2009, 85, 195–204. [Google Scholar] [CrossRef]
- van Holten, T.C.; Bleijerveld, O.B.; Wijten, P.; de Groot, P.G.; Heck, A.J.R.; Barendrecht, A.D.; Merkx, T.H.; Scholten, A.; Roest, M. Quantitative proteomics analysis reveals similar release profiles following specific PAR-1 or PAR-4 stimulation of platelets. Cardiovasc. Res. 2014, 103, 140–146. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 2018, 8, 10647. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Gerdes, N.; Zhu, L.; Ersoy, M.; Hermansson, A.; Hjemdahl, P.; Hu, H.; Hansson, G.K.; Li, N. Platelets regulate CD4+ T-cell differentiation via multiple chemokines in humans. Thromb. Haemost. 2011, 106, 353–362. [Google Scholar] [CrossRef]
- Shi, G.; Field, D.J.; Long, X.; Mickelsen, D.; Ko, K.A.; Ture, S.; Korshunov, V.A.; Miano, J.M.; Morrell, C.N. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood 2013, 121, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, M.; Megens, R.T.; van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef]
- Witte, A.; Chatterjee, M.; Lang, F.; Gawaz, M. Platelets as a novel source of pro-inflammatory chemokine CXCL14. Cell Physiol. Biochem. 2017, 41, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; von Ungern-Sternberg, S.N.I.; Seizer, P.; Schlegel, F.; Büttcher, M.; Sindhu, N.A.; Müller, S.; Mack, A.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnagger, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.M.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Rosinska, J.; Lukasik, M.; Kozubski, W. The impact of vascular disease treatment on platelet-derived microvesicles. Cardiovasc. Drugs Ther. 2017, 31, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, M.; Ishibashi, S.; Ohkubo, T.; Nomura, S.; Sanjo, N.; Yokota, T.; Mizusawa, H. Elevated platelet microparticle levels after acute ischemic stroke with concurrent idiopathic thrombocytopenic purpura. J. Stroke Cerebrovasc. Dis. 2014, 23, 587–589. [Google Scholar] [CrossRef]
- Shirafuji, T.; Hamaguchi, H.; Kanda, F. Measurement of platelet-derived microparticle levels in the chronic phase of cerebral infarction using an enzyme-linked immunosorbent assay. Kobe J. Med. Sci. 2008, 54, E55–E61. [Google Scholar]
- Chen, Y.; Xiao, Y.; Lin, Z.; Xiao, X.; He, C.; Bihl, J.C.; Zhao, B.; Ma, X.; Chen, Y. The role of circulating platelets microparticles and platelet parameters in acute ischemic stroke patients. J. Stroke Cerebrovasc. Dis. 2015, 24, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Imamura, A.; Okuno, M.; Kamiyama, Y.; Fujimura, Y.; Ikeda, Y.; Fukuhara, S. Platelet-derived microparticles in patients with arteriosclerosis obliterans. Thromb. Res. 2000, 98, 257–268. [Google Scholar] [CrossRef]
- Nomura, S.; Shouzu, A.; Omoto, S.; Hayakawa, T.; Kagawa, H.; Nishikawa, M.; Inada, M.; Fujimura, Y.; Ikeda, Y.; Fukuhara, S. Effect of cilostazol on soluble adhesion molecules and platelet-derived microparticles in patients with diabetes. Thromb. Haemost. 1998, 80, 388–392. [Google Scholar]
- Omoto, S.; Nomura, S.; Shouzu, A.; Hayakawa, T.; Shimizu, H.; Miyake, Y.; Yonemoto, T.; Nishikawa, M.; Fukuhara, S.; Inada, M. Significance of platelet-derived microparticles and activated platelets in diabetic nephropathy. Nephron 1999, 81, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Cauwenberghs, S.; Feijge, M.A.H.; Harper, A.G.S.; Sage, S.O.; Curvers, J.; Heemskerk, J.W.M. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006, 580, 5313–5320. [Google Scholar] [CrossRef]
- Tans, G.; Rosing, J.; Thomassen, M.C.; Heeb, M.J.; Zwaal, R.F.; Griffin, J.H. Comparison of anticoagulant and procoagulant activities of stimulated platelets and platelet-derived microparticles. Blood 1991, 77, 2641–2648. [Google Scholar] [CrossRef]
- Muller, I.; Klocke, A.; Alex, M.; Kotzsch, M.; Luther, T.; Morgenstern, E.; Zieseniss, S.; Zahler, S.; Preissner, K.; Engelmann, B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003, 17, 476–478. [Google Scholar] [CrossRef]
- Sims, P.J.; Wiedmer, T.; Esmon, C.T.; Weiss, H.J.; Shattil, S.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: An isolated defect in platelet procoagulant activity. J. Biol. Chem. 1989, 264, 17049–17057. [Google Scholar] [CrossRef]
- Yao, Z.; Wang, L.; Wu, X.; Zhao, L.; Chi, C.; Guo, L.; Tong, D.; Yang, X.; Dong, Z.; Deng, R.; et al. Enhanced procoagulant activity on blood cells after acute ischemic stroke. Transl. Stroke Res. 2017, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Rosińska, J.; Maciejewska, J.; Narożny, R.; Kozubski, W.; Łukasik, M. Association of platelet-derived microvesicles with high on-treatment platelet reactivity in convalescent ischemic stroke patients treated with acetylsalicylic acid. Wiad. Lek. 2019, 72, 1426–1436. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coenen, D.M.; Heinzmann, A.C.A.; Oggero, S.; Albers, H.J.; Nagy, M.; Hagué, P.; Kuijpers, M.J.E.; Vanderwinden, J.-M.; van der Meer, A.D.; Perretti, M.; et al. Inhibition of Phosphodiesterase 3A by Cilostazol Dampens Proinflammatory Platelet Functions. Cells 2021, 10, 1998. https://doi.org/10.3390/cells10081998
Coenen DM, Heinzmann ACA, Oggero S, Albers HJ, Nagy M, Hagué P, Kuijpers MJE, Vanderwinden J-M, van der Meer AD, Perretti M, et al. Inhibition of Phosphodiesterase 3A by Cilostazol Dampens Proinflammatory Platelet Functions. Cells. 2021; 10(8):1998. https://doi.org/10.3390/cells10081998
Chicago/Turabian StyleCoenen, Daniëlle M., Alexandra C. A. Heinzmann, Silvia Oggero, Hugo J. Albers, Magdolna Nagy, Perrine Hagué, Marijke J. E. Kuijpers, Jean-Marie Vanderwinden, Andries D. van der Meer, Mauro Perretti, and et al. 2021. "Inhibition of Phosphodiesterase 3A by Cilostazol Dampens Proinflammatory Platelet Functions" Cells 10, no. 8: 1998. https://doi.org/10.3390/cells10081998
APA StyleCoenen, D. M., Heinzmann, A. C. A., Oggero, S., Albers, H. J., Nagy, M., Hagué, P., Kuijpers, M. J. E., Vanderwinden, J.-M., van der Meer, A. D., Perretti, M., Koenen, R. R., & Cosemans, J. M. E. M. (2021). Inhibition of Phosphodiesterase 3A by Cilostazol Dampens Proinflammatory Platelet Functions. Cells, 10(8), 1998. https://doi.org/10.3390/cells10081998