
UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin

 , , , ,
, , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Constructs
2.2. Antibodies and Proteins
2.3. Protein Expression and Purification
2.4. Cell Culture and Knockdown Experiments
2.5. Animals Overexpression and Knockdown Experiments
2.6. Binding Constant Determination by Fluorescence Quenching
2.7. Yeast Three-Hybrid Experiments
2.8. Statistical Analysis
3. Results
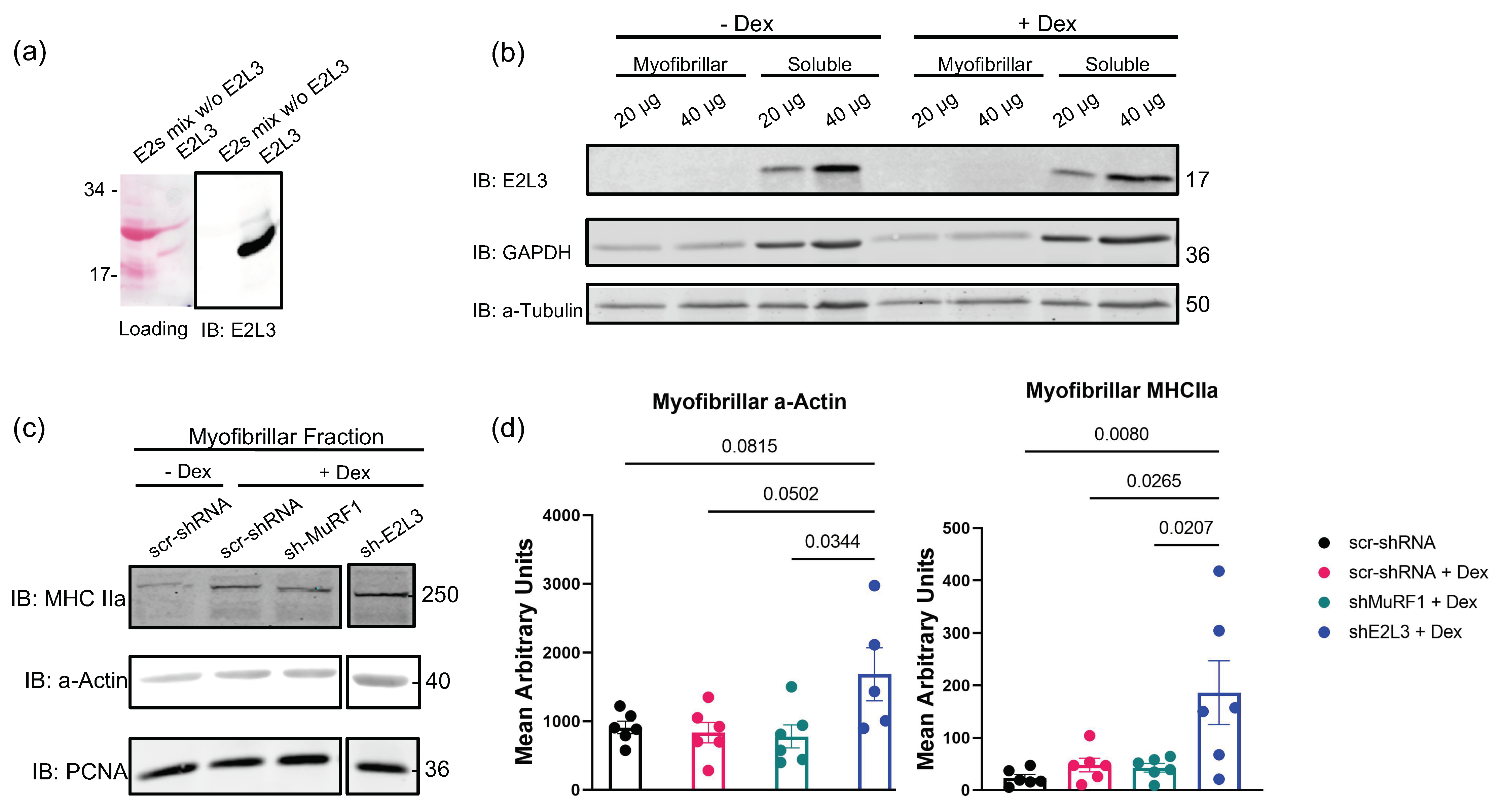
3.1. E2L3 Knockdown Preserves a-Actin and MHCII in Myofibrillar Fractions in Catabolic C2C12 Myotubes
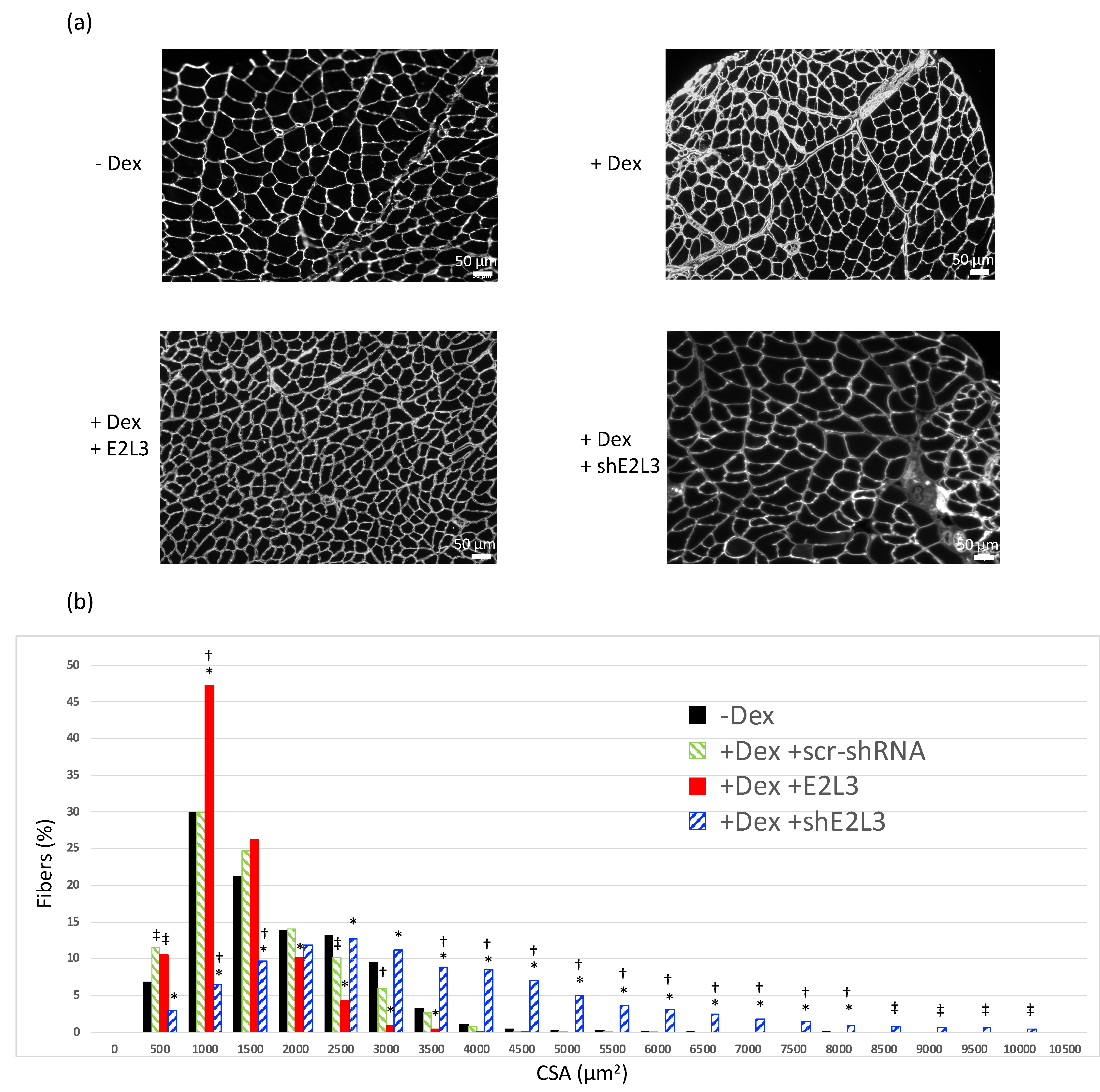
3.2. E2L3 Overexpression Aggravates Muscle Atrophy and E2L3 KD Induces Hypertrophy in Dex-Treated Mice
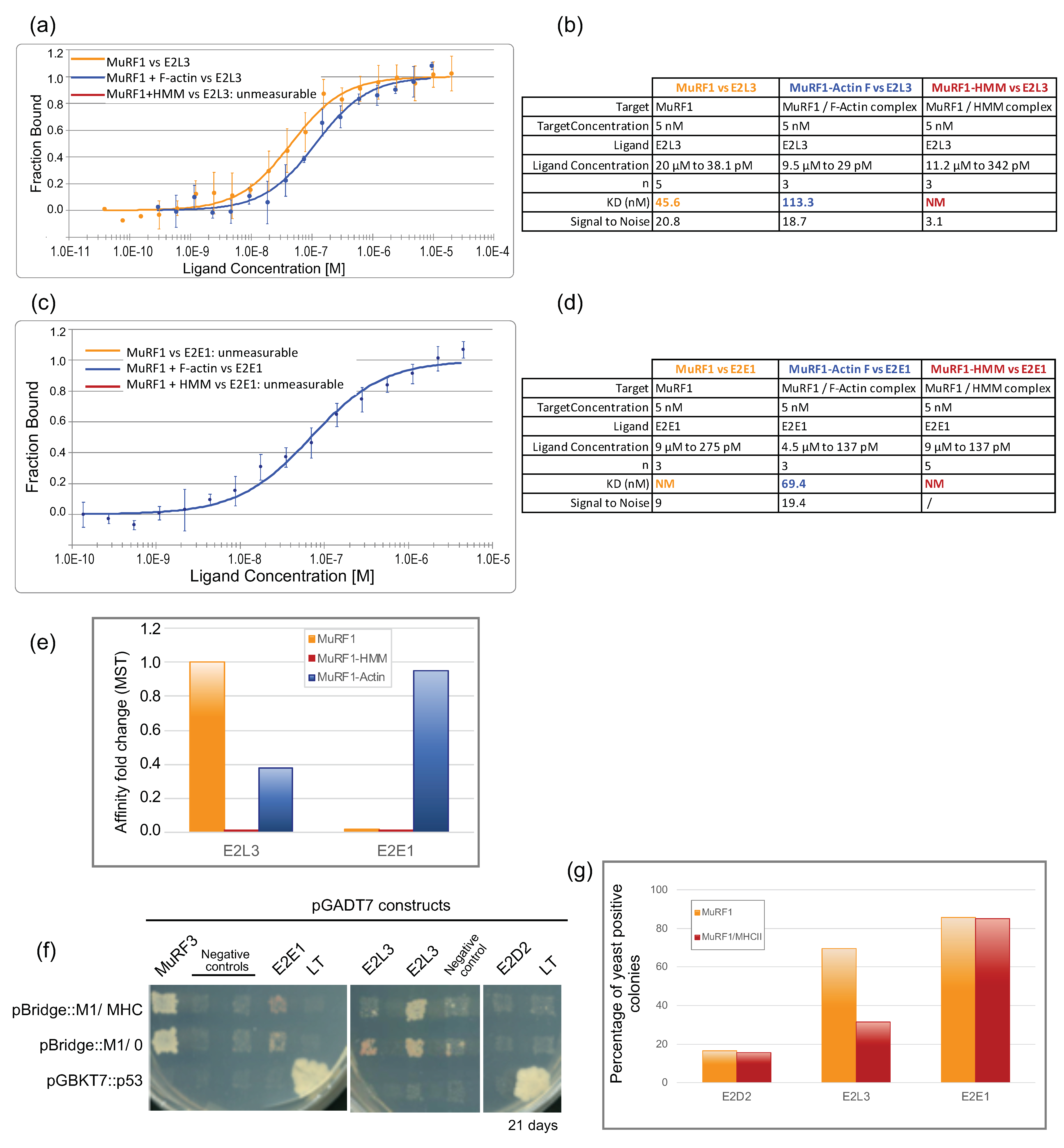
3.3. E2L3 Interaction with MuRF1 Complexed to Filamentous Actin or Myosin
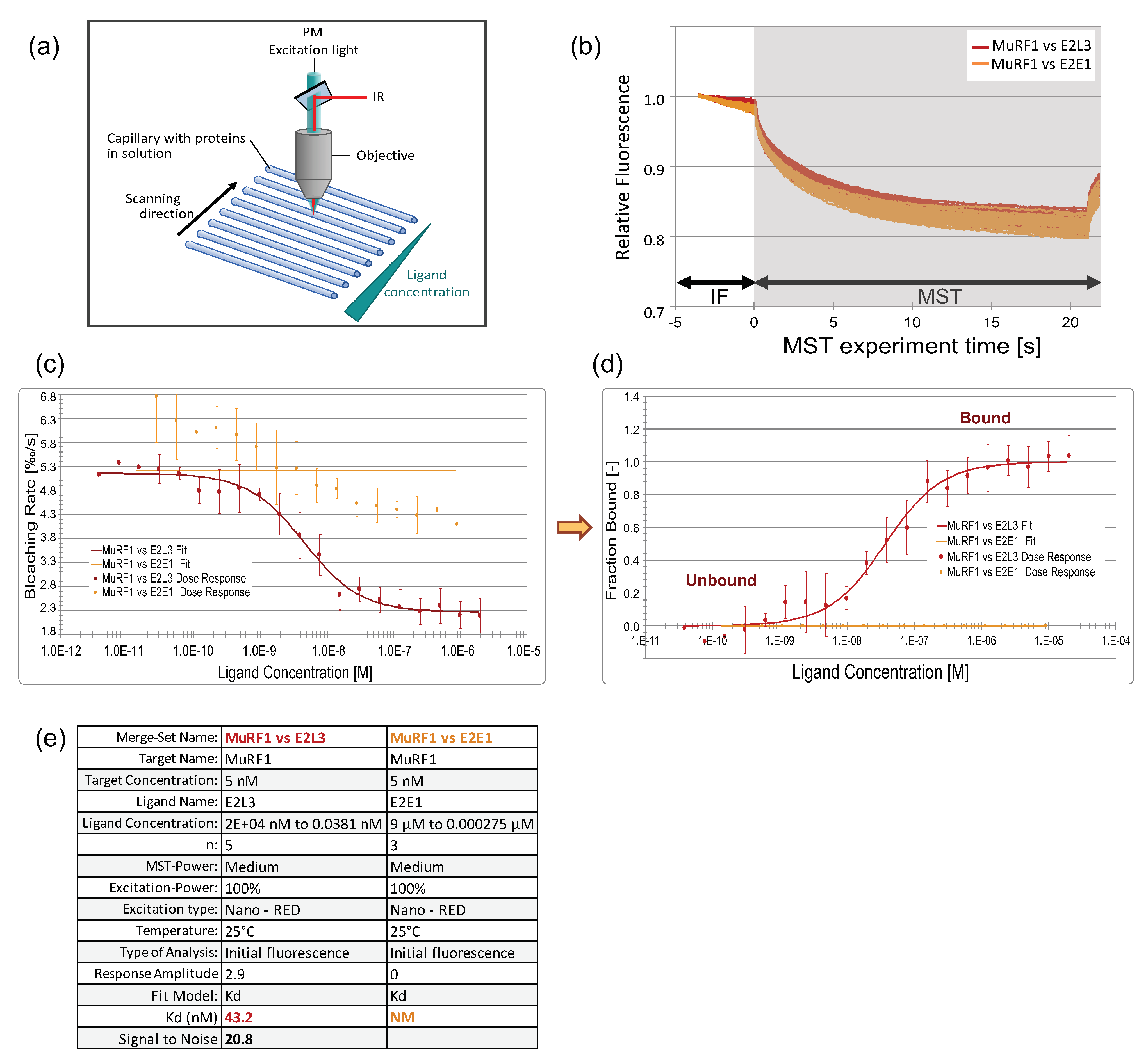
3.3.1. An Innovative Approach to Measure Proteins Interaction in Solution
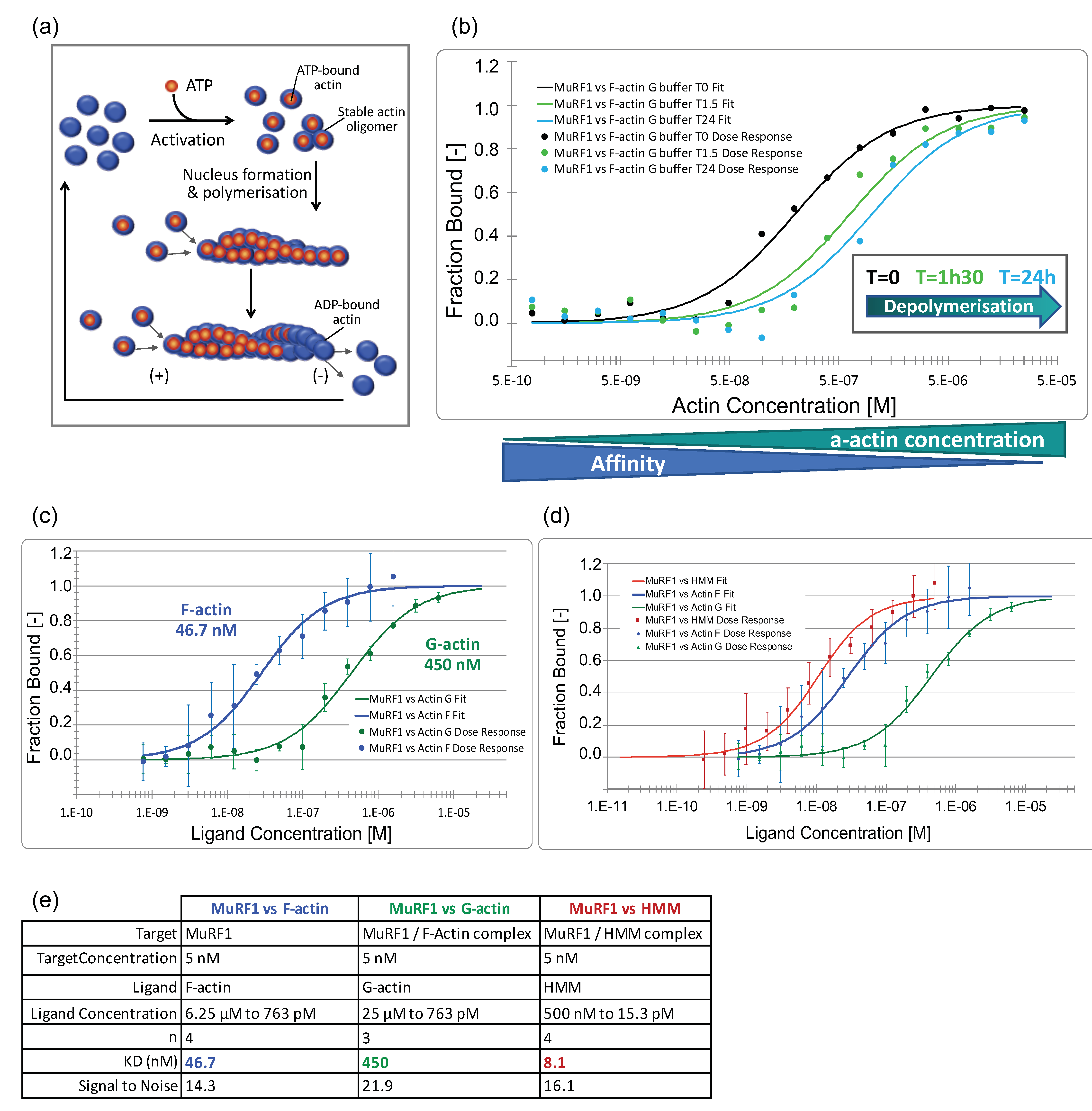
3.3.2. MuRF1 Exhibits a Better Affinity for Filamentous Than for Monomeric Actin
3.3.3. The Affinity between MuRF1 and E2L3 Is Not Enhanced by Actin or Myosin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Von Haehling, S.; Anker, S.D. Cachexia as a Major Underestimated and Unmet Medical Need: Facts and Numbers. J. Cachexia Sarcopenia Muscle 2010, 1, 1–5. [Google Scholar] [CrossRef]
- Von Haehling, S.; Anker, M.S.; Anker, S.D. Prevalence and Clinical Impact of Cachexia in Chronic Illness in Europe, USA, and Japan: Facts and Numbers Update 2016. J. Cachexia Sarcopenia Muscle 2016, 7, 507–509. [Google Scholar] [CrossRef]
- Furuno, K.; Goodman, M.N.; Goldberg, A.L. Role of Different Proteolytic Systems in the Degradation of Muscle Proteins during Denervation Atrophy. J. Biol. Chem. 1990, 265, 8550–8557. [Google Scholar] [CrossRef]
- Solomon, V.; Goldberg, A.L. Importance of the ATP-Ubiquitin-Proteasome Pathway in the Degradation of Soluble and Myofibrillar Proteins in Rabbit Muscle Extracts. J. Biol. Chem. 1996, 271, 26690–26697. [Google Scholar] [CrossRef]
- Tawa, N.E.; Odessey, R.; Goldberg, A.L. Inhibitors of the Proteasome Reduce the Accelerated Proteolysis in Atrophying Rat Skeletal Muscles. J. Clin. Investig. 1997, 100, 197–203. [Google Scholar] [CrossRef]
- Ventadour, S.; Attaix, D. Mechanisms of Skeletal Muscle Atrophy. Curr. Opin. Rheumatol. 2006, 18, 631–635. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin Accumulation and Striated Muscle Myopathy Result from the Loss of Muscle RING Finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle Actin Is Polyubiquitinylated In Vitro and In Vivo and Targeted for Breakdown by the E3 Ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During Muscle Atrophy, Thick, but Not Thin, Filament Components Are Degraded by MuRF1-Dependent Ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef]
- Ikeda, F.; Crosetto, N.; Dikic, I. What Determines the Specificity and Outcomes of Ubiquitin Signaling? Cell 2010, 143, 677–681. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Peris-Moreno, D.; Cussonneau, L.; Combaret, L.; Polge, C.; Taillandier, D. Ubiquitin Ligases at the Heart of Skeletal Muscle Atrophy Control. Molecules 2021, 26, 407. [Google Scholar] [CrossRef] [PubMed]
- Taillandier, D.; Polge, C. Skeletal Muscle Atrogenes: From Rodent Models to Human Pathologies. Biochimie 2019, 166, 251–269. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple Types of Skeletal Muscle Atrophy Involve a Common Program of Changes in Gene Expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of Muscle Atrophy, Fatigue and MLC Phosphorylation by MuRF1 as Indicated by Hindlimb Suspension Studies on MuRF1-KO Mice. J. Biomed. Biotechnol. 2010, 2010, 693741. [Google Scholar] [CrossRef] [PubMed]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle Sparing in Muscle RING Finger 1 Null Mice: Response to Synthetic Glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef]
- Vann, C.G.; Roberson, P.A.; Osburn, S.C.; Mumford, P.W.; Romero, M.A.; Fox, C.D.; Moore, J.H.; Haun, C.T.; Beck, D.T.; Moon, J.R.; et al. Skeletal Muscle Myofibrillar Protein Abundance Is Higher in Resistance-Trained Men, and Aging in the Absence of Training May Have an Opposite Effect. Sports 2020, 8, 7. [Google Scholar] [CrossRef]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.-H.; Rockman, H.A.; Patterson, C. Muscle-Specific RING Finger 1 Is a Bona Fide Ubiquitin Ligase That Degrades Cardiac Troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef]
- Peris-Moreno, D.; Taillandier, D.; Polge, C. MuRF1/TRIM63, Master Regulator of Muscle Mass. Int. J. Mol. Sci. 2020, 21, 6663. [Google Scholar] [CrossRef]
- Alpi, A.F.; Chaugule, V.; Walden, H. Mechanism and Disease Association of E2-Conjugating Enzymes: Lessons from UBE2T and UBE2L3. Biochem. J. 2016, 473, 3401–3419. [Google Scholar] [CrossRef]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A Muscle-Specific MuRF1-E2 Network Requires Stabilization of MuRF1-E2 Complexes by Telethonin, a Newly Identified Substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef]
- Deval, C.; Calonne, J.; Coudy-Gandilhon, C.; Vazeille, E.; Bechet, D.; Polge, C.; Taillandier, D.; Attaix, D.; Combaret, L. Mitophagy and Mitochondria Biogenesis Are Differentially Induced in Rat Skeletal Muscles during Immobilization and/or Remobilization. Int. J. Mol. Sci. 2020, 21, 3691. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 Reactivity Profile Reveals Parkin and HHARI to Be RING/HECT Hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Soss, S.E.; Yue, Y.; Dhe-Paganon, S.; Chazin, W.J. E2 Conjugating Enzyme Selectivity and Requirements for Function of the E3 Ubiquitin Ligase CHIP*. J. Biol. Chem. 2011, 286, 21277–21286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Wang, P.; Jeffrey, P.D.; Pavletich, N.P. Structure of a C-Cbl-UbcH7 Complex: RING Domain Function in Ubiquitin-Protein Ligases. Cell 2000, 102, 533–539. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kondo, T.; Houghton, A.; Bartkiewicz, M.; Horne, W.C.; Zhang, H.; Yoshimura, A.; Baron, R. Ligand-Induced Ubiquitination of the Epidermal Growth Factor Receptor Involves the Interaction of the c-Cbl RING Finger and UbcH7*. J. Biol. Chem. 1999, 274, 31707–31712. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin Ligase Cbl-b Is a Negative Regulator for Insulin-Like Growth Factor 1 Signaling during Muscle Atrophy Caused by Unloading. Mol. Cell. Biol. 2009, 29, 4798–4811. [Google Scholar] [CrossRef]
- Ahel, J.; Lehner, A.; Vogel, A.; Schleiffer, A.; Meinhart, A.; Haselbach, D.; Clausen, T. Moyamoya Disease Factor RNF213 Is a Giant E3 Ligase with a Dynein-like Core and a Distinct Ubiquitin-Transfer Mechanism. eLife 2020, 9, e56185. [Google Scholar] [CrossRef]
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; van Aalten, D.M.F.; Virdee, S. Activity-Based E3 Ligase Profiling Uncovers an E3 Ligase with Esterification Activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef]
- Pao, K.-C.; Stanley, M.; Han, C.; Lai, Y.-C.; Murphy, P.; Balk, K.; Wood, N.T.; Corti, O.; Corvol, J.-C.; Muqit, M.M.K.; et al. Probes of Ubiquitin E3 Ligases Enable Systematic Dissection of Parkin Activation. Nat. Chem. Biol 2016, 12, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-W.; Zheng, H.-F.; Cui, Y.; Sun, L.-D.; Ye, D.-Q.; Hu, Z.; Xu, J.-H.; Cai, Z.-M.; Huang, W.; Zhao, G.-P.; et al. Genome-Wide Association Study in a Chinese Han Population Identifies Nine New Susceptibility Loci for Systemic Lupus Erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef]
- Wang, S.; Adrianto, I.; Wiley, G.B.; Lessard, C.J.; Kelly, J.A.; Adler, A.J.; Glenn, S.B.; Williams, A.H.; Ziegler, J.T.; Comeau, M.E.; et al. A Functional Haplotype of UBE2L3 Confers Risk for Systemic Lupus Erythematosus. Genes Immun. 2012, 13, 380–387. [Google Scholar] [CrossRef]
- Kim, T.; Bae, S.-C.; Kang, C. Synergistic Activation of NF-ΚB by TNFAIP3 (A20) Reduction and UBE2L3 (UBCH7) Augment That Synergistically Elevate Lupus Risk. Arthritis Res. Ther. 2020, 22, 93. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef]
- Whitcomb, E.A.; Dudek, E.J.; Liu, Q.; Taylor, A. Novel Control of S Phase of the Cell Cycle by Ubiquitin-Conjugating Enzyme H7. MBoC 2008, 20, 1–9. [Google Scholar] [CrossRef][Green Version]
- Whitcomb, E.A.; Taylor, A. Ubiquitin Control of S Phase: A New Role for the Ubiquitin Conjugating Enzyme, UbcH7. Cell Div. 2009, 4, 17. [Google Scholar] [CrossRef]
- Whitcomb, E.A.; Tsai, Y.C.; Basappa, J.; Liu, K.; le Feuvre, A.K.; Weissman, A.M.; Taylor, A. Stabilization of P27Kip1/CDKN1B by UBCH7/UBE2L3 Catalyzed Ubiquitinylation: A New Paradigm in Cell-Cycle Control. FASEB J. 2019, 33, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-J.; Kalinina, A.; Wang, J.; Nakayama, K.; Nakayama, K.I.; Bagchi, S. The Papillomavirus E7 Oncoprotein Is Ubiquitinated by UbcH7 and Cullin 1- and Skp2-Containing E3 Ligase. J. Virol. 2004, 78, 5338–5346. [Google Scholar] [CrossRef]
- Anoveros-Barrera, A.; Bhullar, A.S.; Stretch, C.; Dunichand-Hoedl, A.R.; Martins, K.J.B.; Rieger, A.; Bigam, D.; McMullen, T.; Bathe, O.F.; Putman, C.T.; et al. Immunohistochemical Phenotyping of T Cells, Granulocytes, and Phagocytes in the Muscle of Cancer Patients: Association with Radiologically Defined Muscle Mass and Gene Expression. Skelet. Muscle 2019, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, L.; Chung, J.; Pozo, F.M.; Tran, A.; Seachrist, D.D.; Jacobberger, J.W.; Keri, R.A.; Gilmore, H.; Zhang, Y. UbcH7 Regulates 53BP1 Stability and DSB Repair. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Mayca Pozo, F.; Tang, J.; Bonk, K.W.; Keri, R.A.; Yao, X.; Zhang, Y. Regulatory Cross-Talk Determines the Cellular Levels of 53BP1 Protein, a Critical Factor in DNA Repair. J. Biol. Chem. 2017, 292, 5992–6003. [Google Scholar] [CrossRef]
- Devine, M.J.; Plun-Favreau, H.; Wood, N.W. Parkinson’s Disease and Cancer: Two Wars, One Front. Nat. Rev. Cancer 2011, 11, 813–823. [Google Scholar] [CrossRef]
- Polge, C.; Aniort, J.; Armani, A.; Claustre, A.; Coudy-Gandilhon, C.; Tournebize, C.; Deval, C.; Combaret, L.; Béchet, D.; Sandri, M.; et al. UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells 2018, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.J.; Cagnin, S.; Chemello, F.; Silvestrin, M.; Musaro, A.; De Pitta, C.; Lanfranchi, G.; Sandri, M. Involvement of MicroRNAs in the Regulation of Muscle Wasting during Catabolic Conditions*. J. Biol. Chem. 2014, 289, 21909–21925. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-Binding Assays in Biological Liquids Using Microscale Thermophoresis. Nat. Commun 2010, 1, 100. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular Interaction Studies Using Microscale Thermophoresis. ASSAY Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef]
- Polge, C.; Koulmann, N.; Claustre, A.; Jarzaguet, M.; Serrurier, B.; Combaret, L.; Béchet, D.; Bigard, X.; Attaix, D.; Taillandier, D. UBE2D2 Is Not Involved in MuRF1-Dependent Muscle Wasting during Hindlimb Suspension. Int. J. Biochem. Cell Biol. 2016, 79, 488–493. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of Cancer Cachexia and Muscle Wasting by ActRIIB Antagonism Leads to Prolonged Survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Scalabrin, M.; Adams, V.; Labeit, S.; Bowen, T.S. Emerging Strategies Targeting Catabolic Muscle Stress Relief. Int. J. Mol. Sci. 2020, 21, 4681. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Goldberg, A.L. The P97/VCP ATPase Is Critical in Muscle Atrophy and the Accelerated Degradation of Muscle Proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef]
- Volodin, A.; Kosti, I.; Goldberg, A.L.; Cohen, S. Myofibril Breakdown during Atrophy Is a Delayed Response Requiring the Transcription Factor PAX4 and Desmin Depolymerization. Proc. Natl. Acad. Sci. USA 2017, 114, E1375–E1384. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of Linear Polyubiquitylation of NEMO in NF-KappaB Activation. Nat. Cell Biol 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, B.; Rana, R.R.; Koliopoulos, M.G.; Morris-Davies, A.C.; Schaeffer, V.; Christodoulou, E.; Howell, S.; Brown, N.R.; Dikic, I.; Rittinger, K. Structural Basis for Ligase-Specific Conjugation of Linear Ubiquitin Chains by HOIP. Nature 2013, 503, 422–426. [Google Scholar] [CrossRef]
- Nikawa, T.; Ishidoh, K.; Hirasaka, K.; Ishihara, I.; Ikemoto, M.; Kano, M.; Kominami, E.; Nonaka, I.; Ogawa, T.; Adams, G.R.; et al. Skeletal Muscle Gene Expression in Space-Flown Rats. FASEB J. 2004, 18, 522–524. [Google Scholar] [CrossRef]
- Uchida, T.; Sakashita, Y.; Kitahata, K.; Yamashita, Y.; Tomida, C.; Kimori, Y.; Komatsu, A.; Hirasaka, K.; Ohno, A.; Nakao, R.; et al. Reactive Oxygen Species Upregulate Expression of Muscle Atrophy-Associated Ubiquitin Ligase Cbl-b in Rat L6 Skeletal Muscle Cells. Am. J. Physiol. Cell Physiol. 2018, 314, C721–C731. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Vollmer, S.; Golombek, S.; Kahle, P.J. The Ubiquitin-Conjugating Enzymes UBE2N, UBE2L3 and UBE2D2/3 Are Essential for Parkin-Dependent Mitophagy. J. Cell Sci. 2014, 127, 3280–3293. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Moussaud-Lamodière, E.L.; Ando, M.; Springer, W. A Specific Subset of E2 Ubiquitin-Conjugating Enzymes Regulate Parkin Activation and Mitophagy Differently. J. Cell Sci. 2014, 127, 3488–3504. [Google Scholar] [CrossRef] [PubMed]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin Impairs Mitochondrial Function and Leads to Muscle Atrophy. Am. J. Physiol. Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and Is Required for TNF-Mediated Gene Induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Rahighi, S.; Akita, M.; Kato, R.; Sasaki, Y.; Wakatsuki, S.; Iwai, K. Mechanism Underlying IκB Kinase Activation Mediated by the Linear Ubiquitin Chain Assembly Complex. Mol. Cell Biol. 2014, 34, 1322–1335. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peris-Moreno, D.; Malige, M.; Claustre, A.; Armani, A.; Coudy-Gandilhon, C.; Deval, C.; Béchet, D.; Fafournoux, P.; Sandri, M.; Combaret, L.; et al. UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin. Cells 2021, 10, 1974. https://doi.org/10.3390/cells10081974
Peris-Moreno D, Malige M, Claustre A, Armani A, Coudy-Gandilhon C, Deval C, Béchet D, Fafournoux P, Sandri M, Combaret L, et al. UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin. Cells. 2021; 10(8):1974. https://doi.org/10.3390/cells10081974
Chicago/Turabian StylePeris-Moreno, Dulce, Mélodie Malige, Agnès Claustre, Andrea Armani, Cécile Coudy-Gandilhon, Christiane Deval, Daniel Béchet, Pierre Fafournoux, Marco Sandri, Lydie Combaret, and et al. 2021. "UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin" Cells 10, no. 8: 1974. https://doi.org/10.3390/cells10081974
APA StylePeris-Moreno, D., Malige, M., Claustre, A., Armani, A., Coudy-Gandilhon, C., Deval, C., Béchet, D., Fafournoux, P., Sandri, M., Combaret, L., Taillandier, D., & Polge, C. (2021). UBE2L3, a Partner of MuRF1/TRIM63, Is Involved in the Degradation of Myofibrillar Actin and Myosin. Cells, 10(8), 1974. https://doi.org/10.3390/cells10081974