
Epigenetic Drifts during Long-Term Intestinal Organoid Culture

 ,
, 
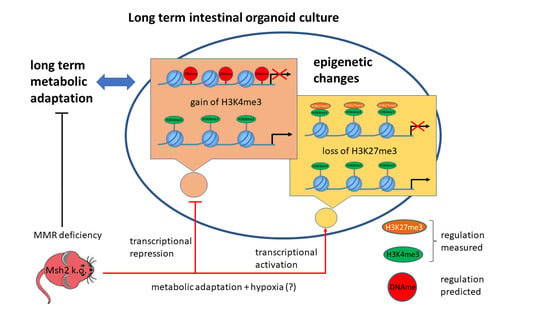
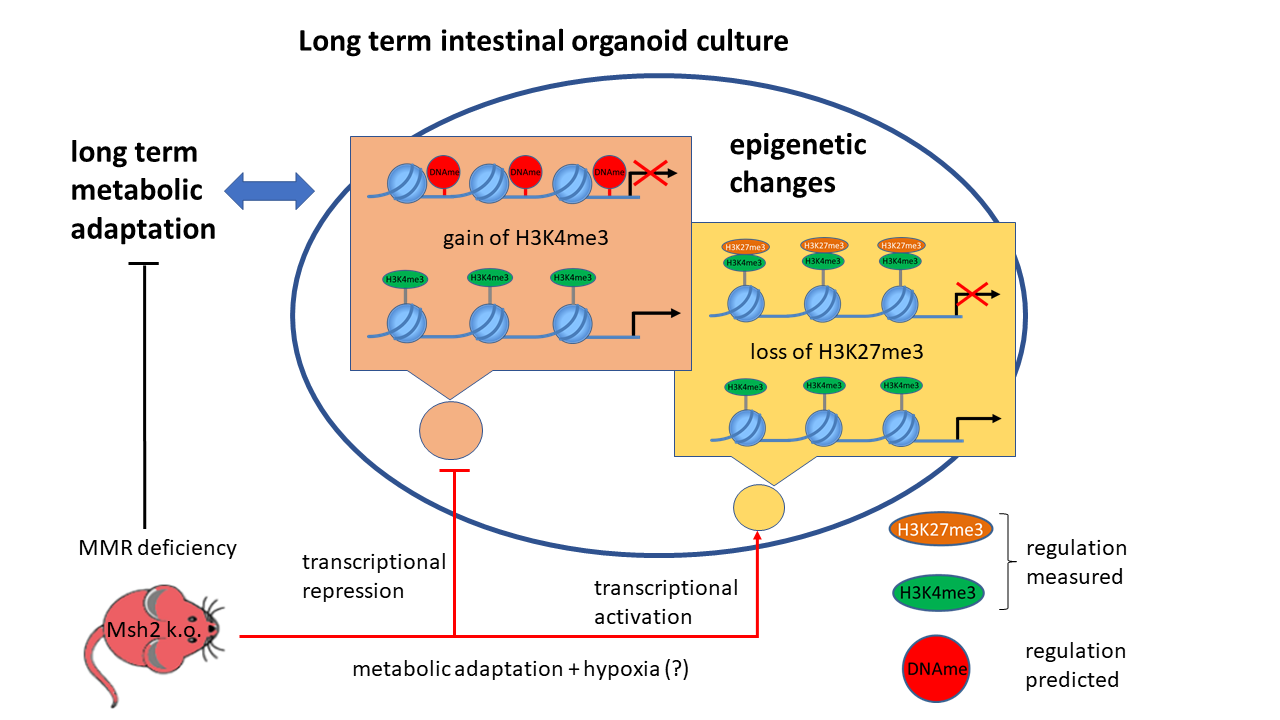
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Organoid Preparation and Culture
2.3. Chromatin Immunoprecipitation (ChIP)
2.4. Quantitative PCR and Illumina Sequencing
2.5. ChIP-Seq Data Preprocessing and Peak Calling
2.6. ChIP-Seq Data Analysis
2.7. RNA-Seq and Data Preprocessing
2.8. Self-Organizing Map (SOM) Analysis
2.9. Gene Set Enrichment Analysis
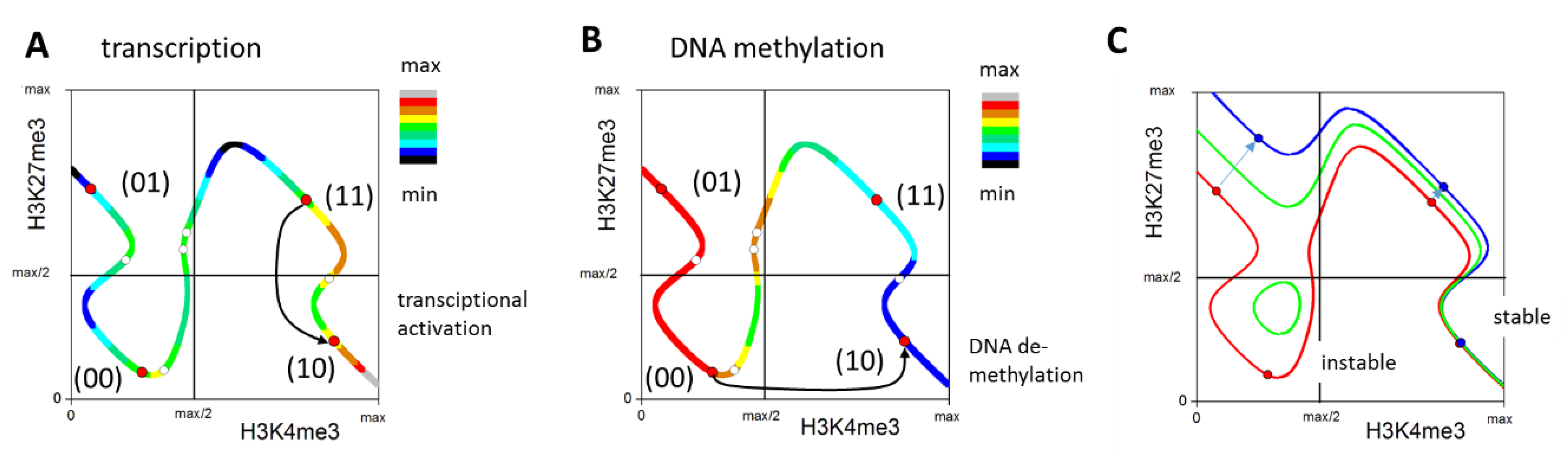
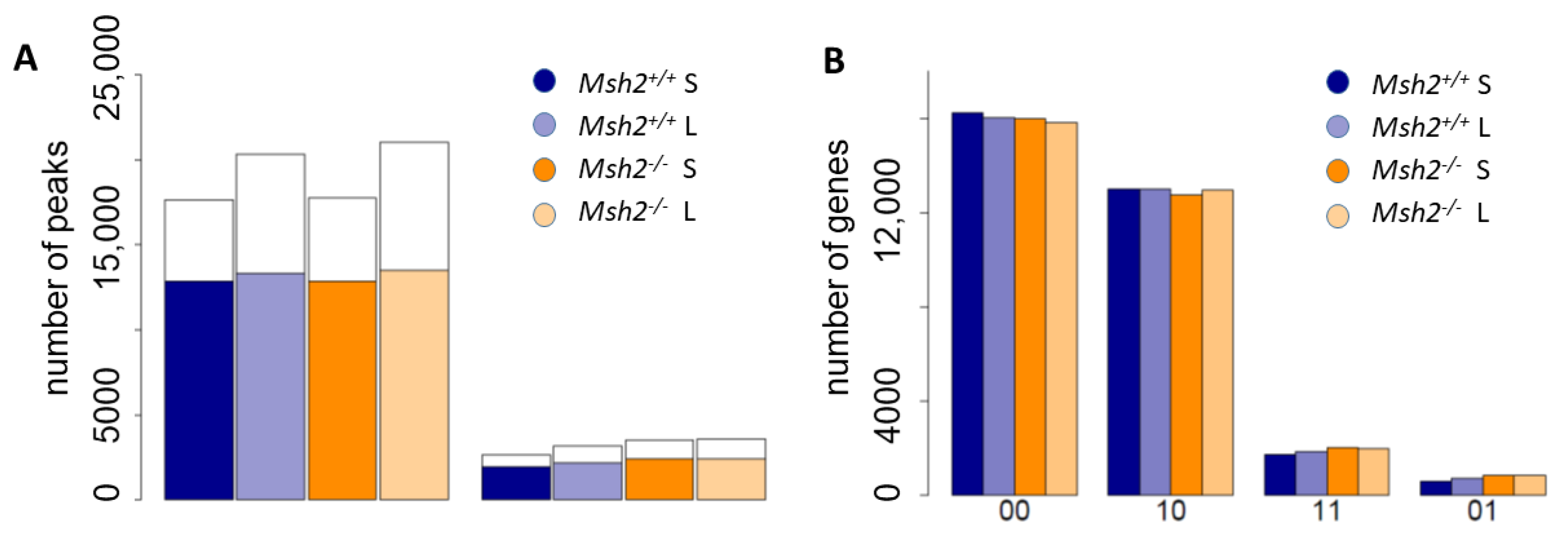
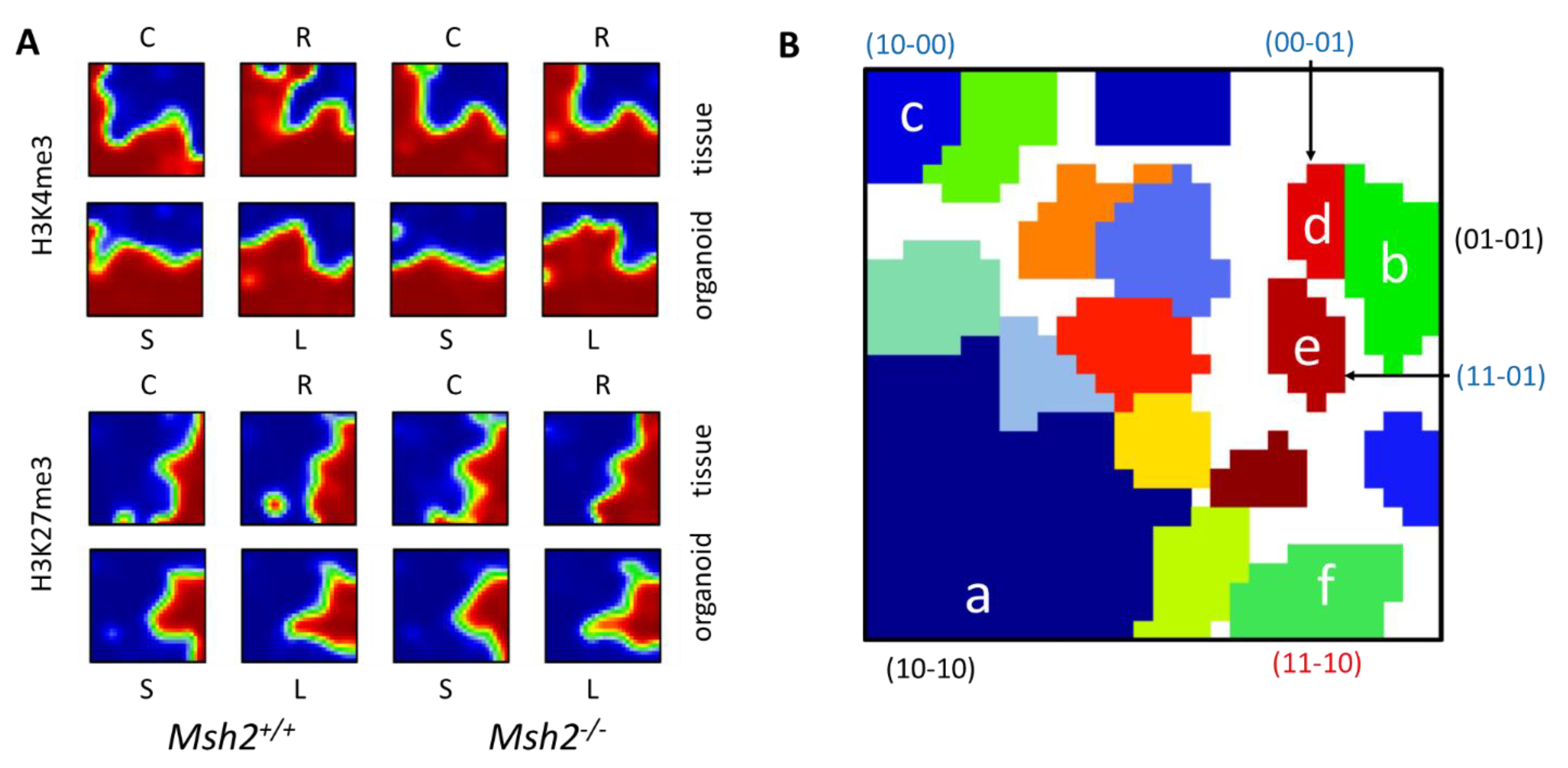
3. Results
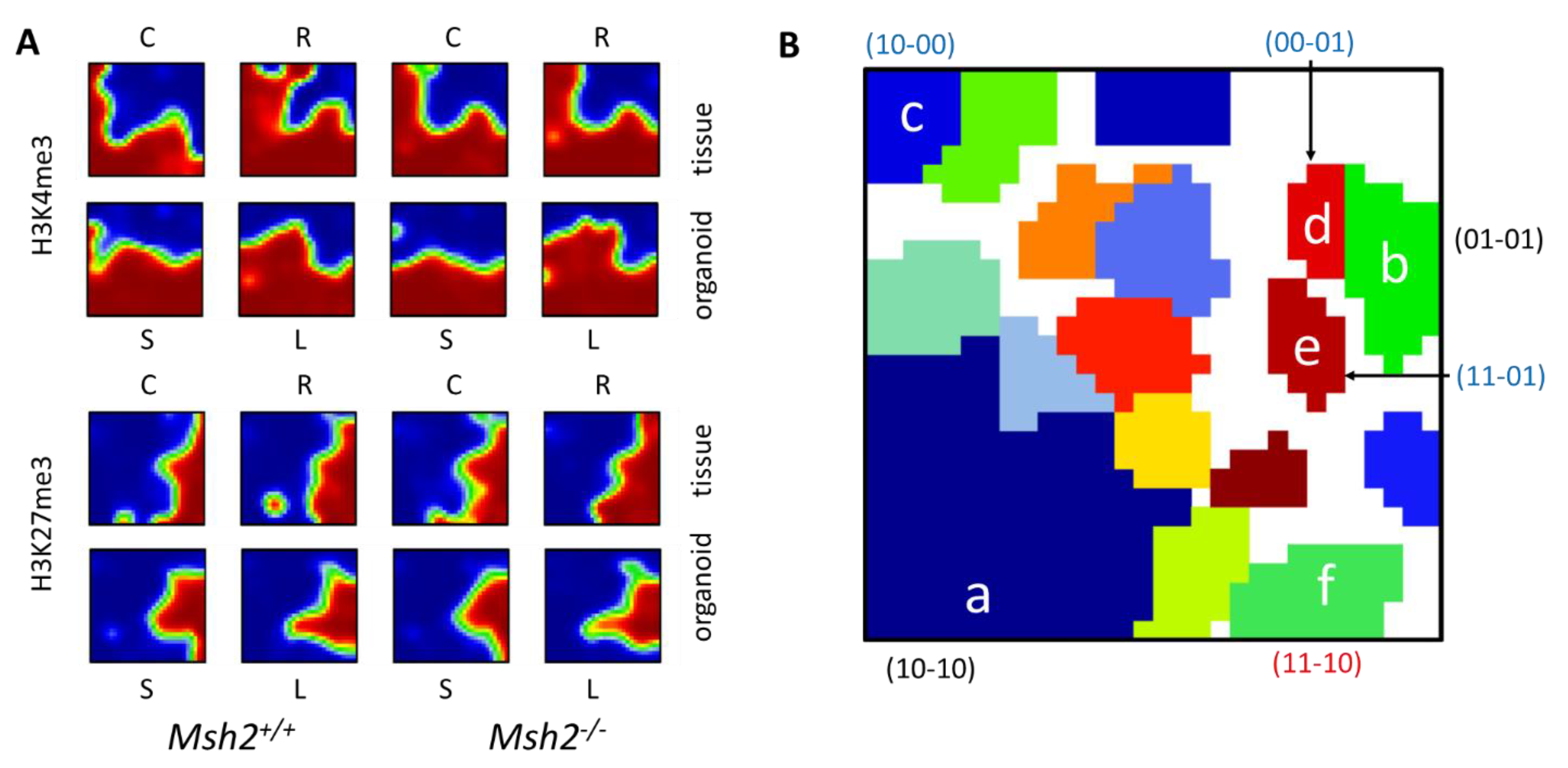
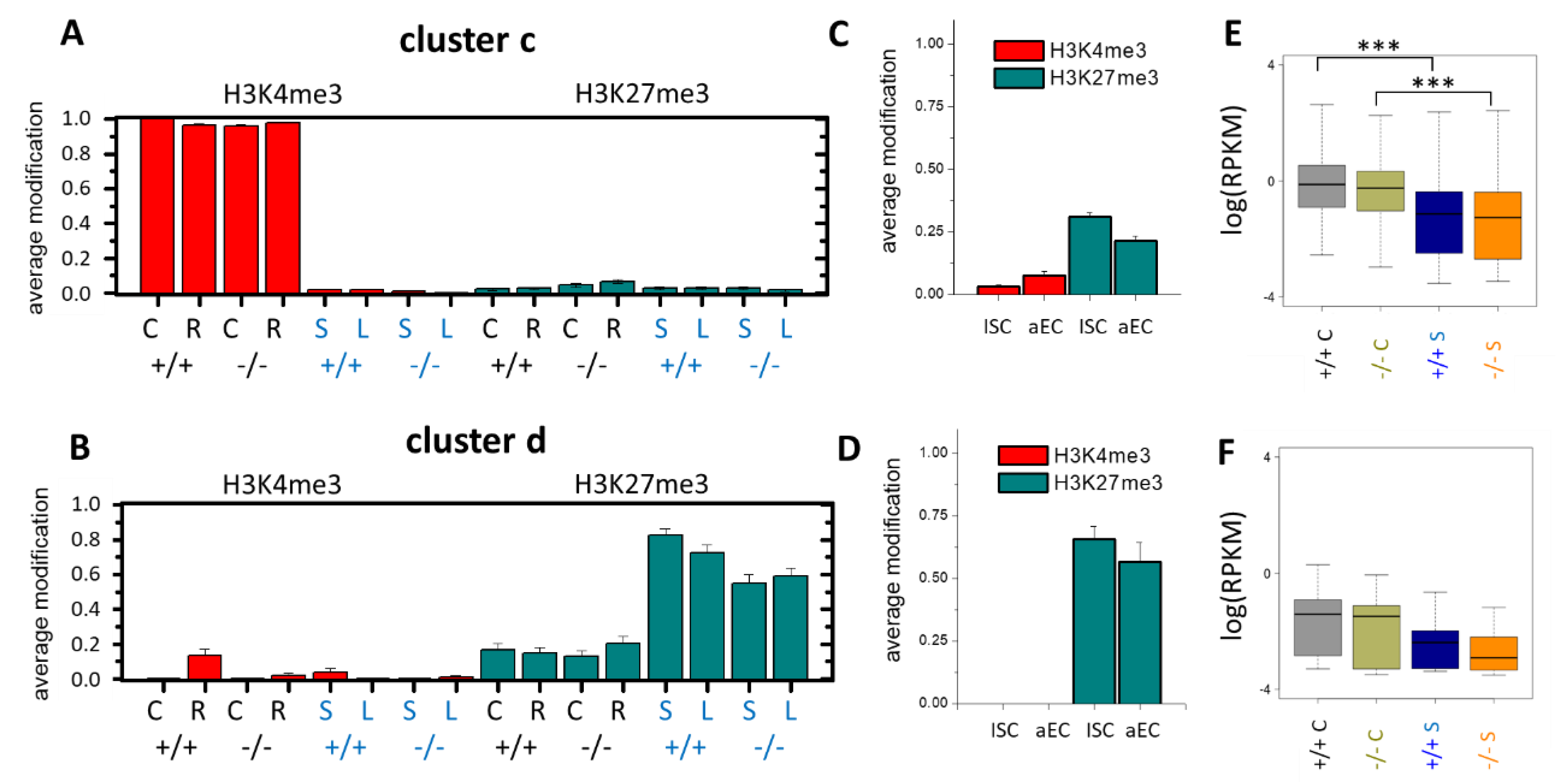
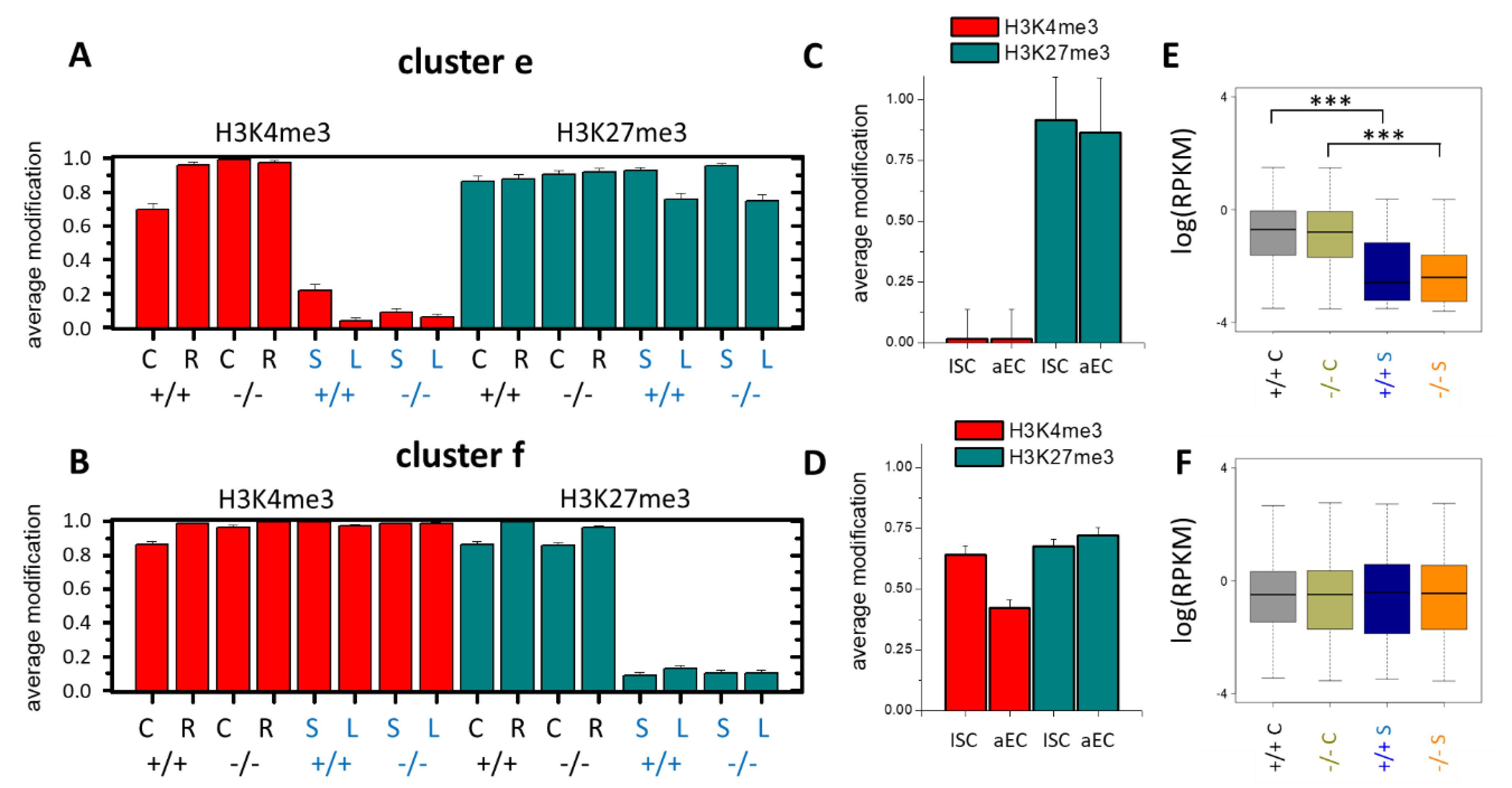
3.1. Histone Modification Profiles in Organoids
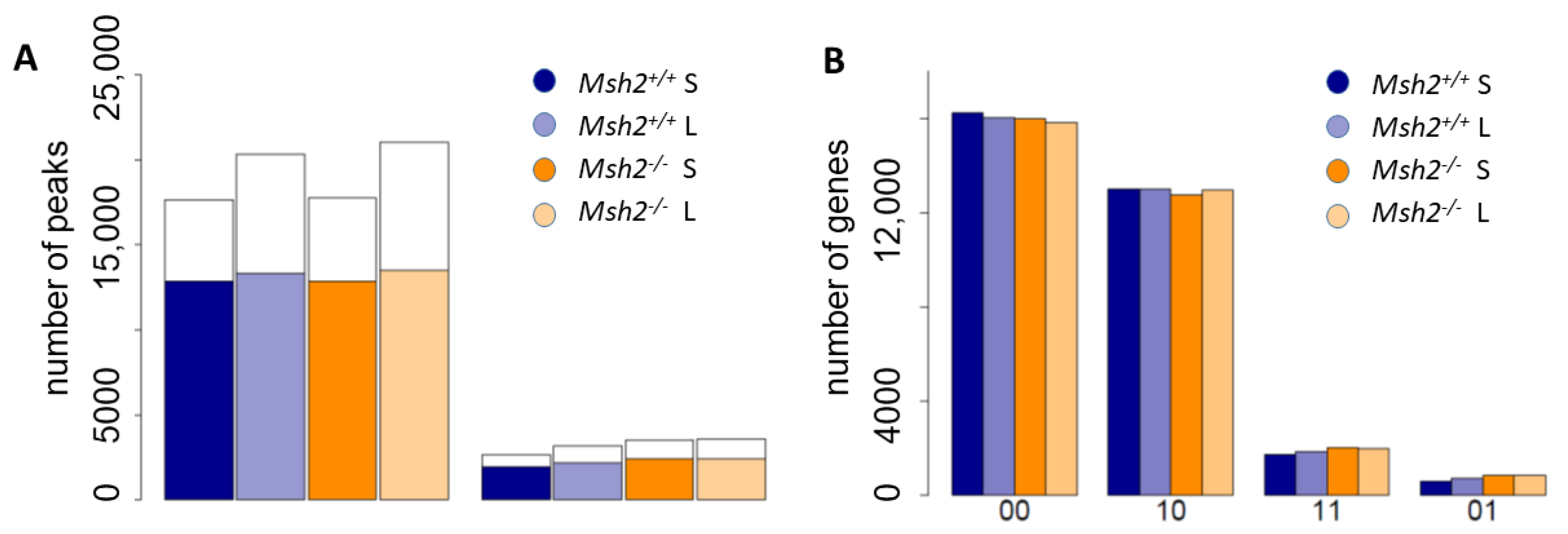
3.2. Epigenetic Profiles of Tissue and Organoids Overlap Considerably
3.3. Differences between the Intestinal Tissue and Organoid Culture
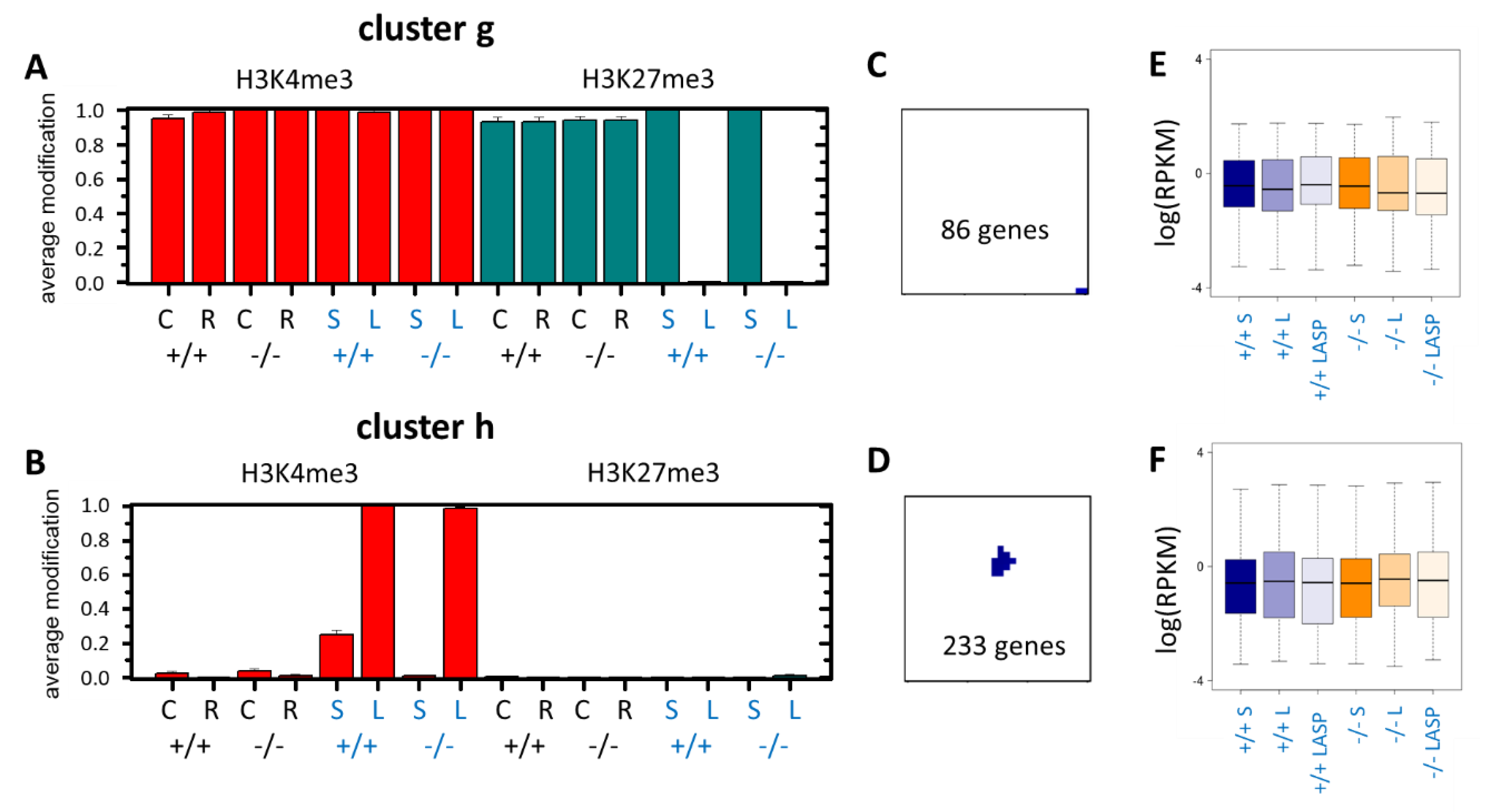
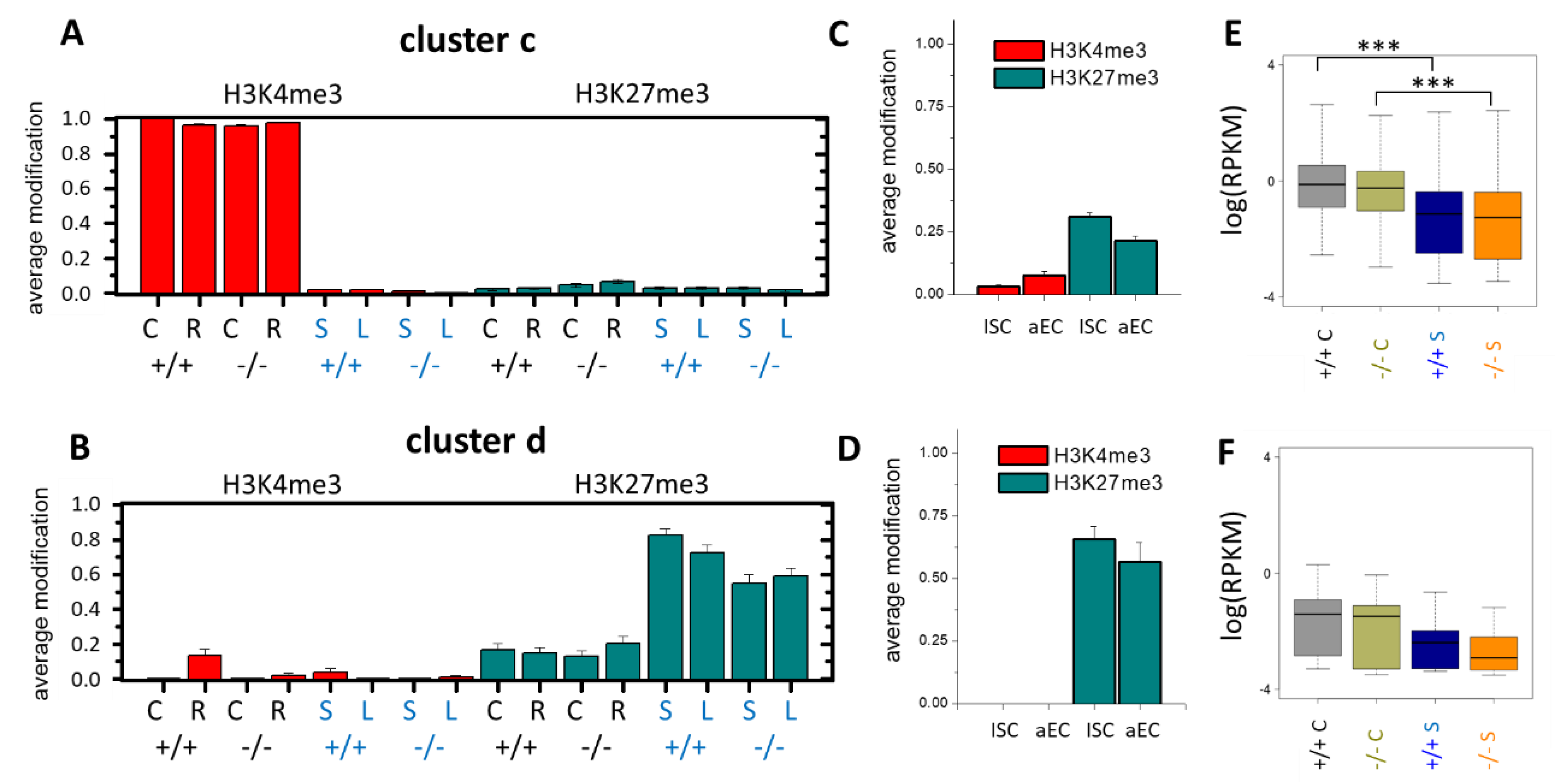
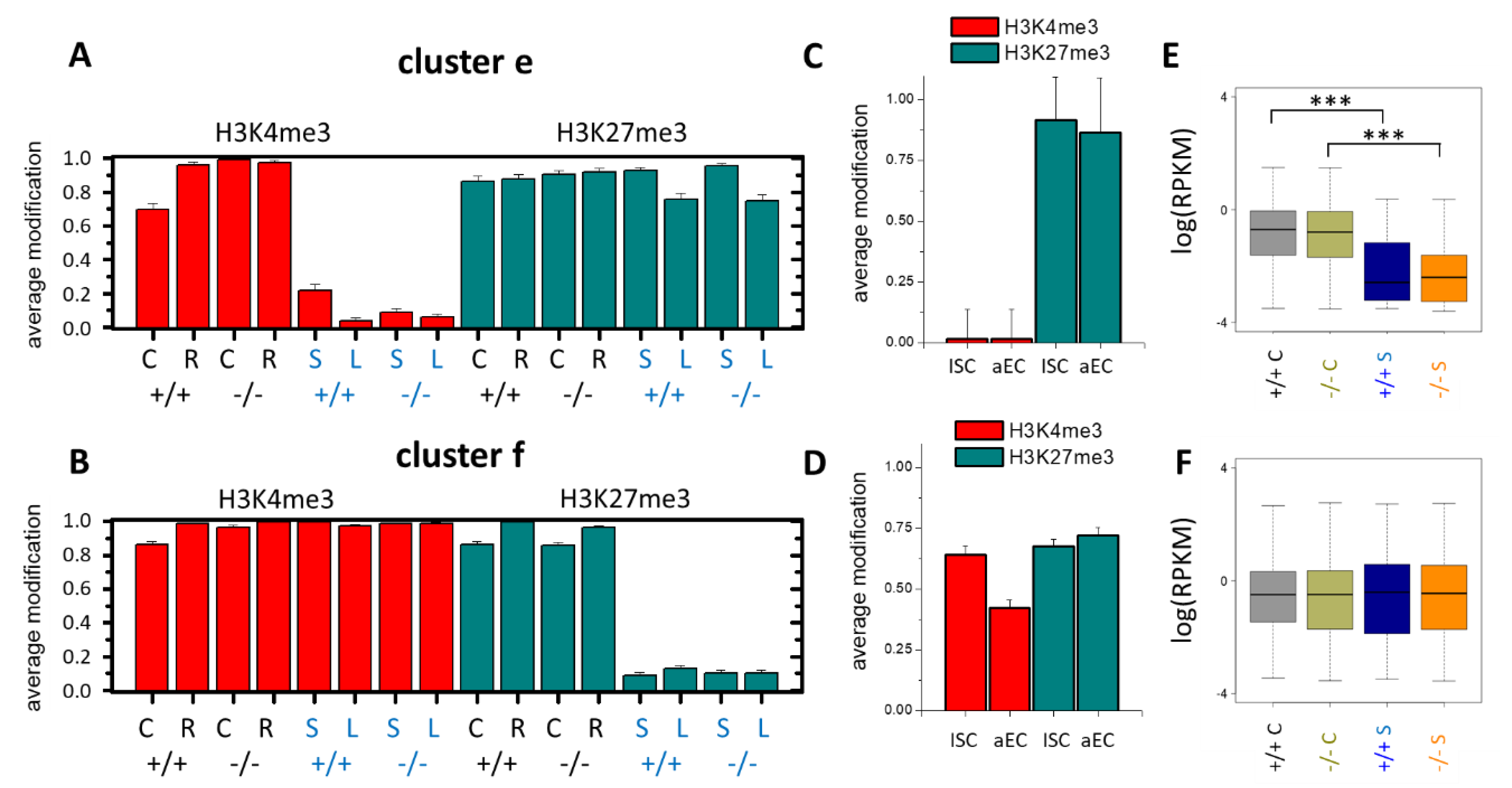
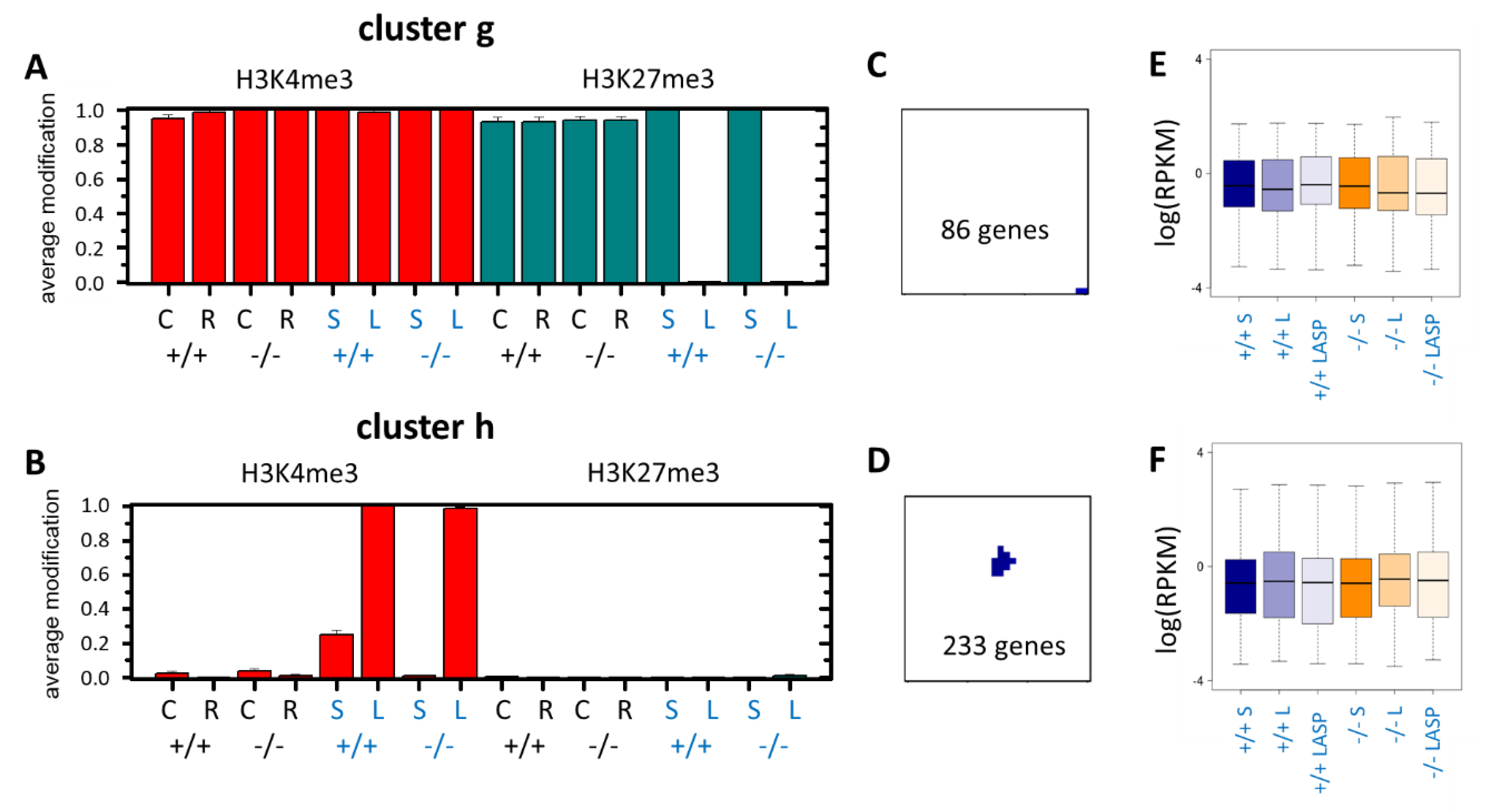
3.4. Epigenetic Activation during the Long-Term Organoid Culture
3.5. Long-Term Organoid Culture-Related Changes in the Transcription
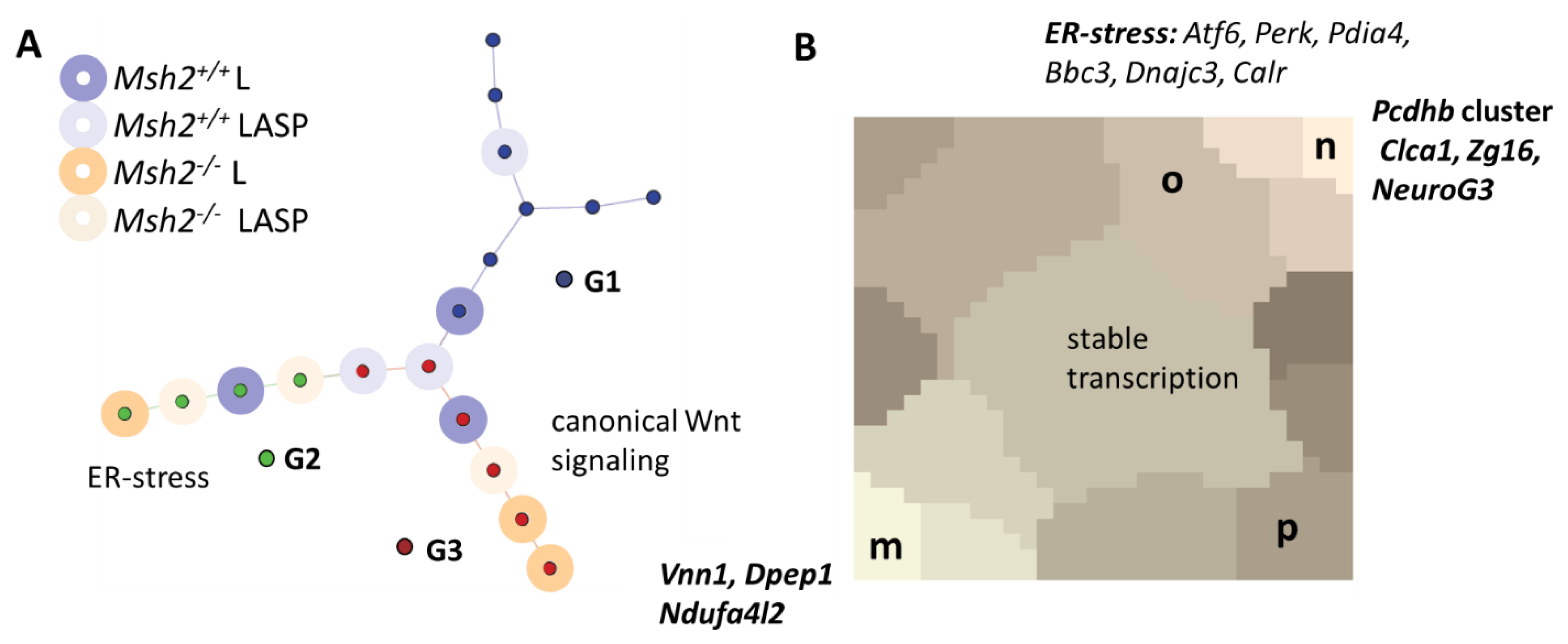
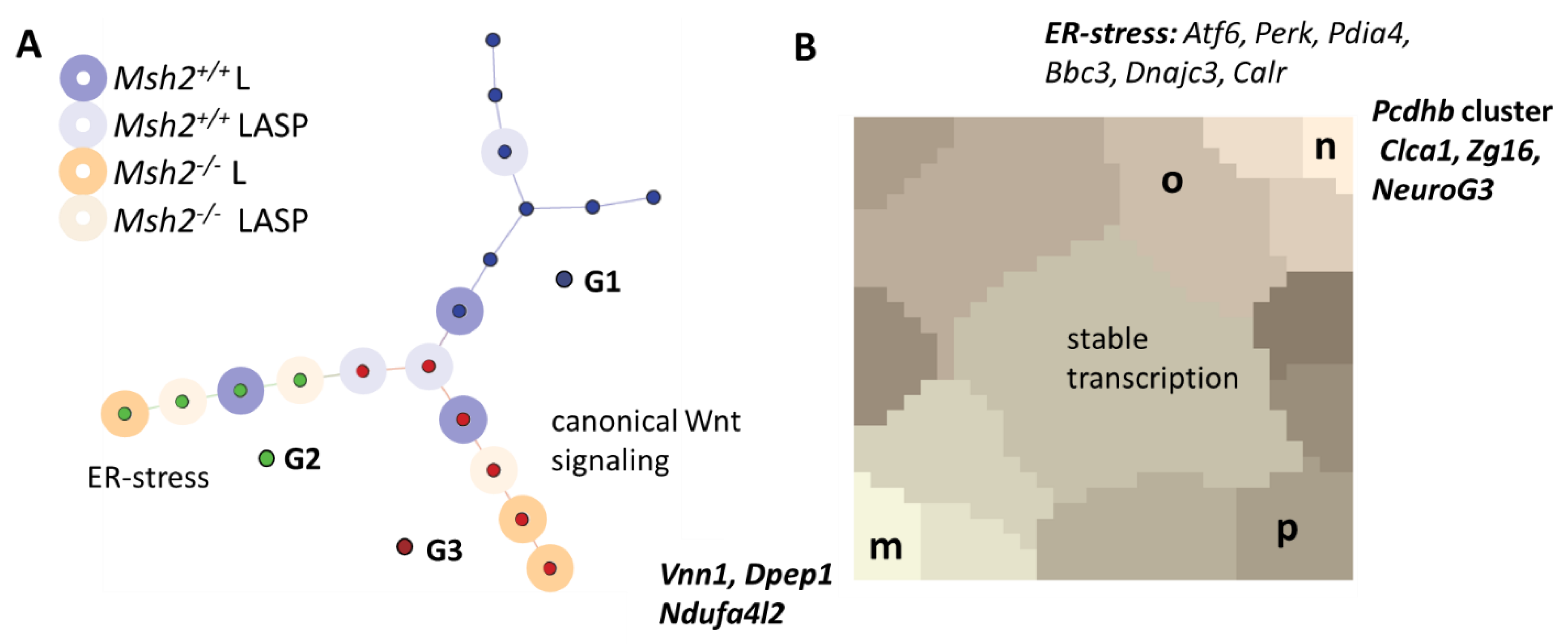
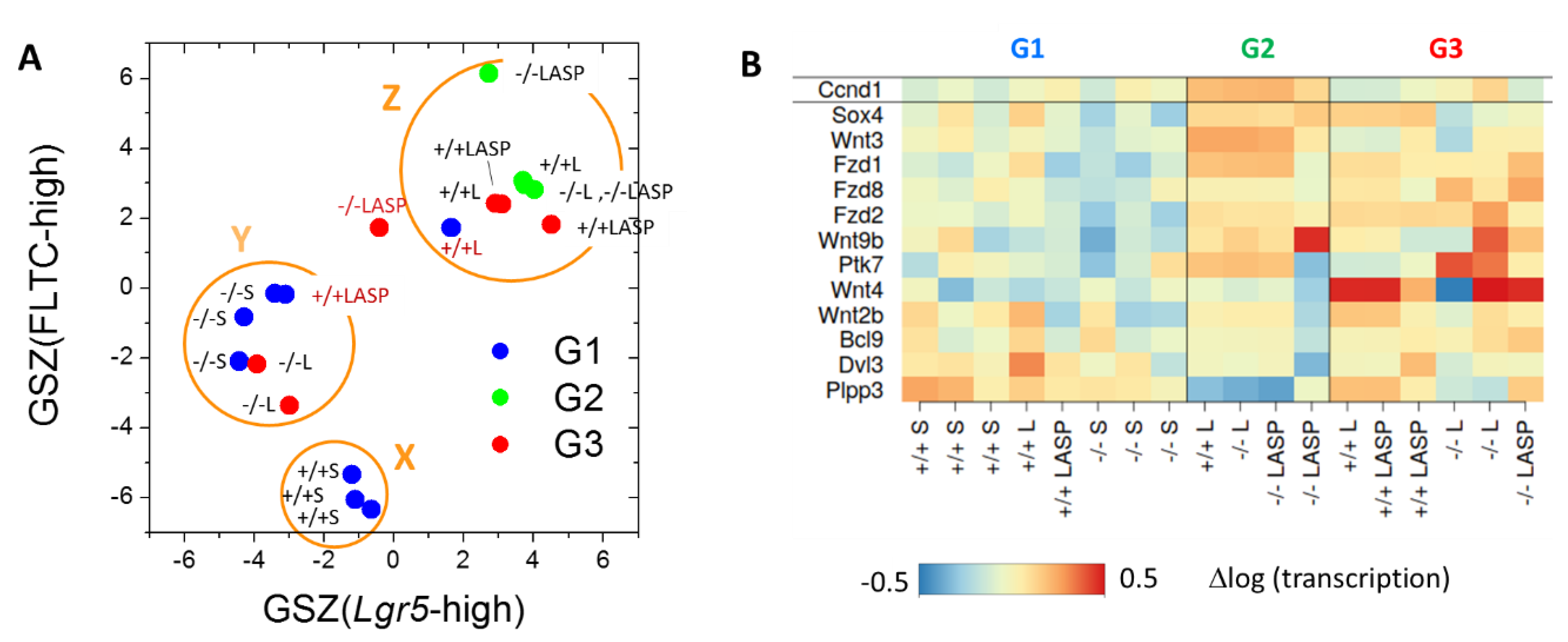
3.6. Maturation and Metabolic Adaptation in Long-Term Organoid Culture
3.7. Epigenetically Activated Genes Respond to Global Transcription
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keysselt, K.; Kreutzmann, T.; Rother, K.; Kerner, C.; Krohn, K.; Przybilla, J.; Buske, P.; Löffler-Wirth, H.; Loeffler, M.; Galle, J.; et al. Different in vivo and in vitro transformation of intestinal stem cells in mismatch repair deficiency. Oncogene 2017, 36, 2750–2761. [Google Scholar] [CrossRef] [PubMed]
- Nalapareddy, K.; Nattamai, K.J.; Kumar, R.S.; Karns, R.; Wikenheiser-Brokamp, K.A.; Sampson, L.L.; Mahe, M.M.; Sundaram, N.; Yacyshyn, M.B.; Yacyshyn, B.; et al. Canonical Wnt signaling ameliorates aging of intestinal stem cells. Cell Rep. 2017, 18, 2608–2621. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.; Saito, Y.; Nogami, K.; Kajiyama, Y.; Suzuki, Y.; Kawase, Y.; Nakaoka, T.; Muramatsu, T.; Kimura, M.; Saito, H. Epigenetic silencing of Lgr5 induces senescence of intestinal epithelial organoids during the process of aging. NPJ Aging Mech. Dis. 2018, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; Clevers, H. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Kang, B.; Petkovich, D.A.; Bhandari, Y.R.; In, J.; Stein-O’Brien, G.; Kong, X.; Xie, W.; Zachos, N.; Maegawa, S.; et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and BrafV600E-Induced Tumorigenesis. Cancer Cell 2019, 35, 315–328.e6. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Farin, H.F.; van Es, J.H.; Clevers, H.; Langer, R.; Karp, J.M. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods 2014, 11, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Thalheim, T.; Quaas, M.; Herberg, M.; Braumann, U.D.; Kerner, C.; Loeffler, M.; Aust, G.; Galle, J. Linking stem cell function and growth pattern of intestinal organoids. Dev. Biol. 2018, 433, 254–261. [Google Scholar] [CrossRef]
- Hahn, M.A.; Li, A.X.; Wu, X.; Yang, R.; Drew, D.A.; Rosenberg, D.W.; Pfeifer, G.P. Loss of the polycomb mark from bivalent promoters leads to activation of cancer-promoting genes in colorectal tumors. Cancer Res. 2014, 74, 3617–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalheim, T.; Herberg, M.; Loeffler, M.; Galle, J. The Regulatory Capacity of Bivalent Genes-A Theoretical Approach. Int. J. Mol. Sci. 2017, 18, 1069. [Google Scholar] [CrossRef] [Green Version]
- Herberg, M.; Siebert, S.; Quaas, M.; Thalheim, T.; Rother, K.; Hussong, M.; Altmüller, J.; Kerner, C.; Galle, J.; Schweiger, M.R.; et al. Loss of Msh2 and a single-radiation hit induce common, genome-wide, and persistent epigenetic changes in the intestine. Clin. Epigenetics 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Thalheim, T.; Hopp, L.; Herberg, M.; Siebert, S.; Kerner, C.; Quaas, M.; Schweiger, M.R.; Aust, G.; Galle, J. Fighting Against Promoter DNA Hyper-Methylation: Protective Histone Modification Profiles of Stress-Resistant Intestinal Stem Cells. Int. J. Mol. Sci. 2020, 21, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucherlapati, M.H.; Lee, K.; Nguyen, A.A.; Clark, A.B.; Hou, H., Jr.; Rosulek, A.; Li, H.; Yang, K.; Fan, K.; Lipkin, M.; et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology 2010, 138, 993–1002.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsch-Haubold, A.G.; Montero, I.; Konrad, K.; Haubold, B. Genome-wide quantitative analysis of histone H3 lysine 4 trimethylation in wild house mouse liver: Environmental change causes epigenetic plasticity. PLoS ONE 2014, 9, e97568. [Google Scholar] [CrossRef]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Bioinform. Action 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2011. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 4 July 2021).
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermuller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Loeffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on Bioconductor. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Flensburg, C.; Kinkel, S.A.; Keniry, A.; Blewitt, M.E.; Oshlack, A. A comparison of control samples for ChIP-seq of histone modifications. Front. Genet. 2014, 5, 329. [Google Scholar] [CrossRef] [Green Version]
- Kazakevych, J.; Sayols, S.; Messner, B.; Krienke, C.; Soshnikova, N. Dynamic changes in chromatin states during specification and differentiation of adult intestinal stem cells. Nucleic Acids Res. 2017, 45, 5770–5784. [Google Scholar] [CrossRef] [PubMed]
- Wirth, H.; von Bergen, M.; Binder, H. Mining SOM expression portraits: Feature selection and integrating concepts of molecular function. BioData Min. 2012, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thalheim, T.; Hopp, L.; Binder, H.; Aust, G.; Galle, J. On the Cooperation between Epigenetics and Transcription Factor Networks in the Specification of Tissue Stem Cells. Epigenomes 2018, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- McIlhatton, M.A.; Tyler, J.; Kerepesi, L.A.; Bocker-Edmonston, T.; Kucherlapati, M.H.; Edelmann, W.; Kucherlapati, R.; Kopelovich, L.; Fishel, R. Aspirin and low-dose nitric oxide-donating aspirin increase life span in a Lynch syndrome mouse model. Cancer Prev. Res. 2011, 4, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, A.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancho, A.; Aerts, T.; Mitsogiannis, M.D.; Seuntjens, E. Protocadherins at the Crossroad of Signaling Pathways. Front. Mol. Neurosci. 2020, 13, 117. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, I.L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Ver Heul, A.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; van den Born, M.; Guryev, V.; Oving, I.; et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Navis, M.; Martins Garcia, T.; Renes, I.B.; Vermeulen, J.L.; Meisner, S.; Wildenberg, M.E.; van den Brink, G.R.; van Elburg, R.M.; Muncan, V. Mouse fetal intestinal organoids: New model to study epithelial maturation from suckling to weaning. EMBO Rep. 2019, 20, e46221. [Google Scholar] [CrossRef]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Dheer, R.; Young, V.B. Stem-cell-derived models: Tools for studying role of microbiota in intestinal homeostasis and disease. Curr. Opin. Gastroenterol. 2021, 37, 15–22. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Okkelman, I.A.; Neto, N.; Papkovsky, D.B.; Monaghan, M.G.; Dmitriev, R.I. A deeper understanding of intestinal organoid metabolism revealed by combining fluorescence lifetime imaging microscopy (FLIM) and extracellular flux analyses. Redox Biol. 2020, 30, 101420. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Live cell imaging of mouse intestinal organoids reveals heterogeneity in their oxygenation. Biomaterials 2017, 146, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, R.; Salvati, A.; Olinga, P.; Boersma, Y.L. Vanin 1: Its Physiological Function and Role in Diseases. Int. J. Mol. Sci. 2019, 20, 3891. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, J.; O’Brien, W.T.; Johnson, R.S.; LaManna, J.C.; Chavez, J.C.; Klein, P.S.; Simon, M.C. O2 regulates stem cells through Wnt/β-catenin signalling. Nat. Cell Biol. 2010, 12, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodríguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018, 14, e8227. [Google Scholar] [CrossRef]
- Jee, J.H.; Lee, D.H.; Ko, J.; Hahn, S.; Jeong, S.Y.; Kim, H.K.; Park, E.; Choi, S.Y.; Jeong, S.; Lee, J.W.; et al. Development of Collagen-Based 3D Matrix for Gastrointestinal Tract-Derived Organoid Culture. Stem Cells Int. 2019, 2019, 8472712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.H.; Shim, S.; Kim, M.J.; Shin, H.Y.; Jang, W.S.; Lee, S.J.; Jin, Y.W.; Lee, S.S.; Lee, S.B.; Park, S. Long-term culture-induced phenotypic difference and efficient cryopreservation of small intestinal organoids by treatment timing of Rho kinase inhibitor. World J. Gastroenterol. 2017, 23, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Buettner, R.; Galle, J.; Loeffler, M. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thalheim, T.; Siebert, S.; Quaas, M.; Herberg, M.; Schweiger, M.R.; Aust, G.; Galle, J. Epigenetic Drifts during Long-Term Intestinal Organoid Culture. Cells 2021, 10, 1718. https://doi.org/10.3390/cells10071718
Thalheim T, Siebert S, Quaas M, Herberg M, Schweiger MR, Aust G, Galle J. Epigenetic Drifts during Long-Term Intestinal Organoid Culture. Cells. 2021; 10(7):1718. https://doi.org/10.3390/cells10071718
Chicago/Turabian StyleThalheim, Torsten, Susann Siebert, Marianne Quaas, Maria Herberg, Michal R. Schweiger, Gabriela Aust, and Joerg Galle. 2021. "Epigenetic Drifts during Long-Term Intestinal Organoid Culture" Cells 10, no. 7: 1718. https://doi.org/10.3390/cells10071718
APA StyleThalheim, T., Siebert, S., Quaas, M., Herberg, M., Schweiger, M. R., Aust, G., & Galle, J. (2021). Epigenetic Drifts during Long-Term Intestinal Organoid Culture. Cells, 10(7), 1718. https://doi.org/10.3390/cells10071718