
RhoA Signaling in Immune Cell Response and Cardiac Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
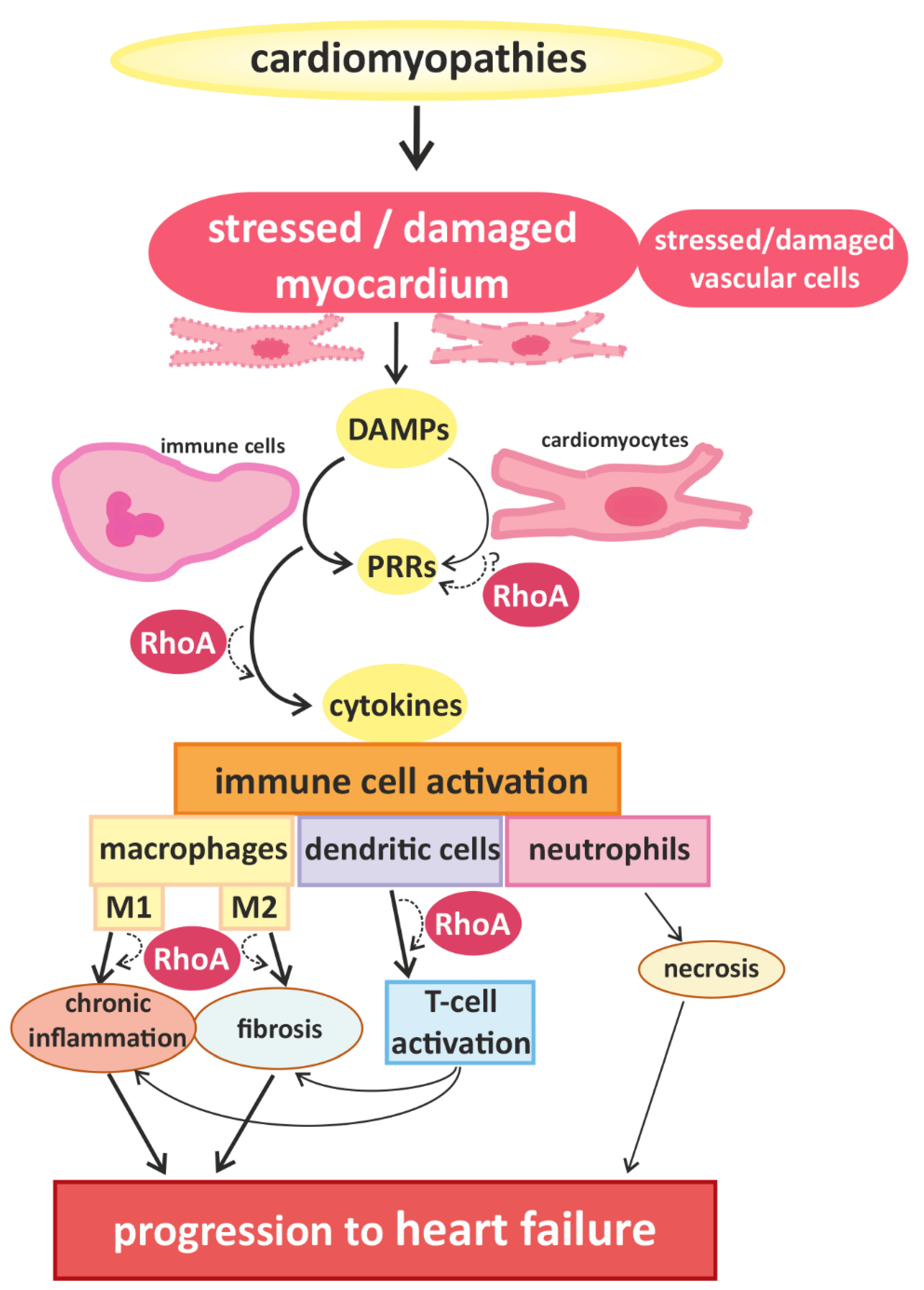
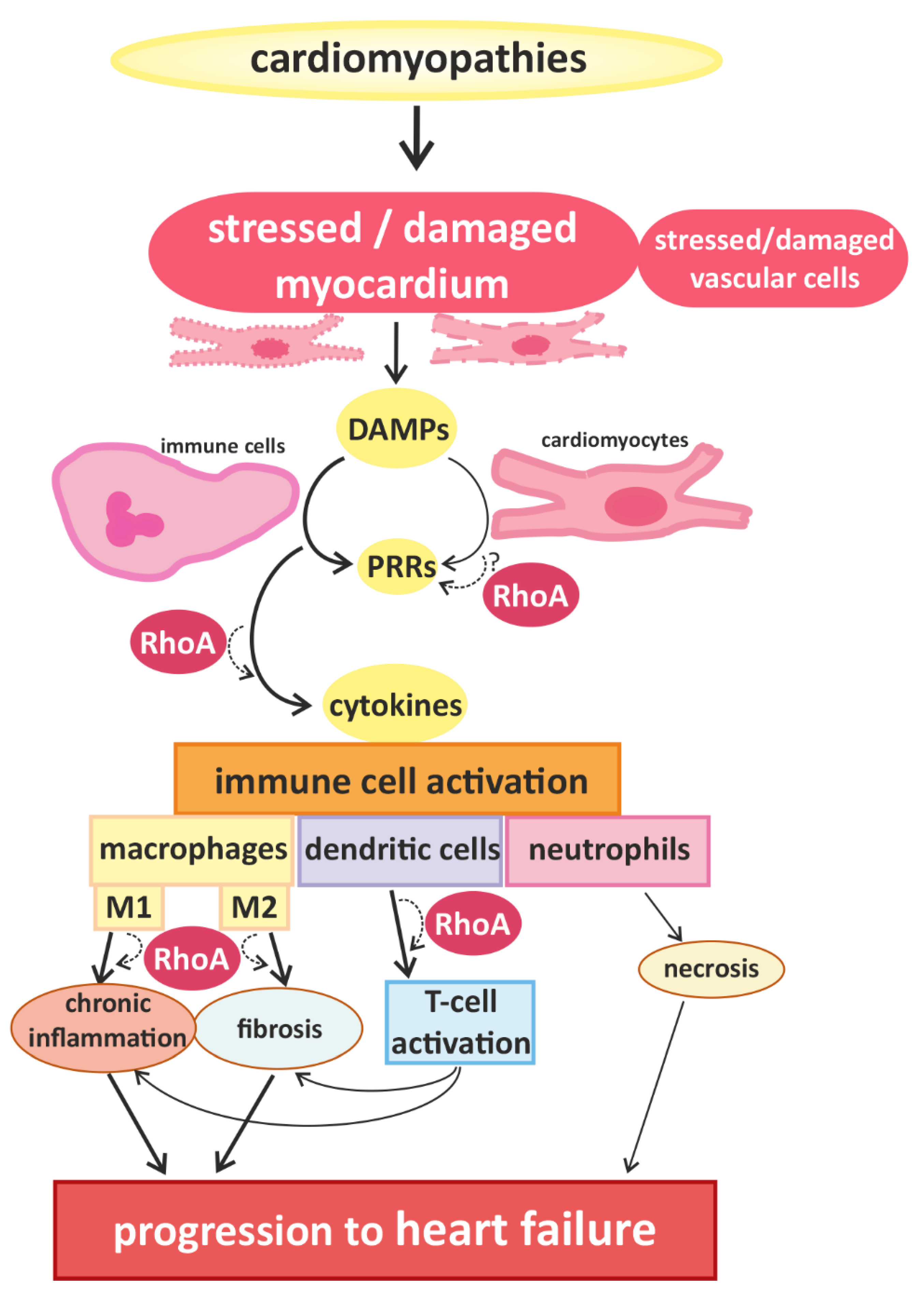
2. Links between Cardiac Hypertrophy, Heart Failure, and Immune Cell Activation
3. RhoA Activation and Signaling in Immune Cells
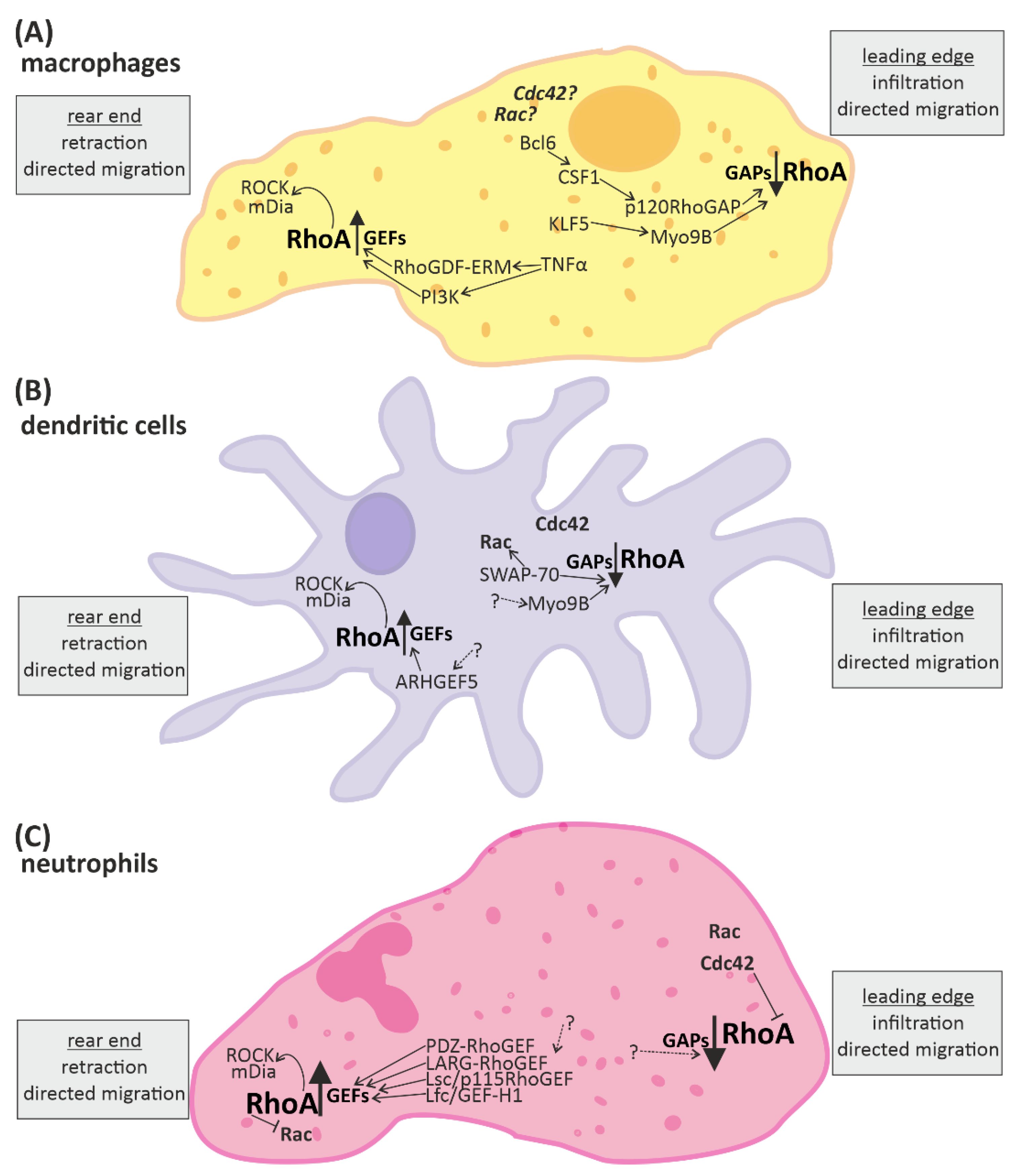
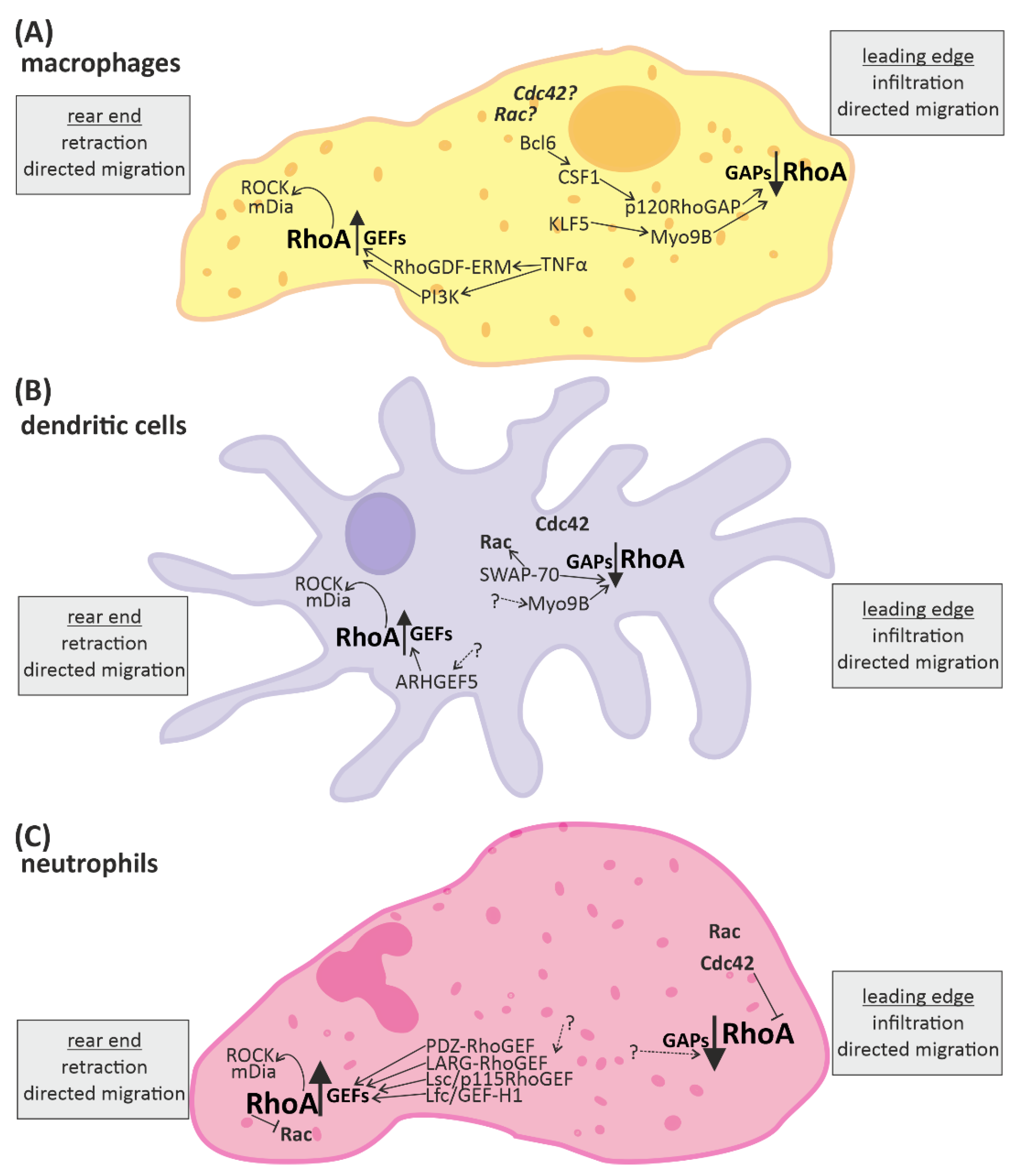
3.1. RhoA Activation in Macrophages, Dendritic Cells, and Granulocytes
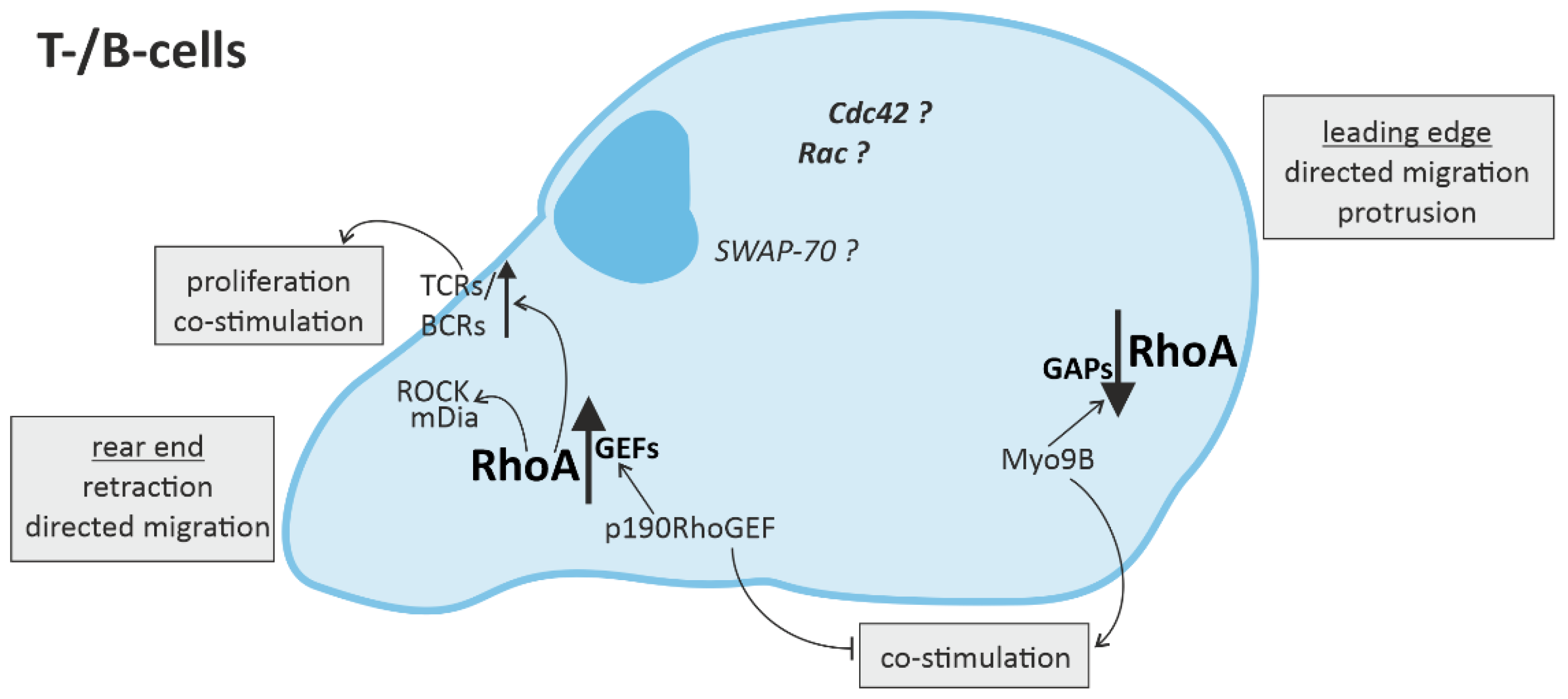
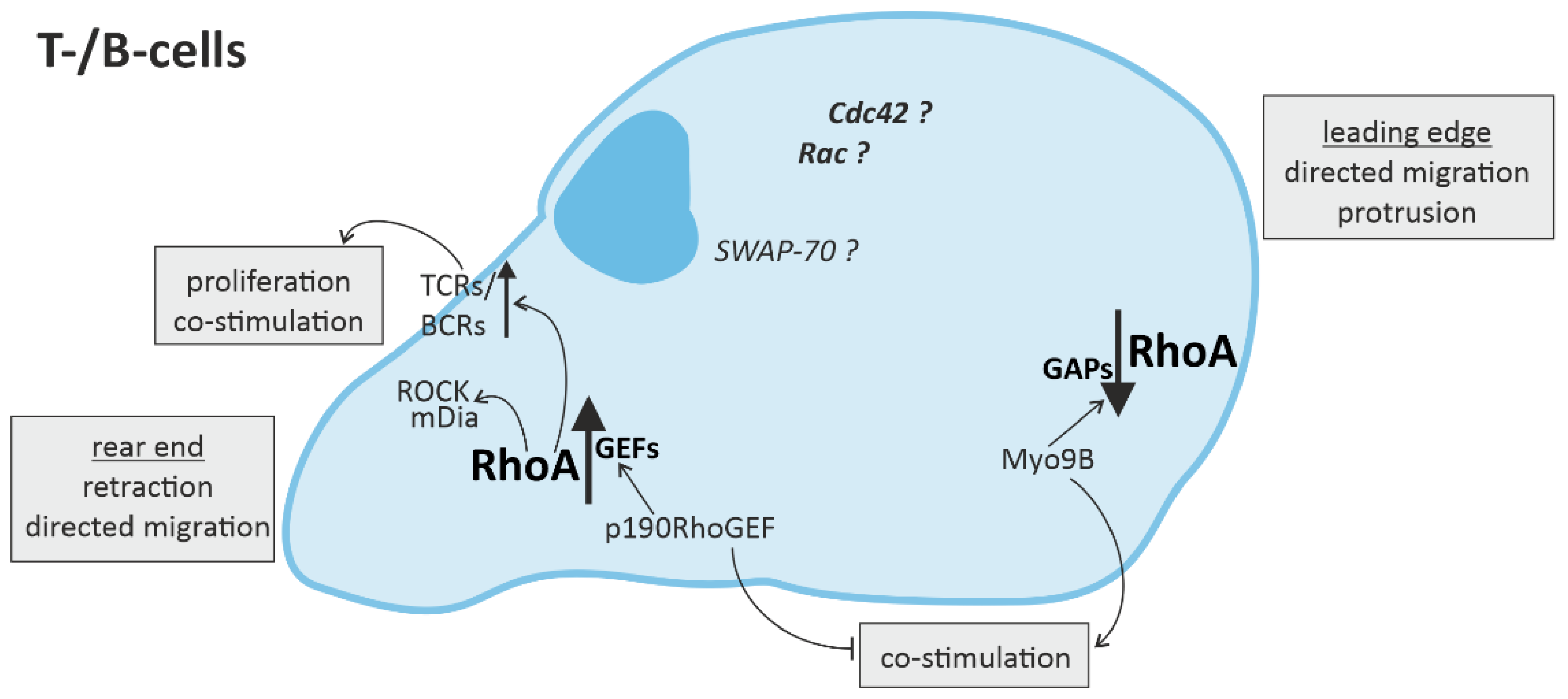
3.2. RhoA Activation in T-Cells and B-Cells
4. Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Elia, E.; Vaduganathan, M.; Gori, M.; Gavazzi, A.; Butler, J.; Senni, M. Role of biomarkers in cardiac structure phenotyping in heart failure with preserved ejection fraction: Critical appraisal and practical use. Eur. J. Heart Fail. 2015, 17, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Thaik, C.M.; Calderone, A.; Takahashi, N.; Colucci, W.S. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J. Clin. Investig. 1995, 96, 1093–1099. [Google Scholar] [CrossRef]
- Yokoyama, T.; Nakano, M.; Bednarczyk, J.L.; McIntyre, B.W.; Entman, M.; Mann, D.L. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation 1997, 95, 1247–1252. [Google Scholar] [CrossRef]
- Purcell, N.H.; Tang, G.; Yu, C.; Mercurio, F.; DiDonato, J.A.; Lin, A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 6668–6673. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef]
- Weisheit, C.; Zhang, Y.; Faron, A.; Kopke, O.; Weisheit, G.; Steinstrasser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS ONE 2014, 9, e112710. [Google Scholar] [CrossRef]
- Liao, X.; Shen, Y.; Zhang, R.; Sugi, K.; Vasudevan, N.T.; Alaiti, M.A.; Sweet, D.R.; Zhou, L.; Qing, Y.; Gerson, S.L.; et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2018, 115, E4661–E4669. [Google Scholar] [CrossRef] [Green Version]
- Kanellakis, P.; Ditiatkovski, M.; Kostolias, G.; Bobik, A. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc. Res. 2012, 95, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Bodin, S.; Welch, M.D. Plasma membrane organization is essential for balancing competing pseudopod- and uropod-promoting signals during neutrophil polarization and migration. Mol. Biol. Cell 2005, 16, 5773–5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilian, L.S.; Voran, J.; Frank, D.; Rangrez, A.Y. RhoA: A dubious molecule in cardiac pathophysiology. J. Biomed. Sci. 2021, 28, 33. [Google Scholar] [CrossRef]
- Frantz, S.; Nahrendorf, M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014, 102, 240–248. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yan, Y.; Jiang, X.; Mai, J.; Chen, N.C.; Wang, H.; Yang, X.F. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 2009, 22, 311–322. [Google Scholar] [CrossRef]
- Lin, L.; Knowlton, A.A. Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 2014, 100, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Rangrez, A.Y.; Eden, M.; Poyanmehr, R.; Kuhn, C.; Stiebeling, K.; Dierck, F.; Bernt, A.; Lullmann-Rauch, R.; Weiler, H.; Kirchof, P.; et al. Myozap Deficiency Promotes Adverse Cardiac Remodeling via Differential Regulation of Mitogen-activated Protein Kinase/Serum-response Factor and beta-Catenin/GSK-3beta Protein Signaling. J. Biol. Chem. 2016, 291, 4128–4143. [Google Scholar] [CrossRef] [Green Version]
- De Vito, P. Atrial natriuretic peptide: An old hormone or a new cytokine? Peptides 2014, 58, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Veinot, J.P.; Davies, R.A.; Haddad, H.; Smith, S.J.; Masters, R.G.; Hendry, P.J.; Starling, R.; de Bold, M.K.; Ponce, A.; et al. Neuroendocrine profiling of humans receiving cardiac allografts. J. Heart Lung Transplant. 2005, 24, 1046–1054. [Google Scholar] [CrossRef]
- Kiemer, A.K.; Weber, N.C.; Furst, R.; Bildner, N.; Kulhanek-Heinze, S.; Vollmar, A.M. Inhibition of p38 MAPK activation via induction of MKP-1: Atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ. Res. 2002, 90, 874–881. [Google Scholar] [CrossRef]
- Mtairag el, M.; Houard, X.; Rais, S.; Pasquier, C.; Oudghiri, M.; Jacob, M.P.; Meilhac, O.; Michel, J.B. Pharmacological potentiation of natriuretic peptide limits polymorphonuclear neutrophil-vascular cell interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1824–1831. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Spiranec, K.; Rock, K.; Fischer, J.W.; Kammerer, U.; et al. Endothelial Actions of ANP Enhance Myocardial Inflammatory Infiltration in the Early Phase After Acute Infarction. Circ. Res. 2016, 119, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Szobi, A.; Goncalvesova, E.; Varga, Z.V.; Leszek, P.; Kusmierczyk, M.; Hulman, M.; Kyselovic, J.; Ferdinandy, P.; Adameova, A. Analysis of necroptotic proteins in failing human hearts. J. Transl. Med. 2017, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic Res. 2019, 25, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, Y.V.; Minasian, S.M.; Demchenko, E.A.; Galagudza, M.M. Study of cardioprotective effects of necroptosis inhibitors on isolated rat heart subjected to global ischemia-reperfusion. Bull. Exp. Biol. Med. 2013, 155, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Koshinuma, S.; Miyamae, M.; Kaneda, K.; Kotani, J.; Figueredo, V.M. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 2014, 28, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Z.H.; Xu, Z.X.; Zhou, X.Y.; Zhang, X.; Shang, L. Implications of Necroptosis for Cardiovascular Diseases. Curr. Med. Sci. 2019, 39, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Miao, K.; Zhou, L.; Ba, H.; Li, C.; Gu, H.; Yin, B.; Wang, J.; Yang, X.P.; Li, Z.; Wang, D.W. Transmembrane tumor necrosis factor alpha attenuates pressure-overload cardiac hypertrophy via tumor necrosis factor receptor 2. PLoS Biol. 2020, 18, e3000967. [Google Scholar] [CrossRef]
- Sharifi, M.; Nazarinia, D.; Ramezani, F.; Azizi, Y.; Naderi, N.; Aboutaleb, N. Necroptosis and RhoA/ROCK pathways: Molecular targets of Nesfatin-1 in cardioprotection against myocardial ischemia/reperfusion injury in a rat model. Mol. Biol. Rep. 2021, 48, 2507–2518. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Sansonetti, M.; Waleczek, F.J.G.; Jung, M.; Thum, T.; Perbellini, F. Resident cardiac macrophages: Crucial modulators of cardiac (patho)physiology. Basic Res. Cardiol. 2020, 115, 77. [Google Scholar] [CrossRef]
- Liu, Y.; Minze, L.J.; Mumma, L.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Mouse macrophage polarity and ROCK1 activity depend on RhoA and non-apoptotic Caspase 3. Exp. Cell Res. 2016, 341, 225–236. [Google Scholar] [CrossRef]
- Hazebroek, M.R.; Moors, S.; Dennert, R.; van den Wijngaard, A.; Krapels, I.; Hoos, M.; Verdonschot, J.; Merken, J.J.; de Vries, B.; Wolffs, P.F.; et al. Prognostic Relevance of Gene-Environment Interactions in Patients with Dilated Cardiomyopathy: Applying the MOGE(S) Classification. J. Am. Coll. Cardiol. 2015, 66, 1313–1323. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.L.t.; Ismahil, M.A.; Jegga, A.G.; Zilliox, M.J.; Troidl, C.; Prabhu, S.D.; Sadayappan, S. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J. Mol. Cell. Cardiol. 2017, 102, 83–93. [Google Scholar] [CrossRef]
- Felix, S.B.; Beug, D.; Dorr, M. Immunoadsorption therapy in dilated cardiomyopathy. Expert Rev. Cardiovasc. Ther. 2015, 13, 145–152. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Krishnamurthy, P.; Barefield, D.; Singh, N.; Gupta, R.; Lambers, E.; Thal, M.; Mackie, A.; Hoxha, E.; Ramirez, V.; et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation 2012, 126, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, R.M. Immune cell modulation of cardiac remodeling. Circulation 2012, 125, 1597–1600. [Google Scholar] [CrossRef] [Green Version]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [Green Version]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velazquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circulation Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Fine, N.; Dimitriou, I.D.; Rottapel, R. Go with the flow: GEF-H1 mediated shear stress mechanotransduction in neutrophils. Small GTPases 2020, 11, 23–31. [Google Scholar] [CrossRef]
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Ormbostad Berre, A.M.; Stolen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680. [Google Scholar] [CrossRef]
- Hind, L.E.; Vincent, W.J.B.; Huttenlocher, A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev. Cell 2016, 38, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, Y.; Kaibuchi, K.; Hori, Y.; Fujioka, H.; Araki, S.; Ueda, T.; Kikuchi, A.; Takai, Y. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 1990, 5, 1321–1328. [Google Scholar]
- Shi, Y.Q.; Zhang, J.Y.; Mullin, M.; Dong, B.X.; Alberts, A.S.; Siminovitch, K.A. The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. J. Immunol. 2009, 182, 3837–3845. [Google Scholar] [CrossRef]
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology 2018, 223, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.A.; Shen, X.; Young, J.B.; Kaul, P.; Lerner, D.J. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood 2006, 107, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Del Re, D.P.; Xiang, S.Y.; Zhao, X.; Florholmen, G.; Brown, J.H. Revisited and revised: Is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 2010, 3, 330–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulinilkunnil, T.; An, D.; Ghosh, S.; Qi, D.; Kewalramani, G.; Yuen, G.; Virk, N.; Abrahani, A.; Rodrigues, B. Lysophosphatidic acid-mediated augmentation of cardiomyocyte lipoprotein lipase involves actin cytoskeleton reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2802–H2810. [Google Scholar] [CrossRef] [PubMed]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhao, Y.; Daaka, Y.; Nie, Z. Acute activation of beta2-adrenergic receptor regulates focal adhesions through betaArrestin2- and p115RhoGEF protein-mediated activation of RhoA. J. Biol. Chem. 2012, 287, 18925–18936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuona, E.; Ridge, K.; Pesce, L.; Batlle, D.; Sznajder, J.I. The GTP-binding protein RhoA mediates Na,K-ATPase exocytosis in alveolar epithelial cells. Mol. Biol. Cell 2003, 14, 3888–3897. [Google Scholar] [CrossRef] [Green Version]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac beta-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N. Engl. J. Med. 1984, 311, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Wakasugi, T.; Nakao, M.; Minamino, T. Catecholamine-Induced Senescence of Endothelial Cells and Bone Marrow Cells Promotes Cardiac Dysfunction in Mice. Int. Heart J. 2018, 59, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnefont-Rousselot, D.; Mahmoudi, A.; Mougenot, N.; Varoquaux, O.; Le Nahour, G.; Fouret, P.; Lechat, P. Catecholamine effects on cardiac remodelling, oxidative stress and fibrosis in experimental heart failure. Redox Rep. 2002, 7, 145–151. [Google Scholar] [CrossRef]
- Myagmar, B.E.; Flynn, J.M.; Cowley, P.M.; Swigart, P.M.; Montgomery, M.D.; Thai, K.; Nair, D.; Gupta, R.; Deng, D.X.; Hosoda, C.; et al. Adrenergic Receptors in Individual Ventricular Myocytes: The Beta-1 and Alpha-1B Are in All Cells, the Alpha-1A Is in a Subpopulation, and the Beta-2 and Beta-3 Are Mostly Absent. Circ. Res. 2017, 120, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Pixley, F.J.; Xiong, Y.; Yu, R.Y.; Sahai, E.A.; Stanley, E.R.; Ye, B.H. BCL6 suppresses RhoA activity to alter macrophage morphology and motility. J. Cell Sci. 2005, 118, 1873–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, P.J.; Xu, Y.; Kronlage, M.; Grobe, K.; Schon, P.; Song, J.; Sorokin, L.; Schwab, A.; Bahler, M. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc. Natl. Acad. Sci. USA 2010, 107, 12145–12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.D.; Das, R.; Goduni, L.; McClellan, S.; Hazlett, L.D.; Mahabeleshwar, G.H. Kruppel-like Factor 6 Promotes Macrophage-mediated Inflammation by Suppressing B Cell Leukemia/Lymphoma 6 Expression. J. Biol. Chem. 2016, 291, 21271–21282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Sasaki, T.; Mammoto, A.; Takaishi, K.; Kameyama, T.; Tsukita, S.; Takai, Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 1997, 272, 23371–23375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncone, E.; Marafini, I.; Stolfi, C.; Monteleone, G. Transforming Growth Factor-beta1/Smad7 in Intestinal Immunity, Inflammation, and Cancer. Front. Immunol. 2018, 9, 1407. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.G.; Moon, M.Y.; Jeon, C.Y.; Won, H.Y.; Kim, H.J.; Jeon, Y.J.; Seo, J.Y.; Kim, J.I.; Kim, J.; et al. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood 2006, 108, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Vargas, P.; Maiuri, P.; Bretou, M.; Saez, P.J.; Pierobon, P.; Maurin, M.; Chabaud, M.; Lankar, D.; Obino, D.; Terriac, E.; et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol. 2016, 18, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Kumamoto, Y.; Wang, P.; Gan, X.; Lehmann, D.; Smrcka, A.V.; Cohn, L.; Iwasaki, A.; Li, L.; Wu, D. Regulation of immature dendritic cell migration by RhoA guanine nucleotide exchange factor Arhgef5. J. Biol. Chem. 2009, 284, 28599–28606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Pektor, S.; Balkow, S.; Hemkemeyer, S.A.; Liu, Z.; Grobe, K.; Hanley, P.J.; Shen, L.; Bros, M.; Schmidt, T.; et al. Dendritic cell motility and T cell activation requires regulation of Rho-cofilin signaling by the Rho-GTPase activating protein myosin IXb. J. Immunol. 2014, 192, 3559–3568. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, M.; Aebischer, D.; Abadier, M.; Haener, S.; Lucic, M.; Vigl, B.; Luche, H.; Fehling, H.J.; Biehlmaier, O.; Lyck, R.; et al. Differential requirement for ROCK in dendritic cell migration within lymphatic capillaries in steady-state and inflammation. Blood 2012, 120, 2249–2258. [Google Scholar] [CrossRef] [Green Version]
- Chacon-Martinez, C.A.; Kiessling, N.; Winterhoff, M.; Faix, J.; Muller-Reichert, T.; Jessberger, R. The switch-associated protein 70 (SWAP-70) bundles actin filaments and contributes to the regulation of F-actin dynamics. J. Biol. Chem. 2013, 288, 28687–28703. [Google Scholar] [CrossRef] [Green Version]
- Seul, H.J.; Ahn, Y.R.; Song, H.M.; Ha, Y.J.; Lee, J.R. Over-expression of a RhoA-specific guanine nucleotide exchange factor, p190RhoGEF, in mouse dendritic cells negatively regulates cellular responses to bacterial lipopolysaccharide. Mol. Cells 2012, 34, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Worthylake, R.A.; Lemoine, S.; Watson, J.M.; Burridge, K. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 2001, 154, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannat, R.A.; Dembo, M.; Hammer, D.A. Traction Forces of Neutrophils Migrating on Compliant Substrates. Biophys J. 2011, 101, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Van Keymeulen, A.; Bourne, H.R. PDZRhoGEF and myosin II localize RhoA activity to the back of polarizing neutrophil-like cells. J. Cell Biol. 2007, 179, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Kitzing, T.; Sahadevan, A.; Brandt, D.T.; Knieing, H.; Hannemann, S.; Fackler, O.T.; Grosse, R. Positive feedback between Dia1, larg and Rhoa is required for cancer cell invasion. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 375, 35. [Google Scholar]
- Xu, J.S.; Wang, F.; Van Keymeulen, A.; Herzmark, P.; Straight, A.; Kelly, K.; Takuwa, Y.; Sugimoto, N.; Mitchison, T.; Bourne, H.R. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 2003, 114, 201–214. [Google Scholar] [CrossRef]
- Wong, K.; Pertz, O.; Hahn, K.; Bourne, H. Neutrophil polarization: Spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 3639–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Collins, S.R.; Meyer, T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 2016, 18, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Ricker, E.; Chowdhury, L.; Yi, W.; Pernis, A.B. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Pollock, J.K.; Verma, N.K.; O’Boyle, N.M.; Carr, M.; Meegan, M.J.; Zisterer, D.M. Combretastatin (CA)-4 and its novel analogue CA-432 impair T-cell migration through the Rho/ROCK signalling pathway. Biochem. Pharm. 2014, 92, 544–557. [Google Scholar] [CrossRef]
- Moalli, F.; Ficht, X.; Germann, P.; Vladymyrov, M.; Stolp, B.; de Vries, I.; Lyck, R.; Balmer, J.; Fiocchi, A.; Kreutzfeldt, M.; et al. The Rho regulator Myosin IXb enables nonlymphoid tissue seeding of protective CD8(+) T cells. J. Exp. Med. 2018, 215, 1869–1890. [Google Scholar] [CrossRef]
- Vielkind, S.; Gallagher-Gambarelli, M.; Gomez, M.; Hinton, H.J.; Cantrell, D.A. Integrin regulation by RhoA in thymocytes. J. Immunol. 2005, 175, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Ocana-Morgner, C.; Wahren, C.; Jessberger, R. SWAP-70 regulates RhoA/RhoB-dependent MHCII surface localization in dendritic cells. Blood 2009, 113, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Ocana-Morgner, C.; Gotz, A.; Wahren, C.; Jessberger, R. SWAP-70 restricts spontaneous maturation of dendritic cells. J. Immunol. 2013, 190, 5545–5558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.M.; Konstantinidis, D.G.; Yang, J.Q.; Mizukawa, B.; Kalim, K.; Lang, R.A.; Kalfa, T.A.; Zheng, Y.; Guo, F.K. Gene Targeting RhoA Reveals Its Essential Role in Coordinating Mitochondrial Function and Thymocyte Development. J. Immunol. 2014, 193, 5973–5982. [Google Scholar] [CrossRef] [Green Version]
- Swat, W.; Fujikawa, K. The Vav family: At the crossroads of signaling pathways. Immunol. Res. 2005, 32, 259–265. [Google Scholar] [CrossRef]
- Goodfellow, H.S.; Frushicheva, M.P.; Ji, Q.; Cheng, D.A.; Kadlecek, T.A.; Cantor, A.J.; Kuriyan, J.; Chakraborty, A.K.; Salomon, A.; Weiss, A. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci. Signal. 2015, 8, ra49. [Google Scholar] [CrossRef] [Green Version]
- Helms, W.S.; Jeffrey, J.L.; Holmes, D.A.; Townsend, M.B.; Clipstone, N.A.; Su, L. Modulation of NFAT-dependent gene expression by the RhoA signaling pathway in T cells. J. Leukoc. Biol. 2007, 82, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Saci, A.; Carpenter, C.L. RhoA GTPase regulates B cell receptor signaling. Mol. Cell 2005, 17, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Ha, Y.J.; Kim, H.J. Induced expression of a RhoA-specifiic guanine nucleotide exchange factor, p190RhoGEF, following CD40 stimulation and WEHI 231 B cell activation. J. Immunol. 2003, 170, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Nagy, T.L.; Weiner, O.D. Joining forces: Crosstalk between biochemical signalling and physical forces orchestrates cellular polarity and dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- McCusker, D. Cellular self-organization: Generating order from the abyss. Mol. Biol. Cell 2020, 31, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Kholodenko, B.N.; von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2018, 9, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronne, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, D.; Aghel, N.; Hillis, C.; Siegal, D.; Karampatos, S.; Rangarajan, S.; Pond, G.; Seow, H. Tyrosine kinase inhibitors in chronic myeloid leukaemia and emergent cardiovascular disease. Heart 2021. [Google Scholar] [CrossRef]
- Amin, E.; Dubey, B.N.; Zhang, S.C.; Gremer, L.; Dvorsky, R.; Moll, J.M.; Taha, M.S.; Nagel-Steger, L.; Piekorz, R.P.; Somlyo, A.V.; et al. Rho-kinase: Regulation, (dys)function, and inhibition. Biol. Chem. 2013, 394, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Al-Lamki, R.S.; Brookes, A.P.; Wang, J.; Reid, M.J.; Parameshwar, J.; Goddard, M.J.; Tellides, G.; Wan, T.; Min, W.; Pober, J.S.; et al. TNF receptors differentially signal and are differentially expressed and regulated in the human heart. Am. J. Transplant. 2009, 9, 2679–2696. [Google Scholar] [CrossRef] [Green Version]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Lechat, P.; Packer, M.; Chalon, S.; Cucherat, M.; Arab, T.; Boissel, J.P. Clinical effects of beta-adrenergic blockade in chronic heart failure: A meta-analysis of double-blind, placebo-controlled, randomized trials. Circulation 1998, 98, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Cohn, J.N. The management of chronic heart failure. N. Engl. J. Med. 1996, 335, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.; Fajardo, G.; Zhao, M. The Role of Beta-Adrenergic Receptors in Heart Failure: Differential Regulation of Cardiotoxicity and Cardioprotection. Prog. Pediatr. Cardiol. 2011, 31, 35–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilian, L.S.; Frank, D.; Rangrez, A.Y. RhoA Signaling in Immune Cell Response and Cardiac Disease. Cells 2021, 10, 1681. https://doi.org/10.3390/cells10071681
Kilian LS, Frank D, Rangrez AY. RhoA Signaling in Immune Cell Response and Cardiac Disease. Cells. 2021; 10(7):1681. https://doi.org/10.3390/cells10071681
Chicago/Turabian StyleKilian, Lucia Sophie, Derk Frank, and Ashraf Yusuf Rangrez. 2021. "RhoA Signaling in Immune Cell Response and Cardiac Disease" Cells 10, no. 7: 1681. https://doi.org/10.3390/cells10071681
APA StyleKilian, L. S., Frank, D., & Rangrez, A. Y. (2021). RhoA Signaling in Immune Cell Response and Cardiac Disease. Cells, 10(7), 1681. https://doi.org/10.3390/cells10071681