
Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis

 , ,
, ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Immunofluorescence Immunohistochemistry
2.3. Selection of Markers for Autophagy, Apoptosis, and the Unfolded Protein Response
2.4. Quantification of Immunofluorescence Intensity and Colocalization
2.5. Pulmonary Function Tests
2.6. Statistical Analysis
3. Results
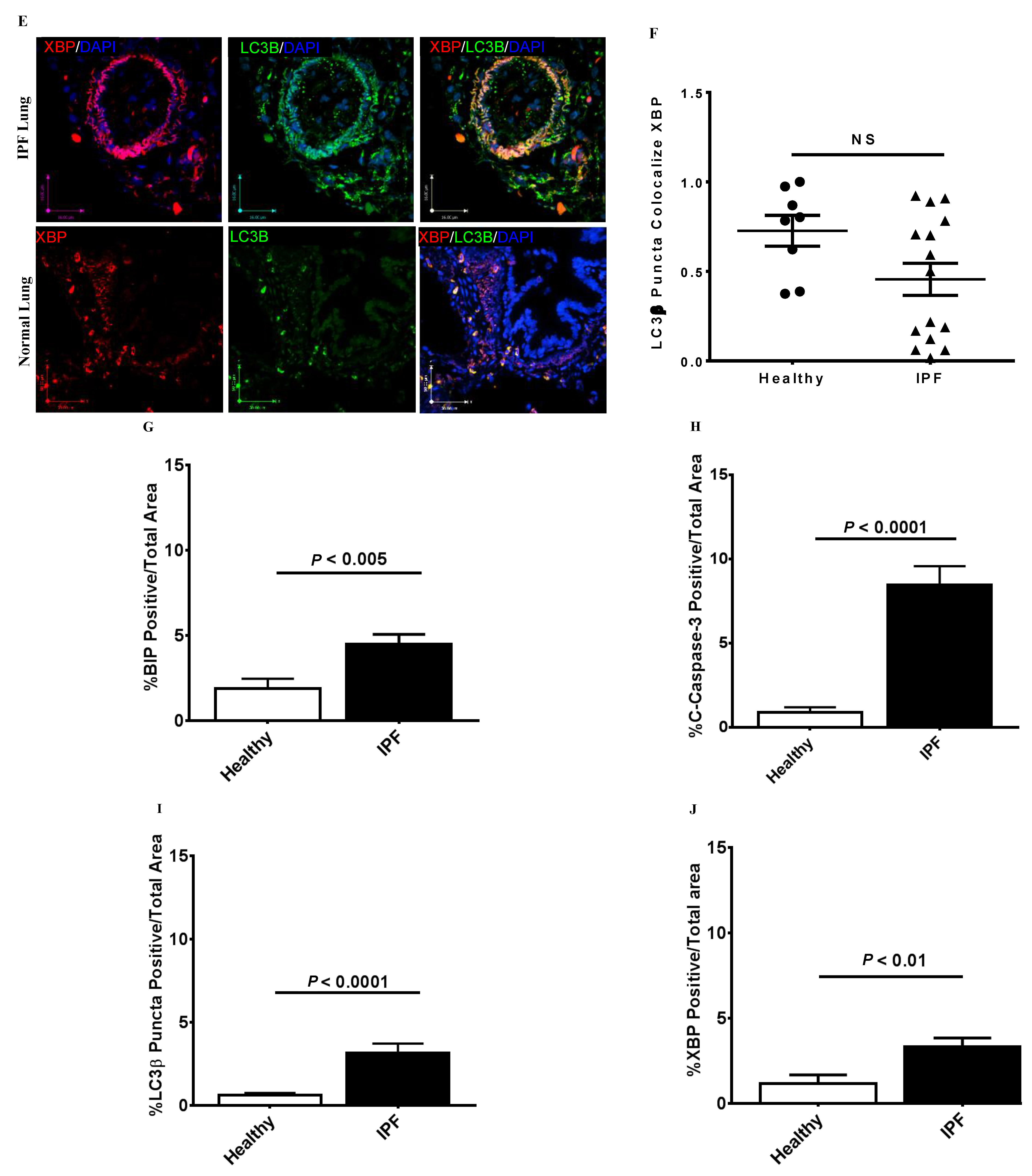
3.1. Co-Localization of Apoptosis, Autophagy, and UPR Markers in IPF Lung Tissues
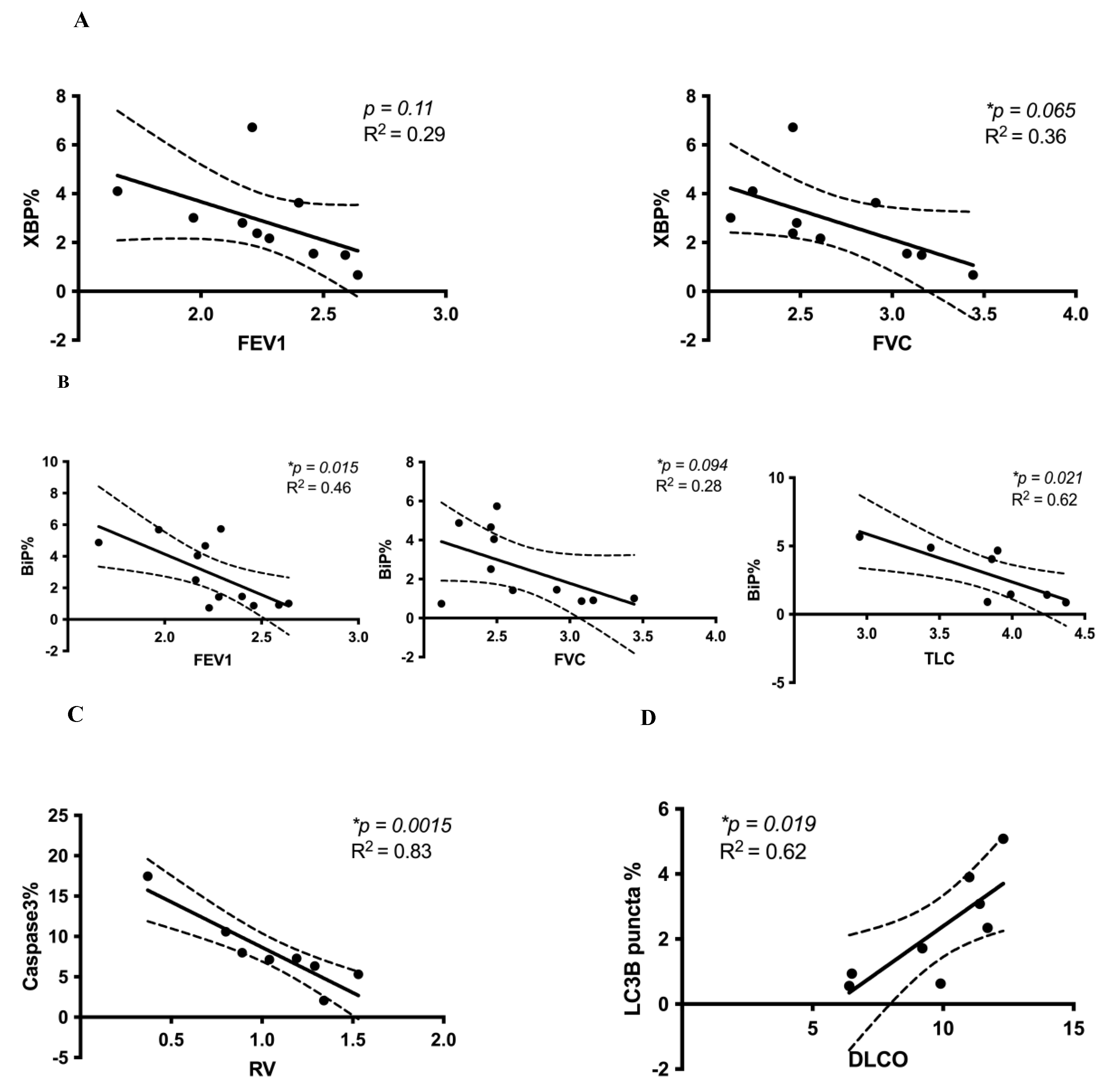
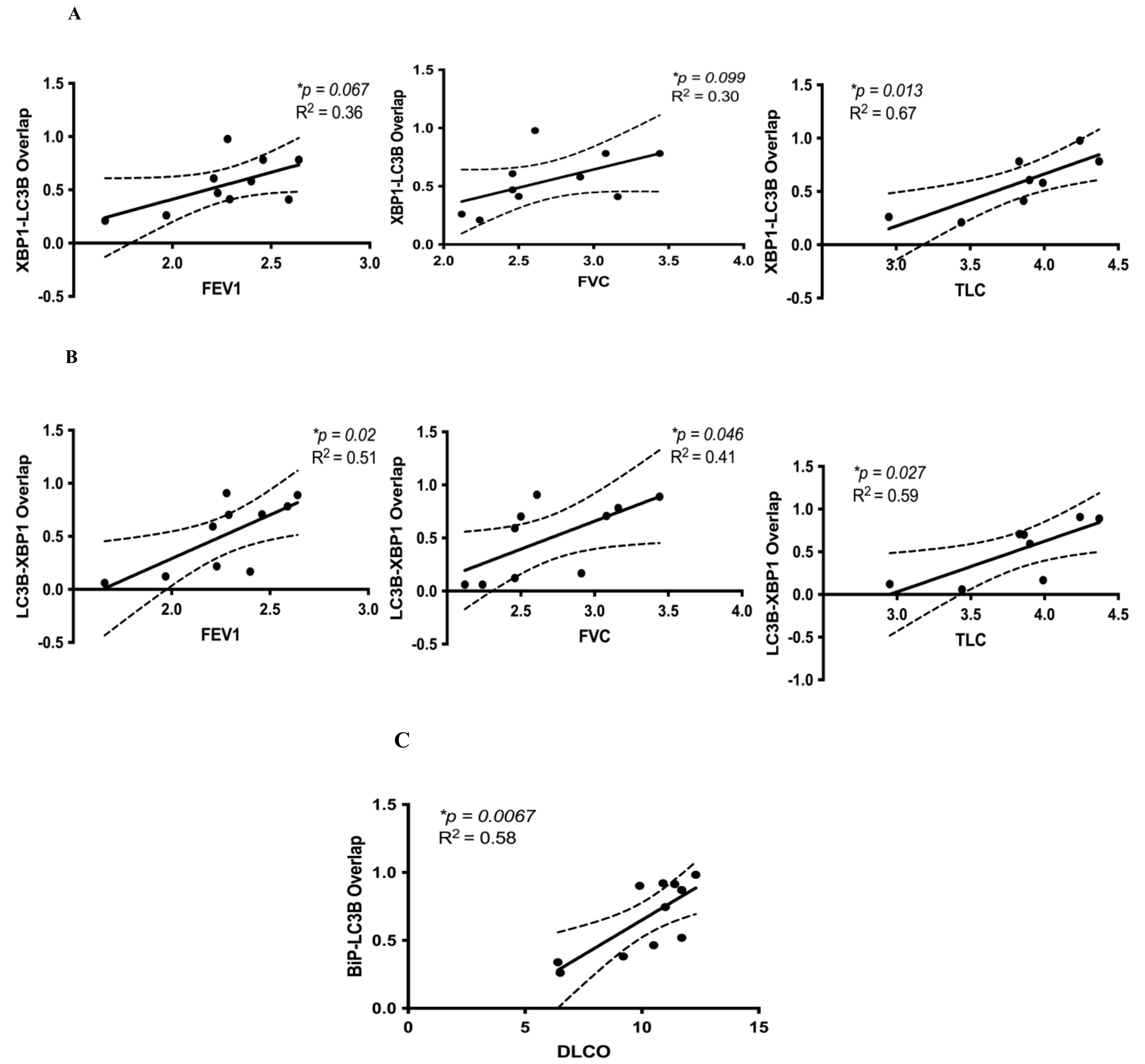
3.2. Correlation between Cell Stress Markers and Lung Function in IPF
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sauleda, J.; Núñez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Haspel, J.A.; Choi, A.M. Autophagy: A core cellular process with emerging links to pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1237–1246. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jablonska, K.; Wiechec, E.; Ghavami, S.; Dziegiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Zeglinski, M.R.; Rattan, S.G.; Landry, N.M.; Ghavami, S.; Wigle, J.T.; Klonisch, T.; Halayko, A.J.; Dixon, I.M. Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac myofibroblasts. Oncotarget 2016, 7, 78516–78531. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ferrell, J.E., Jr. Apoptosis propagates through the cytoplasm as trigger waves. Science 2018, 361, 607–612. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Citrin, D.E.; Shankavaram, U.; Horton, J.A.; Shield, W., 3rd; Zhao, S.; Asano, H.; White, A.; Sowers, A.; Thetford, A.; Chung, E.J. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J. Natl. Cancer. Inst. 2013, 105, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ahmad, I.; Lam, A.; Carlisle, M.A.; Li, C.; Wells, J.M.; Raju, S.V.; Athar, M.; Rowe, S.M.; Dransfield, M.T.; et al. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef]
- Burman, A.; Tanjore, H.; Blackwell, T.S. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018, 68–69, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Yeganeh, B.; Zeki, A.A.; Shojaei, S.; Kenyon, N.J.; Ott, S.; Samali, A.; Patterson, J.; Alizadeh, J.; Moghadam, A.R.; et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-β(1) in human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L493–L504. [Google Scholar] [CrossRef]
- Krempaska, K.; Barnowski, S.; Gavini, J.; Hobi, N.; Ebener, S.; Simillion, C.; Stokes, A.; Schliep, R.; Knudsen, L.; Geiser, T.K.; et al. Azithromycin has enhanced effects on lung fibroblasts from idiopathic pulmonary fibrosis (IPF) patients compared to controls. Respir. Res. 2020, 21, 25, (corrected 28 January 2020). [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Alvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin Induces Unfolded Protein Response and Enhances Temozolomide-Induced Cell Death in Glioblastoma Cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.; et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Behrouj, H.; Seghatoleslam, A.; Mokarram, P.; Ghavami, S. Effect of casein kinase 1alpha inhibition on autophagy flux and the AKT/phospho-beta-catenin (S552) axis in HCT116, a RAS-mutated colorectal cancer cell line. Can. J. Physiol. Pharmacol. 2021, 99, 284–293. [Google Scholar] [CrossRef]
- Du Bois, R.M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Discov. 2010, 9, 129–140. [Google Scholar] [CrossRef]
- McAlinden, K.D.; Deshpande, D.A.; Ghavami, S.; Xenaki, D.; Sohal, S.S.; Oliver, B.G.; Haghi, M.; Sharma, P. Autophagy Activation in Asthma Airways Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 541–553. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Alizadeh, J.; Shojaei, S.; Sepanjnia, A.; Hashemi, M.; Eftekharpour, E.; Ghavami, S. Simultaneous Detection of Autophagy and Epithelial to Mesenchymal Transition in the Non-small Cell Lung Cancer Cells. Methods Mol. Biol. 2019, 1854, 87–103. [Google Scholar]
- Yeganeh, B.; Moghadam, A.R.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Lankarani, K.B.; Post, M.; et al. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225–13239. [Google Scholar] [CrossRef]
- Lara-Reyna, S.; Scambler, T.; Holbrook, J.; Wong, C.; Jarosz-Griffiths, H.H.; Martinon, F.; Savic, S.; Peckham, D.; McDermott, M.F. Metabolic Reprograming of Cystic Fibrosis Macrophages via the IRE1alpha Arm of the Unfolded Protein Response Results in Exacerbated Inflammation. Front. Immunol. 2019, 10, 1789. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Eshraghi, M.; Kadkhoda, K.; Mutawe, M.M.; Maddika, S.; Bay, G.H.; Wesselborg, S.; Halayko, A.J.; Klonisch, T.; Los, M. Role of BNIP3 in TNF-induced cell death--TNF upregulates BNIP3 expression. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wu, Z.; Hergert, P.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Regulation of myofibroblast differentiation by poly(ADP-ribose) polymerase 1. Am. J. Pathol. 2013, 182, 71–83. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Los, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Kasam, R.K.; Reddy, G.B.; Jegga, A.G.; Madala, S.K. Dysregulation of Mesenchymal Cell Survival Pathways in Severe Fibrotic Lung Disease: The Effect of Nintedanib Therapy. Front. Pharmacol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Bressan, C.; Pecora, A.; Gagnon, D.; Snapyan, M.; Labrecque, S.; De Koninck, P.; Parent, M.; Saghatelyan, A. The dynamic interplay between ATP/ADP levels and autophagy sustain neuronal migration in vivo. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Zank, D.C.; Bueno, M.; Mora, A.L.; Rojas, M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front. Med. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Nakahira, K.; Choi, A.M. Autophagy: A potential therapeutic target in lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L93–L107. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Samali, A.; Jager, R. Methods for studying ER stress and UPR markers in human cells. Methods Mol. Biol. 2015, 1292, 3–18. [Google Scholar]
- Dastghaib, S.; Kumar, P.S.; Aftabi, S.; Damera, G.; Dalvand, A.; Sepanjnia, A.; Kiumarsi, M.; Aghanoori, M.R.; Sohal, S.S.; Ande, S.R.; et al. Mechanisms Targeting the Unfolded Protein Response in Asthma. Am. J. Respir. Cell Mol. Biol. 2021, 64, 29–38. [Google Scholar] [CrossRef]
- Talty, A.; Deegan, S.; Ljujic, M.; Mnich, K.; Naicker, S.D.; Quandt, D.; Zeng, Q.; Patterson, J.B.; Gorman, A.M.; Griffin, M.D.; et al. Inhibition of IRE1alpha RNase activity reduces NLRP3 inflammasome assembly and processing of pro-IL1beta. Cell Death Dis. 2019, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Overley-Adamson, B.; Artlett, C.M.; Stephens, C.; Sassi-Gaha, S.; Weis, R.D.; Thacker, J.D. Targeting the unfolded protein response, XBP1, and the NLRP3 inflammasome in fibrosis and cancer. Cancer Biol. Ther. 2014, 15, 452–462. [Google Scholar] [CrossRef][Green Version]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef]
- Liu, A.; Song, Q.; Zheng, Y.; Xu, G.; Huang, C.; Sun, S.; He, L.; Zhao, L.; Zhou, M. Expression of XBP1s in peritoneal mesothelial cells is critical for inflammation-induced peritoneal fibrosis. Sci. Rep. 2019, 9, 19043. [Google Scholar] [CrossRef] [PubMed]
- Maiers, J.L.; Kostallari, E.; Mushref, M.; de Assuncao, T.M.; Li, H.; Jalan-Sakrikar, N.; Huebert, R.C.; Cao, S.; Malhi, H.; Shah, V.H. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology 2017, 65, 983–998. [Google Scholar] [CrossRef]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 6, 39342. [Google Scholar] [CrossRef]
- Heindryckx, F.; Binet, F.; Ponticos, M.; Rombouts, K.; Lau, J.; Kreuger, J.; Gerwins, P. Endoplasmic reticulum stress enhances fibrosis through IRE1alpha-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol. Med. 2016, 8, 729–744. [Google Scholar] [CrossRef]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Uhal, B.D. The role of apoptosis in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 138–144. [Google Scholar] [CrossRef]
- Moodley, Y.P.; Caterina, P.; Scaffidi, A.K.; Misso, N.L.; Papadimitriou, J.M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J. Pathol. 2004, 202, 486–495. [Google Scholar] [CrossRef]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kota, A.; Deshpande, D.; Haghi, M.; Oliver, B.; Sharma, P. Autophagy and airway fibrosis: Is there a link? F1000Research 2017, 6, 409. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID1 | ID2 | ID3 | ID4 | ID1-4 $ | |
|---|---|---|---|---|---|
| Age (deceased, yrs) | 63 | 59 | 85 | 72 | 69.8 ± 11.5 |
| Sex | M | M | F | M | |
| Ethnicity | White | White | White | Hispanic | |
| Lung Function $ | |||||
| FEV1 (L) | 2.52 ± 0.11 | 2.12 ± 0.14 | 1.77 ± 0.10 | 2.16 ± 0.09 | 2.17 ± 0.22 |
| FEV1%p | 71.8 ± 2.6% | 56 ± 3.6% | 95.3 ± 3.8% | 74.8 ± 2.9% | 74.5 ± 10.1% |
| FVC (L) | 3.15 ± 0.22 | 2.33 ± 0.18 | 2.39 ± 0.14 | 2.51 ± 0.08 | 2.58 ± 0.28 |
| FVC%p | 69.8 ± 4.6% | 46.7 ± 3.2% | 93.3 ± 5.0% | 67.4 ± 2.6% | 68.4 ± 11.9% |
| FEV1/FVC | 80% | 90.1% | 74.1% | 86.1% | 84.1% |
| RV (L) | 0.77 ± 0.35 | 0.77 ± 0.05 | 1.54 ± 0.5 | 1.31 ± 0.19 | 1.10 ± 0.42 |
| RV%p | 34 ± 15.9% | 34 ± 2.8% | 65 ± 24.0% | 57.8 ± 7.7% | 48.3 ± 17.9% |
| TLC (L) | 4.06 ± 0.28 | 3.14 ± 0.26 | 3.92 ± 0.68 | 3.91 ± 0.25 | 3.81 ± 0.45 |
| TLC%p | 59.7 ± 4.04% | 43.5 ± 3.5% | 79.5 ± 14.9% | 62.8 ± 2.8% | 61.5 ± 12.7% |
| DLCO | 8.5 ± 1.8 | 6.9 ± 0.71 | 12.9 ± 2.7 | 10.5 ± 1.5 | 9.9 ± 2.3 |
| DLCO%p | 31 ± 6.2% | 22.5 ± 2.12% | 66.5 ± 13.4% | 45.9 ± 5.9% | 42.8 ± 14.1% |
| Lung Pathology # | UIP | UIP | UIP | UIP | - |
| Clinical Diagnosis | IPF | IPF | IPF | IPF | - |
| Comorbidities | AF, CAD, DM, CKD | HIV/AIDS, BPH, SCC (rectal) | HTN, AV dis, MV dis., RBBB, TAH | GERD, HPL, PVD, AF, Divrt, TIA |
| ID1 | ID2 | ID3 | ID1-3 $ | |
|---|---|---|---|---|
| Age (yrs) | 76 | 79 | 73 | 76 ± 3 |
| Sex | M | M | M | - |
| Smoking Status (pack-years) | 60 * | 30 @ | 45 @ | |
| Lung Function $ | ||||
| FEV1 (L) | 1.86 | 2.28 | 3.07 | 2.40 ± 0.61 |
| FEV1%p | 65% | 78% | 99% | 80.67 ± 17.2% |
| FVC (L) | 2.85 | 3.34 | 3.71 | 3.3 ± 0.43 |
| FEV1/FVC | 65% | 68% | 83% | 72 ± 9.6% |
| Diagnosis based on PFT | Moderate Obstructive Disease | Mild Obstructive Disease | Normal | |
| COPD GOLD | ||||
| Classification | Stage II | Stage II | N/A | |
| Tumor | Lung Cancer | Lung Cancer | Lung Cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, P.; Alizadeh, J.; Juarez, M.; Samali, A.; Halayko, A.J.; Kenyon, N.J.; Ghavami, S.; Zeki, A.A. Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis. Cells 2021, 10, 1642. https://doi.org/10.3390/cells10071642
Sharma P, Alizadeh J, Juarez M, Samali A, Halayko AJ, Kenyon NJ, Ghavami S, Zeki AA. Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis. Cells. 2021; 10(7):1642. https://doi.org/10.3390/cells10071642
Chicago/Turabian StyleSharma, Pawan, Javad Alizadeh, Maya Juarez, Afshin Samali, Andrew J. Halayko, Nicholas J. Kenyon, Saeid Ghavami, and Amir A. Zeki. 2021. "Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis" Cells 10, no. 7: 1642. https://doi.org/10.3390/cells10071642
APA StyleSharma, P., Alizadeh, J., Juarez, M., Samali, A., Halayko, A. J., Kenyon, N. J., Ghavami, S., & Zeki, A. A. (2021). Autophagy, Apoptosis, the Unfolded Protein Response, and Lung Function in Idiopathic Pulmonary Fibrosis. Cells, 10(7), 1642. https://doi.org/10.3390/cells10071642