
The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
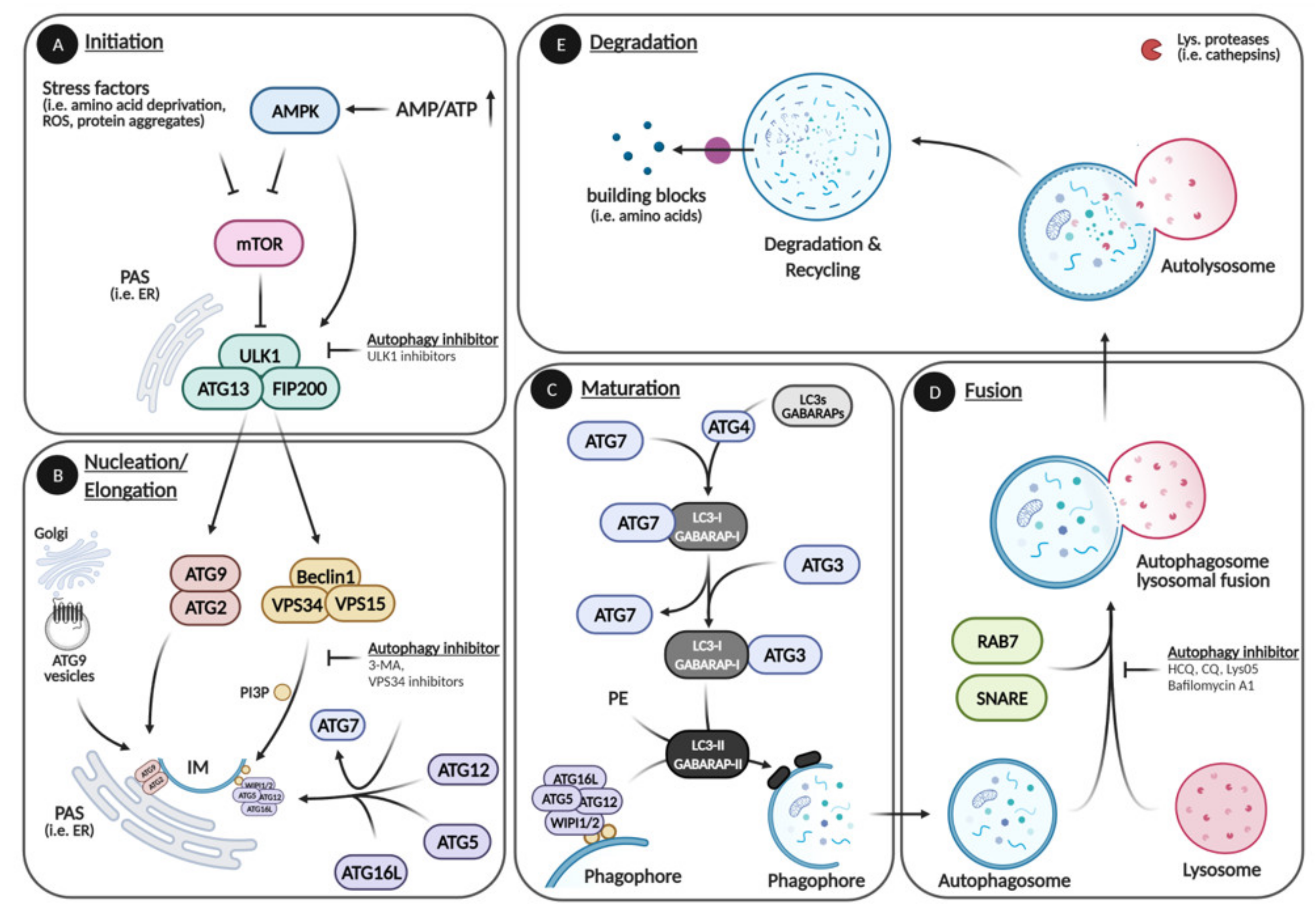
1. Autophagy
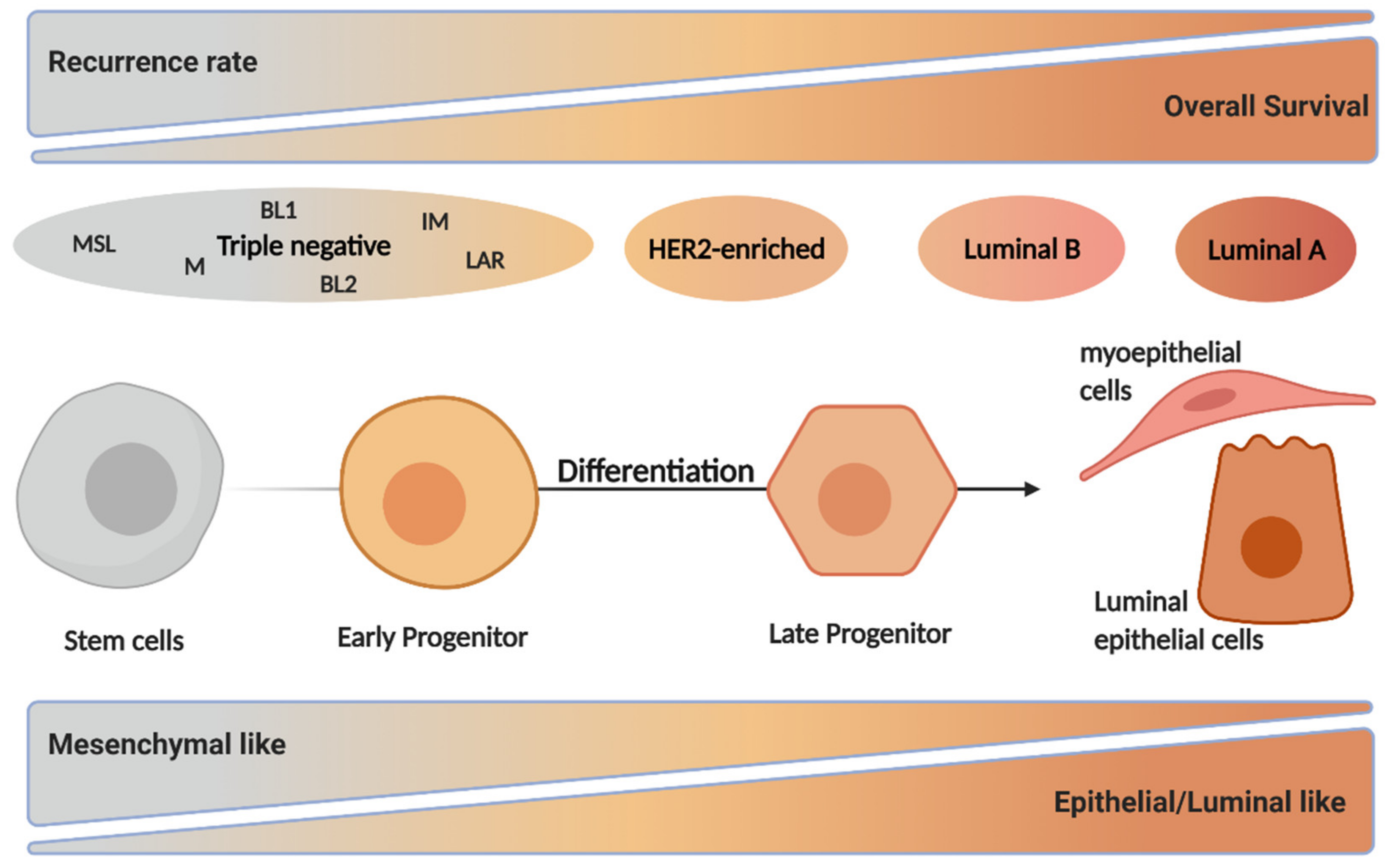
2. Breast Cancer
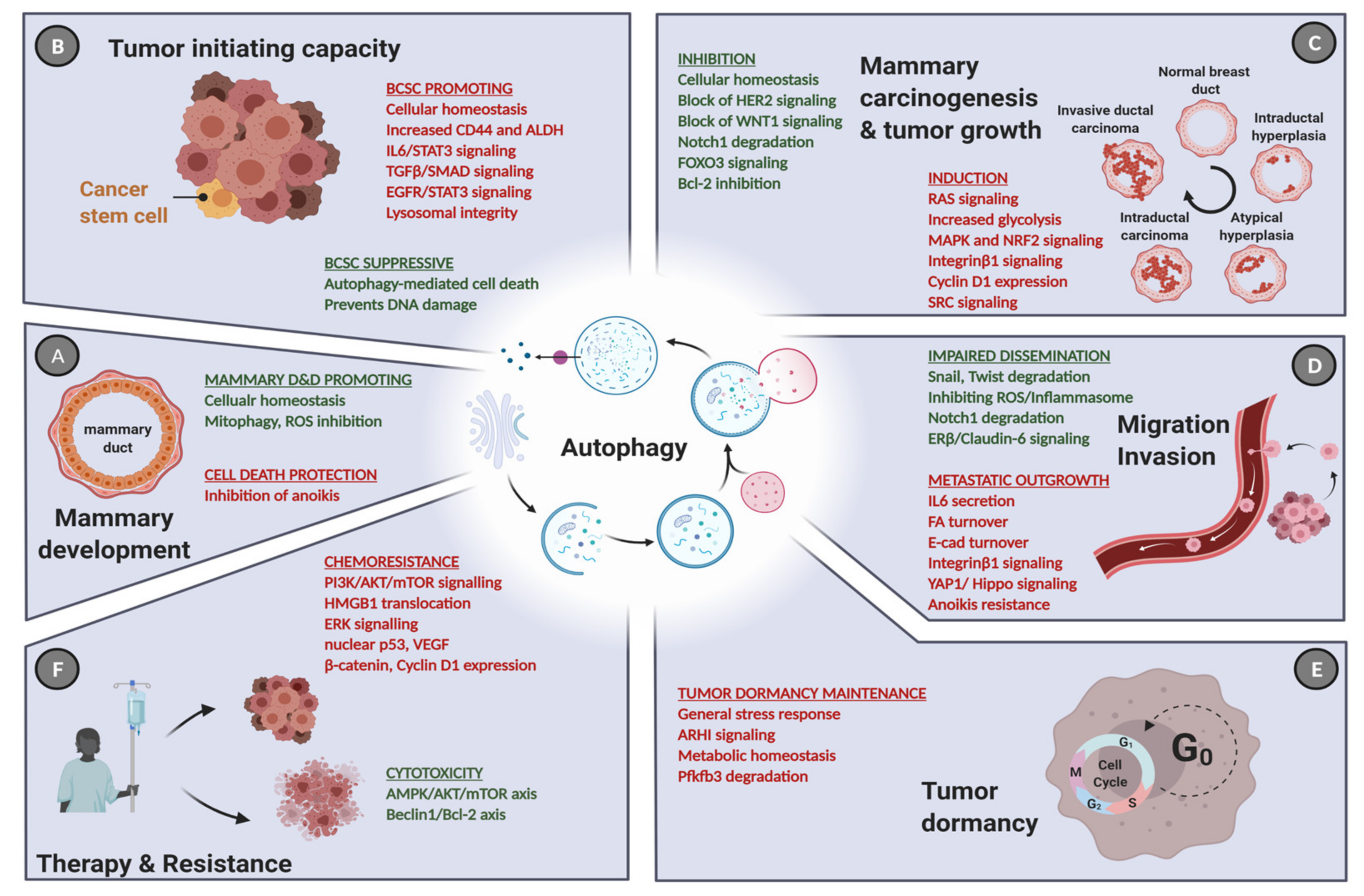
3. Autophagy in Normal Breast Development
4. Autophagy in Breast Cancer Tumorigenesis and Tumor Progression
5. The Role of Autophagy in Breast Cancer Stem Cells
6. Autophagy and Breast Cancer Dormancy
7. Autophagy and Breast Cancer Cell Dissemination
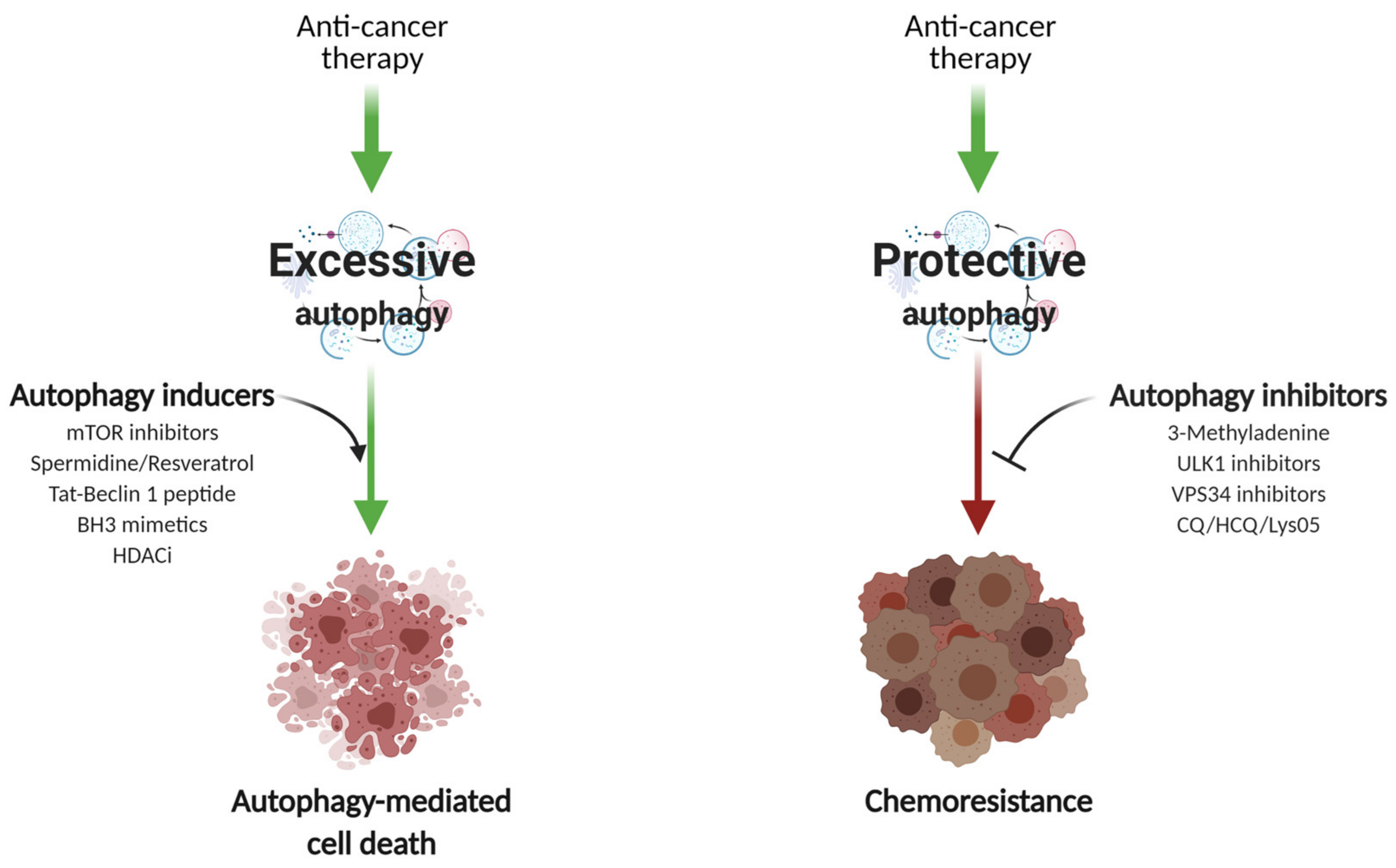
8. Autophagy in Breast Cancer Therapy
8.1. Autophagy Induction
8.2. Autophagy Inhibition
9. Combination Therapies in the Clinic
10. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 Family LC3/GABARAP Proteins Are Crucial for Autophagosome-Lysosome Fusion but Not Autophagosome Formation during PINK1/Parkin Mitophagy and Starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Vaites, L.P.; Paulo, J.A.; Huttlin, E.L.; Harper, J.W. Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef]
- Engedal, N.; Seglen, P.O. Autophagy of Cytoplasmic Bulk Cargo Does Not Require LC3. Autophagy 2016, 12, 439–441. [Google Scholar] [CrossRef]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin-Phosphatidylinositol 3-Kinase Complex Functions at the Trans-Golgi Network. EMBO Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The Autophagosome: Origins Unknown, Biogenesis Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond Autophagy: The Expanding Roles of ATG8 Proteins. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. A SNARE and Specific COPII Requirements Define ER-Derived Vesicles for the Biogenesis of Autophagosomes. Autophagy 2016, 12, 1049–1050. [Google Scholar] [CrossRef][Green Version]
- Whitmarsh-Everiss, T.; Laraia, L. Small Molecule Probes for Targeting Autophagy. Nat. Chem. Biol. 2021, 17, 653–664. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as Dynamic Regulators of Cell and Organismal Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Lin, C.-J.; Tsao, Y.-N.; Shu, C.-W. Autophagy Modulation as a Potential Targeted Cancer Therapy: From Drug Repurposing to New Drug Development. Kaohsiung J. Med. Sci. 2021, 37, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The Mammalian ULK1 Complex and Autophagy Initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Ohashi, Y.; Tremel, S.; Williams, R.L. VPS34 Complexes from a Structural Perspective. J. Lipid Res. 2019, 60, 229–241. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR Motif–Crucial for Selective Autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. In Methods in Enzymology; Autophagy in Mammalian Systems, Part B; Academic Press: Cambridge, MA, USA, 2009; Volume 452, pp. 181–197. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in Mammalian Development and Differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Chang, N.C. Autophagy and Stem Cells: Self-Eating for Self-Renewal. Front. Cell Dev. Biol. 2020, 8, 138. [Google Scholar] [CrossRef]
- Elswood, J.; Pearson, S.J.; Payne, H.R.; Barhoumi, R.; Rijnkels, M.; Porter, W.W. Autophagy Regulates Functional Differentiation of Mammary Epithelial Cells. Autophagy 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer Incidence and Mortality Patterns in Europe: Estimates for 40 Countries and 25 Major Cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.-L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the Molecular Portraits of Breast Cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant Luminal Progenitors as the Candidate Target Population for Basal Tumor Development in BRCA1 Mutation Carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Szymiczek, A.; Lone, A.; Akbari, M.R. Molecular Intrinsic versus Clinical Subtyping in Breast Cancer: A Comprehensive Review. Clin. Genet 2020. [Google Scholar] [CrossRef]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Panel Members Tailoring Therapies--Improving the Management of Early Breast Cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 Index, HER2 Status, and Prognosis of Patients with Luminal B Breast Cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef]
- Vallejos, C.S.; Gómez, H.L.; Cruz, W.R.; Pinto, J.A.; Dyer, R.R.; Velarde, R.; Suazo, J.F.; Neciosup, S.P.; León, M.; de la Cruz, M.A.; et al. Breast Cancer Classification According to Immunohistochemistry Markers: Subtypes and Association with Clinicopathologic Variables in a Peruvian Hospital Database. Clin. Breast Cancer 2010, 10, 294–300. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Van Maaren, M.C.; de Munck, L.; Strobbe, L.J.A.; Sonke, G.S.; Westenend, P.J.; Smidt, M.L.; Poortmans, P.M.P.; Siesling, S. Ten-Year Recurrence Rates for Breast Cancer Subtypes in the Netherlands: A Large Population-Based Study. Int. J. Cancer 2019, 144, 263–272. [Google Scholar] [CrossRef]
- Prat, A.; Perou, C.M. Mammary Development Meets Cancer Genomics. Nat. Med. 2009, 15, 842–844. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic Evaluation of the Basal-like Subtype of Invasive Breast Carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The Role of Apoptosis in Creating and Maintaining Luminal Space within Normal and Oncogene-Expressing Mammary Acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in Malignant Transformation and Cancer Progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Hori, T.; Cooper, T.K.; Liao, J.; Desai, N.; Serfass, J.M.; Young, M.M.; Park, S.; Izu, Y.; Wang, H.-G. Bif-1 Haploinsufficiency Promotes Chromosomal Instability and Accelerates Myc-Driven Lymphomagenesis via Suppression of Mitophagy. Blood 2013, 121, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of P62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of P62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Rello-Varona, S.; Lissa, D.; Shen, S.; Niso-Santano, M.; Senovilla, L.; Mariño, G.; Vitale, I.; Jemaá, M.; Harper, F.; Pierron, G.; et al. Autophagic Removal of Micronuclei. Cell Cycle 2012, 11, 170–176. [Google Scholar] [CrossRef]
- Guo, H.; Chitiprolu, M.; Gagnon, D.; Meng, L.; Perez-Iratxeta, C.; Lagace, D.; Gibbings, D. Autophagy Supports Genomic Stability by Degrading Retrotransposon RNA. Nat. Commun. 2014, 5, 5276. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary Downregulation of Mutant P53 Levels via Glucose Restriction: Mechanisms and Implications for Tumor Therapy. Cell Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef]
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the Pathways That Destabilize Mutant P53: The Proteasome or Autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of Mutant P53H175 Protein by Zn(II) through Autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Greim, H.; Kaden, D.A.; Larson, R.A.; Palermo, C.M.; Rice, J.M.; Ross, D.; Snyder, R. The Bone Marrow Niche, Stem Cells, and Leukemia: Impact of Drugs, Chemicals, and the Environment. Ann. N.Y. Acad. Sci. 2014, 1310, 7–31. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and Cellular Immune Responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D Receptor Regulates Autophagy in the Normal Mammary Gland and in Luminal Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186–E2194. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Vega-Rubín-de-Celis, S.; Zou, Z.; Fernández, Á.F.; Ci, B.; Kim, M.; Xiao, G.; Xie, Y.; Levine, B. Increased Autophagy Blocks HER2-Mediated Breast Tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4176–4181. [Google Scholar] [CrossRef]
- Cicchini, M.; Chakrabarti, R.; Kongara, S.; Price, S.; Nahar, R.; Lozy, F.; Zhong, H.; Vazquez, A.; Kang, Y.; Karantza, V. Autophagy Regulator BECN1 Suppresses Mammary Tumorigenesis Driven by WNT1 Activation and Following Parity. Autophagy 2014, 10, 2036–2052. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal Structure of the Bcl-XL-Beclin 1 Peptide Complex BECLIN 1 IS A NOVEL BH3-ONLY PROTEIN. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against Fatal Sindbis Virus Encephalitis by Beclin, a Novel Bcl-2-Interacting Protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Ann, E.-J.; Kim, M.-Y.; Yoon, J.-H.; Lee, H.-J.; Jo, E.-H.; Lee, K.; Lee, J.S.; Park, H.-S. Autophagy Negatively Regulates Tumor Cell Proliferation through Phosphorylation Dependent Degradation of the Notch1 Intracellular Domain. Oncotarget 2016, 7, 79047–79063. [Google Scholar] [CrossRef]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V.; et al. Protein and MRNA Expression of Autophagy Gene Beclin 1 in Human Brain Tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar]
- Smit, L.; Berns, K.; Spence, K.; Ryder, W.D.; Zeps, N.; Madiredjo, M.; Beijersbergen, R.; Bernards, R.; Clarke, R.B. An Integrated Genomic Approach Identifies That the PI3K/AKT/FOXO Pathway Is Involved in Breast Cancer Tumor Initiation. Oncotarget 2016, 7, 2596–2610. [Google Scholar] [CrossRef]
- Schäffner, I.; Minakaki, G.; Khan, M.A.; Balta, E.-A.; Schlötzer-Schrehardt, U.; Schwarz, T.J.; Beckervordersandforth, R.; Winner, B.; Webb, A.E.; DePinho, R.A.; et al. FoxO Function Is Essential for Maintenance of Autophagic Flux and Neuronal Morphogenesis in Adult Neurogenesis. Neuron 2018, 99, 1188–1203.e6. [Google Scholar] [CrossRef]
- Audesse, A.J.; Dhakal, S.; Hassell, L.-A.; Gardell, Z.; Nemtsova, Y.; Webb, A.E. FOXO3 Directly Regulates an Autophagy Network to Functionally Regulate Proteostasis in Adult Neural Stem Cells. PLoS Genet. 2019, 15, e1008097. [Google Scholar] [CrossRef]
- Li, M.; Liu, J.; Li, S.; Feng, Y.; Yi, F.; Wang, L.; Wei, S.; Cao, L. Autophagy-Related 7 Modulates Tumor Progression in Triple-Negative Breast Cancer. Lab. Investig. 2019, 99. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy Facilitates Glycolysis during Ras-Mediated Oncogenic Transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-Mediated Autophagy Dependence Identifies Subtypes of Breast Cancer Where Autophagy Inhibition Can Be Efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of Autophagy by FIP200 Deletion Inhibits Mammary Tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Delibaşı, N.; Geçene, S.; Şener, E.F.; Dönmez-Altuntaş, H.; Özkul, Y.; Canatan, H.; Ozpolat, B. Targeting LC3 and Beclin-1 Autophagy Genes Suppresses Proliferation, Survival, Migration and Invasion by Inhibition of Cyclin-D1 and UPAR/Integrin Β1/ Src Signaling in Triple Negative Breast Cancer Cells. J. Cancer Res. Clin. Oncol. 2018, 144, 415–430. [Google Scholar] [CrossRef]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 Enables Autophagy-Dependent Focal Adhesion Turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.; Kenific, C.M.; Suresh, D.; Gonzalez, H.; Shamir, E.R.; Mei, W.; Tankka, A.; Leidal, A.M.; Kalavacherla, S.; Woo, K.; et al. Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev. Cell 2020, 52, 591–604.e6. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 Pathway by P62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 Stimulation of P62 Phosphorylation for Cancer Progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and Autophagy Are Required for the Tumorigenicity of Breast Cancer Stem-like/Progenitor Cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, Q.; Zou, Y.; Chen, H.; Qi, L.; Chen, Y. Stem Cells and Cellular Origins of Breast Cancer: Updates in the Rationale, Controversies, and Therapeutic Implications. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the Roles of CD44/CD24 and ALDH1 as Cancer Stem Cell Markers in Tumorigenesis and Metastasis. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Parlato, M.; Souza-Fonseca-Guimaraes, F.; Philippart, F.; Misset, B.; Captain Study Group; Adib-Conquy, M.; Cavaillon, J.-M. CD24-Triggered Caspase-Dependent Apoptosis via Mitochondrial Membrane Depolarization and Reactive Oxygen Species Production of Human Neutrophils Is Impaired in Sepsis. J. Immunol. 2014, 192, 2449–2459. [Google Scholar] [CrossRef]
- Gilliam, D.T.; Menon, V.; Bretz, N.P.; Pruszak, J. The CD24 Surface Antigen in Neural Development and Disease. Neurobiol. Dis. 2017, 99, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Goodison, S.; Urquidi, V.; Tarin, D. CD44 Cell Adhesion Molecules. Mol. Pathol. 1999, 52, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde Dehydrogenases in Cancer Stem Cells: Potential as Therapeutic Targets. Ann. Transl. Med. 2016, 4. [Google Scholar] [CrossRef]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of Autophagy in Breast Cancer and Breast Cancer Stem Cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Müller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A Mammosphere Formation RNAi Screen Reveals That ATG4A Promotes a Breast Cancer Stem-like Phenotype. Breast Cancer Res. 2013, 15, R109. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy Positively Regulates the CD44+CD24-/Low Breast Cancer Stem-like Phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.H.; Eom, M.; Choi, J. Interleukin-6/STAT3 Signalling Regulates Adipocyte Induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Lieblein, J.C.; Ball, S.; Hutzen, B.; Sasser, A.K.; Lin, H.-J.; Huang, T.H.; Hall, B.M.; Lin, J. STAT3 Can Be Activated through Paracrine Signaling in Breast Epithelial Cells. BMC Cancer 2008, 8, 302. [Google Scholar] [CrossRef]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.-L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The Autophagy Inhibitor Chloroquine Targets Cancer Stem Cells in Triple Negative Breast Cancer by Inducing Mitochondrial Damage and Impairing DNA Break Repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef]
- Yue, W.; Hamaï, A.; Tonelli, G.; Bauvy, C.; Nicolas, V.; Tharinger, H.; Codogno, P.; Mehrpour, M. Inhibition of the Autophagic Flux by Salinomycin in Breast Cancer Stem-like/Progenitor Cells Interferes with Their Maintenance. Autophagy 2013, 9, 714–729. [Google Scholar] [CrossRef]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin-Induced Autophagy Leads to the Apoptosis in Breast Cancer Stem Cells: Molecular Mechanisms. Mol. Cancer 2013, 12, 171. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy Promotes the Survival of Dormant Breast Cancer Cells and Metastatic Tumour Recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Maishman, T.; Cutress, R.I.; Hernandez, A.; Gerty, S.; Copson, E.R.; Durcan, L.; Eccles, D.M. Local Recurrence and Breast Oncological Surgery in Young Women With Breast Cancer: The POSH Observational Cohort Study. Ann. Surg. 2017, 266, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Zucchinetti, C.; Buda, G.; Orciuolo, E. Tumor Dormancy as an Alternative Step in the Development of Chemoresistance and Metastasis-Clinical Implications. Cell Oncol. 2020, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, Mechanisms and Clinical Evidence for Cancer Dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Green, J.E.; Chambers, A.F. Extracellular Matrix: A Gatekeeper in the Transition from Dormancy to Metastatic Growth. Eur. J. Cancer 2010, 46, 1181–1188. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From Transformation to Metastasis: Deconstructing the Extracellular Matrix in Breast Cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, Cancer Stem Cells and Drug Resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The Tumor Suppressor Gene ARHI Regulates Autophagy and Tumor Dormancy in Human Ovarian Cancer Cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.-F.; Jia, L.; Jin, H.; Yao, M.; Zhao, N.; Huan, J.; Lu, Z.; Bast, R.C.; Feng, Y.; Yu, Y. Re-Expression of ARHI (DIRAS3) Induces Autophagy in Breast Cancer Cells and Enhances the Inhibitory Effect of Paclitaxel. BMC Cancer 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy Inhibition Elicits Emergence from Metastatic Dormancy by Inducing and Stabilizing Pfkfb3 Expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of Cell Migration: A Multiscale Tuning Model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-Cell Invasion and Migration: Diversity and Escape Mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Zhang, W.; Bado, I.; Wang, H.; Lo, H.-C.; Zhang, X.H.-F. Bone Metastasis: Find Your Niche and Fit In. Trends Cancer 2019, 5, 95–110. [Google Scholar] [CrossRef]
- Zhang, M.; Lee, A.V.; Rosen, J.M. The Cellular Origin and Evolution of Breast Cancer. Cold. Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Mohd Sobri, S.N.; Abdul Sani, S.F.; Sabtu, S.N.; Looi, L.M.; Chiew, S.F.; Pathmanathan, D.; Chio-Srichan, S.; Bradley, D.A. Structural Studies of Epithelial Mesenchymal Transition Breast Tissues. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-T.; Liu, H.; Mao, M.-J.; Tan, Y.; Mo, X.-Q.; Meng, X.-J.; Cao, M.-T.; Zhong, C.-Y.; Liu, Y.; Shan, H.; et al. Crosstalk between Autophagy and Epithelial-Mesenchymal Transition and Its Application in Cancer Therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fässler, R.; Geiger, B.; Legate, K.R. The Integrin Adhesome: From Genes and Proteins to Human Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Danen, E.H.J. Adhesion Signaling–Crosstalk between Integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Bijian, K.; Qiu, D.; Su, J.; Saad, A.; Dahabieh, M.S.; Miller, W.H.; Alaoui-Jamali, M.A. Endosomal Sorting and C-Cbl Targeting of Paxillin to Autophagosomes Regulate Cell-Matrix Adhesion Turnover in Human Breast Cancer Cells. Oncotarget 2017, 8, 31199–31214. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bai, Y.; Patel, C.; Geng, F. Autophagy Promotes Triple Negative Breast Cancer Metastasis via YAP Nuclear Localization. Biochem Biophys Res Commun 2019, 520, 263–268. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.-W. DEDD Interacts with PI3KC3 to Activate Autophagy and Attenuate Epithelial-Mesenchymal Transition in Human Breast Cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zhang, H.-L.; Huang, J.-H.; Cai, R.-Z.; Wang, Y.; Chen, Y.-H.; Hu, B.-X.; Ye, Z.-P.; Li, Z.-L.; Mai, J.; et al. MAPK1/3 Kinase-Dependent ULK1 Degradation Attenuates Mitophagy and Promotes Breast Cancer Bone Metastasis. Autophagy 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, Y.; Dong, Y.; Liang, Y.; Qu, H.; Qi, D.; Lu, Y.; Jin, X.; Guo, Y.; Jia, Y.; et al. Estrogen Receptor β Inhibits Breast Cancer Cells Migration and Invasion through CLDN6-Mediated Autophagy. J. Exp. Clin. Cancer Res. 2019, 38, 354. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Zhu, J.-S.; Zhang, Z.; Shen, W.-J.; Jiang, S.; Long, Y.-F.; Wu, B.; Ding, T.; Huan, F.; Wang, S.-L. Hydroxytyrosol and Oleuropein Inhibit Migration and Invasion of MDA-MB-231 Triple-Negative Breast Cancer Cell via Induction of Autophagy. Anticancer Agents Med. Chem. 2019, 19, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The Chemotherapeutic Agent Paclitaxel Inhibits Autophagy through Two Distinct Mechanisms That Regulate Apoptosis. Oncogene 2013, 32, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.J.; Gorski, S.M. Molecular Mechanisms Underlying Autophagy-Mediated Treatment Resistance in Cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288. [Google Scholar] [CrossRef]
- Mandlekar, S.; Kong, A.N. Mechanisms of Tamoxifen-Induced Apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Chloroquine Inhibits Autophagy to Potentiate Antiestrogen Responsiveness in ER+ Breast Cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef]
- Zambrano, J.; Yeh, E.S. Autophagy and Apoptotic Crosstalk: Mechanism of Therapeutic Resistance in HER2-Positive Breast Cancer. Breast Cancer 2016, 10, 13–23. [Google Scholar] [CrossRef]
- FDA Approves First PI3K Inhibitor for Breast Cancer. Available online: https://www.cancer.org/latest-news/fda-approves-first-pi3k-inhibitor-for-breast-cancer.html (accessed on 10 April 2021).
- Xu, B.; Fan, Y. CDK4/6 Inhibition in Early-Stage Breast Cancer: How Far Is It from Becoming Standard of Care? Lancet Oncol. 2021, 22, 159–160. [Google Scholar] [CrossRef]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef]
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A Tale of Two Approaches: Complementary Mechanisms of Cytotoxic and Targeted Therapy Resistance May Inform next-Generation Cancer Treatments. Carcinogenesis 2013, 34, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Chiu, S.; Oleinick, N.L. Atg7 Deficiency Increases Resistance of MCF-7 Human Breast Cancer Cells to Photodynamic Therapy. Autophagy 2010, 6, 248–255. [Google Scholar] [CrossRef]
- Gong, C.; Hu, C.; Gu, F.; Xia, Q.; Yao, C.; Zhang, L.; Qiang, L.; Gao, S.; Gao, Y. Co-Delivery of Autophagy Inhibitor ATG7 SiRNA and Docetaxel for Breast Cancer Treatment. J. Control. Release 2017, 266, 272–286. [Google Scholar] [CrossRef]
- Chen, G.; Ding, X.-F.; Bouamar, H.; Pressley, K.; Sun, L.-Z. Everolimus Induces G1 Cell Cycle Arrest through Autophagy-Mediated Protein Degradation of Cyclin D1 in Breast Cancer Cells. Am. J. Physiol. Cell Physiol. 2019, 317, C244–C252. [Google Scholar] [CrossRef] [PubMed]
- Lui, A.; New, J.; Ogony, J.; Thomas, S.; Lewis-Wambi, J. Everolimus Downregulates Estrogen Receptor and Induces Autophagy in Aromatase Inhibitor-Resistant Breast Cancer Cells. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, J.; Feng, X.; Zhou, H.; Guo, S.; Zhou, W. Autophagy-Related Genes Are Induced by Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid via the Activation of Cathepsin B in Human Breast Cancer Cells. Oncotarget 2017, 8, 53352–53365. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Sun, Y.; Snyder, R.B.; Berger, S.M.; Yu, J.J.; Holz, M.K. Resveratrol Prevents Rapamycin-Induced Upregulation of Autophagy and Selectively Induces Apoptosis in TSC2-Deficient Cells. Cell Cycle 2014, 13, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol Inhibits Breast Cancer Stem-Like Cells and Induces Autophagy via Suppressing Wnt/β-Catenin Signaling Pathway. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K.W. Natural Small-Molecule Enhancers of Autophagy Induce Autophagic Cell Death in Apoptosis-Defective Cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Gordillo-Martínez, F.; Qu, Y.Q.; Zhang, N.; Xu, S.W.; Coghi, P.S.; Fai Mok, S.W.; Guo, J.; Zhang, W.; Leung, E.L.H.; et al. Thalidezine, a Novel AMPK Activator, Eliminates Apoptosis-Resistant Cancer Cells through Energy-Mediated Autophagic Cell Death. Oncotarget 2017, 8, 30077–30091. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Mok, S.W.F.; Chan, W.K.; Xu, S.W.; Wu, A.G.; Yao, X.J.; Wang, J.R.; Liu, L.; Wong, V.K.W. Hernandezine, a Novel AMPK Activator Induces Autophagic Cell Death in Drug-Resistant Cancers. Oncotarget 2016, 7, 8090–8104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-W.; Hu, J.-J.; Fu, R.-Q.; Liu, X.; Zhang, Y.-H.; Li, J.; Liu, L.; Li, Y.-N.; Deng, Q.; Luo, Q.-S.; et al. Flavonoids Inhibit Cell Proliferation and Induce Apoptosis and Autophagy through Downregulation of PI3Kγ Mediated PI3K/AKT/MTOR/P70S6K/ULK Signaling Pathway in Human Breast Cancer Cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis Is a Na+,K+-ATPase-Regulated Form of Cell Death Triggered by Autophagy-Inducing Peptides, Starvation, and Hypoxia-Ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, Z.; Simon, H.-U. Chapter 14—The Role of Autophagy in Cancer and Chemotherapy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 253–265. ISBN 978-0-12-802937-4. [Google Scholar]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/MTOR-Mediated Autophagy for Tumor Therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Patel, N.H.; Gewirtz, D.A. Triangular Relationship between P53, Autophagy, and Chemotherapy Resistance. Int. J. Mol. Sci. 2020, 21, 8991. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and Chemotherapy Resistance: A Promising Therapeutic Target for Cancer Treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The Challenge of Drug Resistance in Cancer Treatment: A Current Overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Zhu, C.; Qi, X.; Chen, Y.; Sun, B.; Dai, Y.; Gu, Y. PI3K/Akt and MAPK/ERK1/2 Signaling Pathways Are Involved in IGF-1-Induced VEGF-C Upregulation in Breast Cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 1587–1594. [Google Scholar] [CrossRef]
- Park, J.-H.; Kim, K.P.; Ko, J.-J.; Park, K.-S. PI3K/Akt/MTOR Activation by Suppression of ELK3 Mediates Chemosensitivity of MDA-MB-231 Cells to Doxorubicin by Inhibiting Autophagy. Biochem. Biophys. Res. Commun. 2016, 477, 277–282. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Morita, T.Y.; Ohashi, A.; Haeno, H.; Hakozaki, Y.; Fujii, M.; Kashima, Y.; Kobayashi, S.S.; Mukohara, T. Combination Treatment with a PI3K/Akt/MTOR Pathway Inhibitor Overcomes Resistance to Anti-HER2 Therapy in PIK3CA -Mutant HER2-Positive Breast Cancer Cells. Sci. Rep. 2020, 10, 21762. [Google Scholar] [CrossRef]
- Kong, S.-Y.; Kim, K.-S.; Kim, J.; Kim, M.K.; Lee, K.H.; Lee, J.-Y.; Oh, N.; Park, J.-I.; Park, J.-H.; Heo, S.-H.; et al. The ELK3-GATA3 Axis Orchestrates Invasion and Metastasis of Breast Cancer Cells in Vitro and in Vivo. Oncotarget 2016, 7, 65137–65146. [Google Scholar] [CrossRef]
- Qadir, M.A.; Kwok, B.; Dragowska, W.H.; To, K.H.; Le, D.; Bally, M.B.; Gorski, S.M. Macroautophagy Inhibition Sensitizes Tamoxifen-Resistant Breast Cancer Cells and Enhances Mitochondrial Depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403. [Google Scholar] [CrossRef]
- Sun, W.-L.; Chen, J.; Wang, Y.-P.; Zheng, H. Autophagy Protects Breast Cancer Cells from Epirubicin-Induced Apoptosis and Facilitates Epirubicin-Resistance Development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef]
- Amani, N.; Shaki, F.; Shokrzadeh, M. Contribution of Autophagy in Acquired Drug Resistance of Human Breast Cancer Cells MCF7 to Doxorubicin. Appl. In Vitro Toxicol. 2019, 5, 173–179. [Google Scholar] [CrossRef]
- Ajabnoor, G.M.A.; Crook, T.; Coley, H.M. Paclitaxel Resistance Is Associated with Switch from Apoptotic to Autophagic Cell Death in MCF-7 Breast Cancer Cells. Cell Death Dis. 2012, 3, e260. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of Autophagy Is an Early Response to Gefitinib and a Potential Therapeutic Target in Breast Cancer. PLoS ONE 2013, 8, e76503. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy Facilitates the Development of Breast Cancer Resistance to the Anti-HER2 Monoclonal Antibody Trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castillo, B.; Menendez, J.A. Autophagy-Related Gene 12 (ATG12) Is a Novel Determinant of Primary Resistance to HER2-Targeted Therapies: Utility of Transcriptome Analysis of the Autophagy Interactome to Guide Breast Cancer Treatment. Oncotarget 2012, 3, 1600–1614. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; López-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The Anti-Malarial Chloroquine Overcomes Primary Resistance and Restores Sensitivity to Trastuzumab in HER2-Positive Breast Cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef]
- Wang, P.; Du, Y.; Wang, J. Indentification of Breast Cancer Subtypes Sensitive to HCQ-Induced Autophagy Inhibition. Pathol. Res. Pract. 2019, 215, 152609. [Google Scholar] [CrossRef]
- Negri, T.; Tarantino, E.; Orsenigo, M.; Reid, J.F.; Gariboldi, M.; Zambetti, M.; Pierotti, M.A.; Pilotti, S. Chromosome Band 17q21 in Breast Cancer: Significant Association between Beclin 1 Loss and HER2/NEU Amplification. Genes Chromosomes Cancer 2010, 49, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-L.; Liu, J.-F.; Liu, Y.; Wang, Y.-X.; Fu, K.-F.; Yu, X.-J.; Pu, Q.; Chen, X.-X.; Zhou, L.-J. Beclin1 Inhibition Enhances Paclitaxel-mediated Cytotoxicity in Breast Cancer in Vitro and in Vivo. Int. J. Mol. Med. 2019, 43, 1866–1878. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, T.; Li, G.; Xu, C.; Xu, Z.; Zhang, J.; He, K.; Zheng, L.; Guan, Z.; Su, X.; et al. Lower Beclin 1 Downregulates HER2 Expression to Enhance Tamoxifen Sensitivity and Predicts a Favorable Outcome for ER Positive Breast Cancer. Oncotarget 2016, 8, 52156–52177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-C.; Wu, A.-G.; Huang, Y.-Z.; Shao, G.-L.; Ji, S.-F.; Wang, R.-W.; Yuan, H.-J.; Fan, X.-L.; Zheng, L.-H.; Jiao, Q.-L. Autophagic Regulation of Cell Growth by Altered Expression of Beclin 1 in Triple-Negative Breast Cancer. Int. J. Clin. Exp. Med. 2015, 8, 7049–7058. [Google Scholar]
- Xu, T.; Jiang, L.; Wang, Z. The Progression of HMGB1-Induced Autophagy in Cancer Biology. Onco Targets Ther. 2019, 12, 365–377. [Google Scholar] [CrossRef]
- Ladoire, S.; Enot, D.; Senovilla, L.; Ghiringhelli, F.; Poirier-Colame, V.; Chaba, K.; Semeraro, M.; Chaix, M.; Penault-Llorca, F.; Arnould, L.; et al. The Presence of LC3B Puncta and HMGB1 Expression in Malignant Cells Correlate with the Immune Infiltrate in Breast Cancer. Autophagy 2016, 12, 864–875. [Google Scholar] [CrossRef]
- Pan, B.; Chen, D.; Huang, J.; Wang, R.; Feng, B.; Song, H.; Chen, L. HMGB1-Mediated Autophagy Promotes Docetaxel Resistance in Human Lung Adenocarcinoma. Mol. Cancer 2014, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 Promotes Drug Resistance in Osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef]
- Khambu, B.; Hong, H.; Liu, S.; Liu, G.; Chen, X.; Dong, Z.; Wan, J.; Yin, X.-M. The HMGB1-RAGE Axis Modulates the Growth of Autophagy-Deficient Hepatic Tumors. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qi, X.; Zhang, X.; Gao, D.; Fang, K.; Guo, Z.; Li, L. Med19 Is Involved in Chemoresistance by Mediating Autophagy through HMGB1 in Breast Cancer. J. Cell. Biochem. 2019, 120, 507–518. [Google Scholar] [CrossRef]
- García-Miranda, A.; Solano-Alcalá, K.A.; Montes-Alvarado, J.B.; Rosas-Cruz, A.; Reyes-Leyva, J.; Navarro-Tito, N.; Maycotte, P.; Castañeda-Saucedo, E. Autophagy Mediates Leptin-Induced Migration and ERK Activation in Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Chang, C.; Chen, Y.; Bi, F.; Ji, C.; Liu, W. EGCG Overcomes Gefitinib Resistance by Inhibiting Autophagy and Augmenting Cell Death through Targeting ERK Phosphorylation in NSCLC. Onco Targets Ther. 2019, 12, 6033–6043. [Google Scholar] [CrossRef]
- Bimonte, S.; Cascella, M.; Barbieri, A.; Arra, C.; Cuomo, A. Current Shreds of Evidence on the Anticancer Role of EGCG in Triple Negative Breast Cancer: An Update of the Current State of Knowledge. Infect. Agent Cancer 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine Sensitizes Breast Cancer Cells to Chemotherapy Independent of Autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef]
- Rebecca, V.; Amaravadi, R. Emerging Strategies to Effectively Target Autophagy in Cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nuñez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II Randomized, Double-Blind, Placebo-Controlled Study of Whole-Brain Irradiation with Concomitant Chloroquine for Brain Metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Wang, P.; Burikhanov, R.; Jayswal, R.; Weiss, H.L.; Arnold, S.M.; Villano, J.L.; Rangnekar, V.M. Neoadjuvant Administration of Hydroxychloroquine in a Phase 1 Clinical Trial Induced Plasma Par-4 Levels and Apoptosis in Diverse Tumors. Genes Cancer 2018, 9, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting Autophagy by Small Molecule Inhibitors of Vacuolar Protein Sorting 34 (Vps34) Improves the Sensitivity of Breast Cancer Cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). Hydroxychloroquine, Abemaciclib and Endocrine Therapy in Hormone Receptor Positive (HR+)/Her2 Negative Breast Cancer. Identifier NCT04316169. March 2020. Available online: https://clinicaltrials.gov/ct2/show/study/NCT04316169 (accessed on 10 April 2021).
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.-H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy Inhibitor Lys05 Has Single-Agent Antitumor Activity and Reproduces the Phenotype of a Genetic Autophagy Deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Dröse, S.; Altendorf, K. Bafilomycins and Concanamycins as Inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997, 200, 1–8. [Google Scholar] [CrossRef]
- Li, Z.; Du, L.; Zhang, W.; Zhang, X.; Jiang, Y.; Liu, K.; Men, P.; Xu, H.; Fortman, J.L.; Sherman, D.H.; et al. Complete Elucidation of the Late Steps of Bafilomycin Biosynthesis in Streptomyces Lohii. J. Biol. Chem. 2017, 292, 7095–7104. [Google Scholar] [CrossRef]
- Giovannelli, P.; Donato, M.D.; Galasso, G.; Zazzo, E.D.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast Cancer Stem Cells: The Role of Sex Steroid Receptors. WJSC 2019, 11, 594–603. [Google Scholar] [CrossRef]
- Ramesh, P.; Medema, J.P. BCL-2 Family Deregulation in Colorectal Cancer: Potential for BH3 Mimetics in Therapy. Apoptosis 2020, 25, 305–320. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-Only Proteins and BH3 Mimetics Induce Autophagy by Competitively Disrupting the Interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubín-de-Celis, S.; Kinch, L.; Peña-Llopis, S. Regulation of Beclin 1-Mediated Autophagy by Oncogenic Tyrosine Kinases. Int. J. Mol. Sci. 2020, 21, 9210. [Google Scholar] [CrossRef]
- Alhoshani, A.; Alatawi, F.O.; Al-Anazi, F.E.; Attafi, I.M.; Zeidan, A.; Agouni, A.; El Gamal, H.M.; Shamoon, L.S.; Khalaf, S.; Korashy, H.M. BCL-2 Inhibitor Venetoclax Induces Autophagy-Associated Cell Death, Cell Cycle Arrest, and Apoptosis in Human Breast Cancer Cells. Onco Targets Ther. 2020, 13, 13357–13370. [Google Scholar] [CrossRef] [PubMed]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 Inhibition Sensitizes Triple-Negative Human Breast Cancer Cells to Doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-Expressing Breast Tumors to Chemotherapy by the BH3 Mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.E.; Smyth, G.K.; Christie, M.; Phillipson, L.J.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2–Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef]
- Lindeman, G.J.; Bardia, A.; Bowen, R.; Flechais, A.; Lei, G.; Hogea, A.; Mobasher, M.; Rafii, S. Randomized Phase II Trial of Venetoclax + Fulvestrant versus Fulvestrant in Estrogen Receptor+, HER2– Locally Advanced or Metastatic Breast Cancer Following Recurrence or Progression during or after a CDK4/6 Inhibitor: VERONICA. JCO 2019, 37, TPS1108. [Google Scholar] [CrossRef]
- Lee, J.S.; Yost, S.E.; Blanchard, S.; Schmolze, D.; Yin, H.H.; Pillai, R.; Robinson, K.; Tang, A.; Martinez, N.; Portnow, J.; et al. Phase I Clinical Trial of the Combination of Eribulin and Everolimus in Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2019, 21, 119. [Google Scholar] [CrossRef]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for Women with Trastuzumab-Resistant, HER2-Positive, Advanced Breast Cancer (BOLERO-3): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Vinayak, S.; Carlson, R.W. MTOR Inhibitors in the Treatment of Breast Cancer. Oncology 2013, 27, 38–44, 46, 48 passim. [Google Scholar] [PubMed]
- Yusuf, I.H.; Sharma, S.; Luqmani, R.; Downes, S.M. Hydroxychloroquine Retinopathy. Eye 2017, 31, 828–845. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zou, Z. Targeting Autophagy to Overcome Drug Resistance: Further Developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.S.C.; Gonçalves, L.B.; Lepique, A.P.; de Araujo-Souza, P.S. The Role of Autophagy in Tumor Immunology—Complex Mechanisms That May Be Explored Therapeutically. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Gilardini Montani, M.S.; Granato, M.; Garufi, A.; Faggioni, A.; D’Orazi, G. Autophagy Manipulation as a Strategy for Efficient Anticancer Therapies: Possible Consequences. J. Exp. Clin. Cancer Res. 2019, 38, 262. [Google Scholar] [CrossRef]
- Ladoire, S.; Enot, D.; Andre, F.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death-Related Biomarkers: Impact on the Survival of Breast Cancer Patients after Adjuvant Chemotherapy. Oncoimmunology 2015, 5. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niklaus, N.J.; Tokarchuk, I.; Zbinden, M.; Schläfli, A.M.; Maycotte, P.; Tschan, M.P. The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells 2021, 10, 1447. https://doi.org/10.3390/cells10061447
Niklaus NJ, Tokarchuk I, Zbinden M, Schläfli AM, Maycotte P, Tschan MP. The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells. 2021; 10(6):1447. https://doi.org/10.3390/cells10061447
Chicago/Turabian StyleNiklaus, Nicolas J., Igor Tokarchuk, Mara Zbinden, Anna M. Schläfli, Paola Maycotte, and Mario P. Tschan. 2021. "The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment" Cells 10, no. 6: 1447. https://doi.org/10.3390/cells10061447
APA StyleNiklaus, N. J., Tokarchuk, I., Zbinden, M., Schläfli, A. M., Maycotte, P., & Tschan, M. P. (2021). The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells, 10(6), 1447. https://doi.org/10.3390/cells10061447