
The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise



Abstract
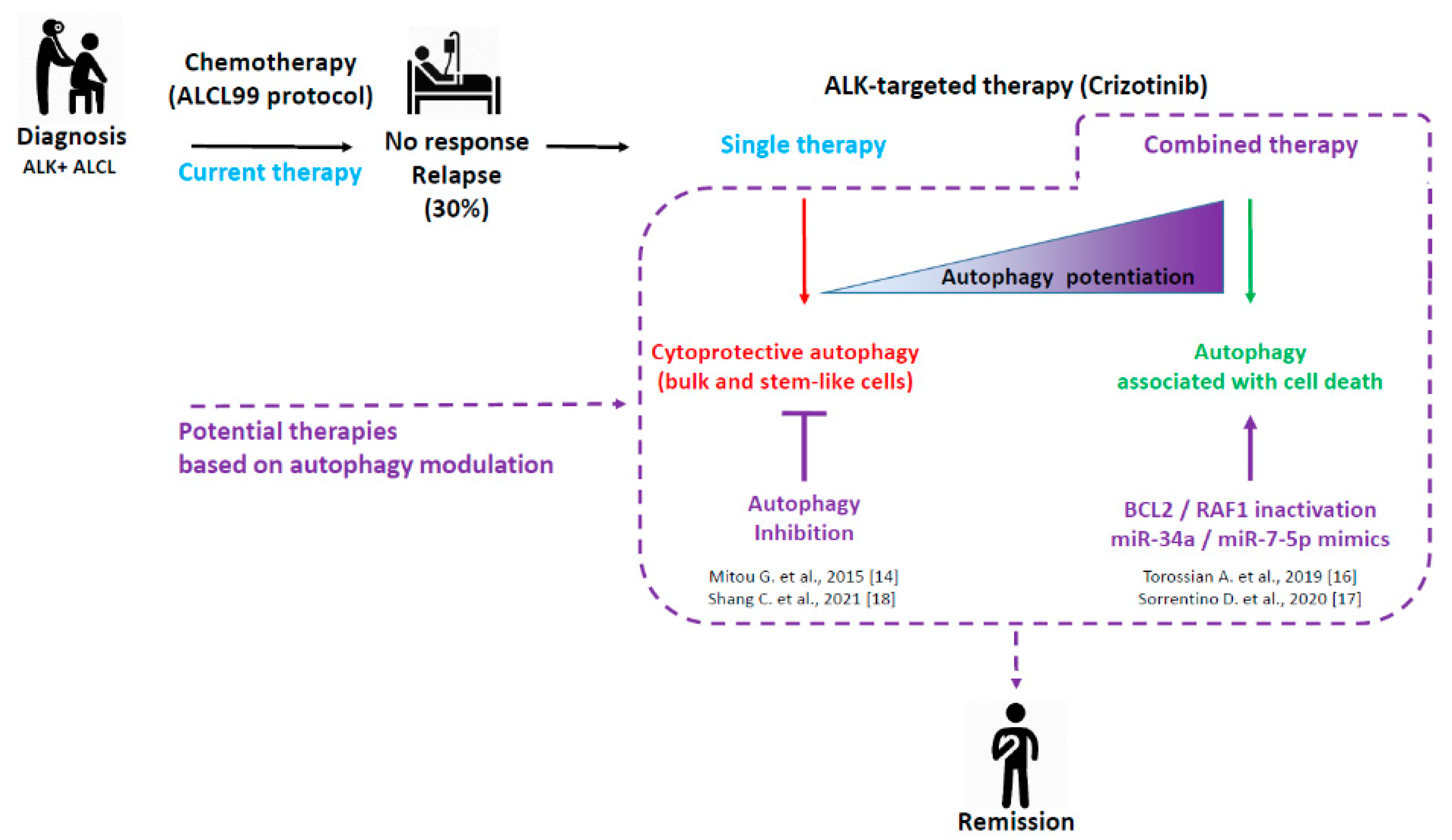
1. Introduction
2. Pro-Survival versus Pro-Death Autophagy in Crizotinib-Treated ALK+ ALCL
2.1. Cytoprotective Autophagy in Crizotinib-Treated ALK+ ALCL
2.1.1. Initial Findings
- Autophagy induction upon ALK inactivation. We performed a set of experiments to study whether autophagy was induced upon ALK pharmacological or molecular inactivation [14]. We first visualized and quantified an increase in the number of autophagosomes, both by electron microscopy and by LC3B immunohistological or immunofluorescent staining. These results were confirmed by performing classical LC3B turn-over assays and finally by using mRFP-eGFP-LC3 stably expressing ALK+ ALCL Karpas-299 cells (generated in our laboratory), which allowed the quantification of the autophagic flux as an increase in the RFP/GFP ratio [30]. Altogether, these methods demonstrated the ability of therapeutically stressed ALK+ ALCL cells to mount an autophagic response.
- Autophagy induction following the inactivation of mTOR signaling. Mammalian target of rapamycin (mTOR) is an ubiquitously expressed serine/threonine kinase that controls a wide range of key cell functions, including protein synthesis, cell proliferation, and autophagy. The ALK oncogene, acting as a trophic factor, activates mTOR through the MEK/ERK, and to a lesser extent the PI3K/Akt, pathways [31]. As expected, we found that ALK inhibition induced through crizotinib treatment, led to the inactivation of the mTOR pathway, as attested to by the strong reduction in the phosphorylation of the S6 ribosomal protein and 4EBP1 protein, which were both used as read-outs for mTOR activity (unpublished data). In regards to the well-known role of mTOR in the inhibition of the autophagic process, we proposed that mTOR inactivation, as a consequence of ALK inhibition, may account for the activation of the autophagy process [14].
- Characterization of the cytoprotective function of the crizotinib-induced autophagy. To decipher whether this crizotinib-induced autophagy affected cell death or cell survival, we tested the cell viability, clonogenic survival, apoptosis, and ability to form xenografted tumors in vivo. We found that upon combined ALK and autophagy pharmacological inhibition, these drugs had a synergistic effect on the reduction of cell viability; they drove cells towards apoptotic/necrotic cell death, strongly reduced ALK+ Karpas-299 clonogenic survival, and impaired xenograft tumor growth [14]. Thus, we believe that autophagy is activated upon crizotinib treatment as a stress response, and that its protective function relies on the partial impairment of crizotinib-induced concomitant apoptosis. Indeed, we found, both in vitro and in vivo, that autophagy inhibition potentiated crizotinib-induced apoptosis. These results in ALK+ ALCL cells are in line with findings in other cancers and support that the inhibition of cytoprotective autophagy could improve therapeutic outcomes for cancer patients [32,33].
2.1.2. Bulk and Stem-like Cells in ALK+ ALCL
- Intra-tumoral heterogeneity of ALK+ ALCL. Accumulating evidence supports the existence of intra-tumoral heterogeneity in many types of cancer. In ALK+ ALCL cell lines, we found a small but phenotypically distinct cell population that are characterized by their responsiveness to a Sox2 reporter, which we labeled as reporter responsive or RR cells [34]. The bulk cell population is reporter unresponsive and thus labeled RU cells. As the readout for the Sox2 reporter is based on the expression of GFP, RU and RR cells derived from ALK+ ALCL cell lines stably transduced with the Sox2 reporter were readily separated and purified using flow cytometry. RR cells consistently showed a higher level of stem-like features, such as chemo-resistance and tumorigenicity. At the molecular level, RR cells are characterized by a high protein level of MYC, as well as evidence of constitutive activation of the Wnt canonical pathway [35]. We have also provided evidence that this RU/RR dichotomy exists in ALK+ ALCL tumors, since the MYC expression level detectable by immunostaining is heterogeneous among tumor cells, and its level correlates significantly with that of active β-catenin, a marker of the activated Wnt canonical pathway.
- Stem-like cells display higher crizotinib-induced autophagic flux. Using the RU/RR study model, we asked if the cytoprotective effect of crizotinib-induced autophagy is different between the bulk RU cells and stem-like RR cells. To address this question, we first established that the inhibitory concentration of crizotinib at 50% is significantly higher in RR cells than in RU cells (409 nM versus 326 nM). Correlating with this finding, we found that crizotinib triggered a significantly higher autophagic flux in RR cells, as evidenced by some of the assays described in Section 2.1.1. Furthermore, using quantitative RT-PCR, we found that RR cells expressed significantly higher levels of several key autophagy genes, including ULK1, WIPI1, and MAP1LC3B. Importantly, inhibition of autophagy using chloroquine significantly sensitized RR cells to crizotinib, suggesting that autophagy is directly responsible for the cytoprotection against crizotinib. Details of these experiments can be found in reference [18].
- MYC is a key regulator of the RU/RR dichotomy. MYC, one of the four inducible pluripotent stem cell (iPS) factors, is known to be frequently deregulated in human cancers. Nonetheless, its role in the pathobiology of ALK+ ALCL has not been extensively studied. Our previous studies revealed that the protein level of MYC is a key regulator of the RU/RR phenotype [35]. Thus, overexpression of MYC in RU cells effectively resulted in a gain of Sox2 reporter responsiveness, as well as increased chemoresistance and tumorigenicity (i.e., RR-like). Conversely, knockdown of MYC in RR cells using shRNA or pharmacologic agents resulted in a phenotypic conversion into RU-like cells. With this background, we tested if the different protein levels of MYC between RU and RR cells contributed to their difference in the autophagic flux triggered by crizotinib. This turned out to be the case [18]. Specifically, inhibition of MYC in RR cells significantly dampened the crizotinib-induced autophagic response and its cytoprotective effect. The opposite was observed when RU cells were transduced with a MYC expression vector.
2.2. Autophagy Associated with Cell Death in Crizotinib-Treated ALK+ ALCL
2.2.1. Combined ALK and BCL2 Inactivation
- It has been observed for many years, in many studies, that ALK+ ALCL has characteristically low expression levels of B-cell Lymphoma 2 (BCL2) proteins [36,37,38], whereas BCL2 overexpression is a classical feature of cancers, including hematopoietic tumors. Our study was the first to report that either crizotinib-mediated inhibition of ALK or its molecular inactivation by specific ALK-targeted siRNA caused an increase in BCL2 levels, highlighting an ALK-dependent BCL2 repression mechanism that is not yet currently elucidated.
- As the BCL2-family proteins are well-known regulators of all major types of cell death, including apoptosis, necroptosis, and autophagy, we next investigated the role of this increase in BCL2 levels on the cellular response to NPM-ALK inactivation or following NPM-ALK downregulation. To address this question, we specifically downregulated BCL2 by RNA interference and showed, as expected, that BCL2 knockdown (BCL2 KD) led to an increase in crizotinib-induced apoptosis, and also to an increase in the autophagic flux [16].
- To decipher whether apoptosis and autophagy were interconnected or occurred independently following combined ALK and BCL2 inactivation in ALK+ ALCL cells, we first used the pan-caspase inhibitor Z-VAD-FMK. Although the autophagic flux in the presence of crizotinib was not impaired by the addition of Z-VAD-FMK, we observed that this inhibitor partially rescued cell viability in the siCTL-transfected cells and led to a more pronounced, but still not complete, rescue of cell viability in BCL2 KD cells (unpublished data). Moreover, we observed similar results in response to the inhibition of autophagy using a siRNA targeting the ULK1 mRNA. All these data led us to propose that the excessive autophagy observed in BCL2-depleted cells drives cell death through multiple modalities, which include apoptosis. This assumption was corroborated by another set of experiments that we performed with a combination of crizotinib and rapamycin (a well-known mTOR inhibitor and strong inducer of autophagy) to amplify the autophagic response without interfering with the BCL2 protein content. We found that the potentiation of autophagy (upon combined treatment) resulted in a strong loss of viability, but with no potentiation of apoptosis, suggesting that another cell death pathway may account for cell killing.
- The mechanistic link between BCL2 inhibition and the overactivation of autophagy is complex. Multiple observations have provided strong support for the prevailing model, in which BCL2 inhibits autophagy through direct interaction with a BH3-like domain of BECN1 [39,40,41]. More recently, Vaux and co-workers [42,43] demonstrated that the inhibition of autophagy by BCL2 is indirect and due to activation of BAX and BAK and thus depends on the presence of an intact intrinsic apoptosis pathway. We therefore performed an array of preliminary experiments to understand which one of these mechanisms could account for the overactivation of autophagy observed in our model. To test the model proposed by Vaux’s group, we first used different agents to interfere with the apoptotis pathway (use of the Z-VAD-FMK compound or use of a siRNA targeting the BAK mRNA) and found that inhibiting apoptosis did not impair the potentiation of the autophagic flux observed in cells that received a combined crizonitib/BCL2 KD treatment (unpublished data). We also revealed that, chronologically, autophagic flux induction and potentiation of this flux in response to combined rapamycin/crizotinib treatment is observed prior to the marked decrease in cell viability and the detection of apoptotic cell death, as revealed by Annexin V staining. Altogether, these data suggest a model in which pro-apoptotic effectors (such as BAK) and a functional apoptosis pathway are not a prerequisite for autophagy induction upon crizotinib treatment and autophagy potentiation, either through BCL2 molecular downregulation or rapamycin combined treatment.
2.2.2. Combined ALK and RAF1 Inactivation
- Rapidly Accelerated Fibrosarcoma 1 (RAF1) is a key signal transduction protein with serine/threonine kinase activity. A previous study reported that RAF1 downregulation using a RNA interference approach did not impair ALK+ ALCL cell growth and proliferation [44], a result in line with emerging literature attributing MEK/ERK independent functions to RAF1 [45,46]. Of interest, RAF1 downregulation in a conditional mouse model for RAF1-induced lung tumorigenesis led to tumor regression associated with an enhanced autophagic flux [47]. Based on this literature, we formulated a hypothesis in which RAF1 could restrain crizotinib-induced autophagy in ALK+ ALCL through the serine/threonine inhibitory phosphorylation of a key initiator of autophagy, such as ULK1, as it works for the serine/threonine kinase mTOR.
- To investigate whether RAF1 could phosphorylate ULK1, we first performed specific phospho-serine757 Western blotting on lysates from Karpas-299 cells treated or not with single or combined ALK (using crizotinib) and RAF1 inhibitions (using vemurafenib or a siRNA targeting the RAF1 mRNA (KD approach)), or using Karpas-299 cells invalidated for RAF1 expression using the CRISPR/Cas9 system (knockout (KO) approach). Second, we performed in vitro kinase assays using recombinant human RAF1 and ULK1 proteins, in the presence or absence of vemurafenib. Collectively, our results produced evidence supporting the involvement of RAF1 in the inhibitory phosphorylation of ULK1 on its serine 757 residue [17].
- We then evaluated how RAF1 inactivation could affect the autophagic flux, viability, and apoptosis of crizotinib-treated ALK+ ALCL cells. We found that RAF1 pharmacological inhibition or molecular downregulation potentiated the crizotinib-induced autophagic flux, which was associated with a loss in cell viability and increased apoptosis [17].
3. Autophagy as a Great Orchestrator of Cell Fate
3.1. Molecular Mechanisms Potentially Underlying Pro-Survival Autophagy in Crizotinib-Treated ALK+ ALCL
3.1.1. Impairment of Apoptosis
- In line with our findings, several studies have highlighted the molecular interplay between autophagy and apoptosis, which provides explanations for the synergy of anti-cancer therapies combining apoptosis-inducing drugs and autophagy inhibitors. As an example, Hou et al. reported the sequestration and degradation of active caspase-8 through autophagy in apoptosis-deficient colon cancer cells submitted to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Autophagy inhibition in these settings increased caspase 8 enzymatic activity and promoted apoptotic cell death [56].
- The emerging literature on mitophagy, which is a selective form of autophagy allowing the clearance of defective mitochondria, highlights its role in tumor suppression. Indeed, by limiting the production of pro-apoptotic ROS, mitophagy acts as a survival mechanism, in both growing and therapeutically challenged tumors [57]. This is reminiscent of previous studies showing that impairing macro-autophagy led to defects in mitochondrial homeostasis, thereby increasing ROS levels and DNA damages, and resulting in apoptosis [58,59]. Consequently, inhibiting cytoprotective autophagy/mitophagy in cancer cells leads to oxidative stress-induced apoptotic cell death.
- Recently, a global understanding of how autophagy inhibition could sensitize tumor cells to apoptosis was provided by the group of A. Thorburn [60]. Autophagy, through degradation of the FOXO3a transcription factor, regulates the levels of the pro-apoptotic PUMA protein [61]. A high autophagy activity therefore protects cells by lowering the cellular levels of PUMA, whereas a low autophagy or inhibition of autophagy renders the cells prone to apoptosis. Thus, autophagy may control cancer cell fate by regulating their apoptosis threshold.
3.1.2. Autophagy and Cancer Stemness
- Autophagy is important in the maintenance of cancer stem cells. Cancer stem cells (CSCs) are known for their propensity to maintain a dormant state and a high level of chemoresistance, which is believed to contribute to treatment failure and disease relapses in cancer patients. The link between cancer stemness and autophagy has been described in a few cancer types [62]. High levels of autophagic flux have been identified in CSCs isolated from cancers of the endometrium, brain, ovary, colon, and breast [63,64,65,66,67]. Importantly, there is accumulating evidence that autophagy plays a crucial role in the maintenance and survival of CSCs. For instance, depletion of Beclin1 in breast CSCs was found to significantly decrease their tumorigenicity in a xenograft mouse model [68]. In another study using ovarian CSCs, the authors found that pharmacologic inhibition of autophagy resulted in decreased chemoresistance, self-renewal ability, and tumorigenicity [65]. In chronic myeloid leukemia, knockdown of ATG4B in CD34-positive stem cell population sensitized these cells to the tyrosine kinase inhibitor imatinib [69]. Similar observations were made in a study using acute myeloid leukemia [70]. As mentioned in Section 2.1.2, inhibition of autophagy using pharmacologic agents (such as chloroquine) and specific siRNA targeting the ATG7 or Beclin1 mRNAs in ALK+ ALCL significantly lowered the level of crizotinib resistance in the stem-like cell subset.
- Stem-like cells in ALK+ ALCL can be induced. Autophagic flux can be triggered in response to a diversity of stressful conditions, such as nutrient shortage, hypoxia, chemotherapeutic challenge, and oxidative stress, with the goals of survival and self-preservation. Using the RU/RR study model of ALK+ ALCL, we previously found that adverse conditions such as oxidative stress can induce the conversion of RU cells into RR-like cells [71]. Specifically, a small fraction of RU cells treated with hydrogen peroxide acquired Sox2 reporter responsiveness, with a higher level of chemoresistance. Correlating with these phenotypic changes, the MYC protein level substantially increased in these RR-like cells. Although autophagic flux was not assessed in this study of cancer plasticity, it is highly likely that autophagic flux also increased along with the acquisition of cancer stemness. It would be of great interest to examine if autophagy is necessary for this phenotypic conversion in ALK+ ALCL. Future experiments will test if the inhibition of autophagy can abrogate the conversion of RU cells into RR-like cells.
- MYC as a regulator of autophagy and cancer stemness. While the protein level of MYC is a key regulator of the RU/RR dichotomy in ALK+ ALCL, accumulating evidence also suggests that MYC is a regulator of autophagy. As mentioned in Section 2.1.2, modulation of the MYC protein level significantly changed the autophagic response in ALK+ ALCL cells. In keeping with our findings, a handful of published studies that directly examined the link between MYC and autophagy shared the same conclusion. One of the first such studies was published by Toh et al. in 2013 [72] and showed that depletion of MYC in HeLa cells impaired autophagosome formation and decreased the level of LC3B-II. The same study also highlighted the importance of JNK1/BCL2 phosphorylation in the regulation of autophagy by MYC. Specifically, MYC promotes the phosphorylation/activation of JNK, which then phosphorylates BCL2 and facilitates the release of Beclin1 from the sequestration of BCL2. A more recent study showed that miR-27b-3p, a microRNA species that is regulated by MYC, can increase autophagy and chemoresistance in colorectal cancer cells via ATG10 [73]. Taken together, we hypothesize that increased autophagic flux induced by adverse conditions promotes cancer stemness and a high protein level of MYC in ALK+ ALCL. The resulting high MYC level stimulates further autophagic activity via the JNK/BCL2 and miRNA pathways, thus creating a positive feedback loop.
3.2. Molecular Mechanisms Potentially Underlying Pro-Death Autophagy in Crizotinib-Treated ALK+ ALCL
3.2.1. Selective Removal of Pro-Survival Substrates or Anti-Apoptotic Factors
- The first autophagic substrate we could consider is the NPM-ALK oncogene itself, since it drives and sustains lymphomagenesis. The hypothesis of NPM-ALK degradation through excessive autophagy is interesting and relevant, since previous reports did indeed show the autophagosomal relocation and degradation of other fusion oncogenes (BCR-ABL, PML-RARA, FLT3-ITD) in different hematological malignancies upon anti-cancer treatment [74,75,76].
- Another interesting substrate, which autophagic degradation was reported to lead to tumor cell death, is the reactive oxygen species (ROS) scavenger catalase [77]. Indeed, such autophagy-mediated ROS accumulation accounts for membrane lipid oxidation, loss of membrane integrity, and subsequent cell demise. Interestingly, ALK+ ALCL cells were found to produce a high level of ROS by a pathway involving lipoxygenases (LOX) [78]. Thus, it is tempting to speculate that excessive autophagy by degrading ROS catalase could further increase the ROS content in ALK+ ALCL cells until reaching toxic levels responsible for the subsequent cell death. It would be interesting to determine if ROS catalase could be detected in autophagosomes purified from ALK+ ALCL cells.
- The same reasoning could apply to the excessive autophagy-mediated degradation of ferritin, which could induce ferroptosis cell death through the accumulation of labile iron and ROS [79,80], as described in erastin-treated MEFs [81]. Thus, the occurrence of autophagy and subsequent degradation of ferroptosis repressors in ALK+ ALCL should be investigated. This could be particularly relevant, since ALK+ ALCL cell lines and primary tumors have been shown to be resistant to ferroptosis because of the accumulation of squalene, a lipophilic metabolite, in cell membranes and lipid droplets [82].
- Finally, the autophagic degradation of anti-apoptotic proteins, such as Fap-1, which is an inhibitor of Fas-mediated apoptosis (as described in BJAB lymphoma cells [83]), could account for cell death and its occurrence in ALK+ ALCL should be investigated.
3.2.2. Scaffold for Cell Death Complexes
- Scaffold for apoptosis. Components of the autophagy machinery have been shown to serve as a platform for the apoptotis machinery. As an example, Young et al. demonstrated in MEFs treated with sphingosine kinase inhibitor (SKI) that ATG5- and ATG16L-positive autophagosomal membranes were required to form an efficient intracellular death-inducing signaling complex (iDISC) containing Fas-associated protein with death domain (FADD) and caspase-8 homocomplex [85]. In the same vein, Laussman et al. reported that ATG5 and autophagosome formation contributed to caspase-8 activation following proteasome inhibition in BCL2-overexpressing HeLa cervical cancer cells [86].
- Scaffold for necroptosis. A similar scaffold function was described for the necroptosis machinery. Indeed, two studies reported that Obatoclax, an antagonist of BCL2 family proteins, triggers cell death via autophagy through the recruitment of components of the necrosome, such as FADD, RIP1, and RIP3 to autophagosomal membranes [87,88]. Another study demonstrated that Sorafenib, a multi-tyrosine kinase inhibitor, induced autophagy-dependent cell death in ATG5-deficient prostate cancer cells [89]. In this model Sorafenib induced the interaction between p62 and RIPK1, leading to cell death by necroptosis.
3.2.3. Reconstitution of Immune Surveillance
- NPM-ALK is an oncoantigen. Over the last two decades, several publications have demonstrated the immunogenicity of the NPM-ALK oncogene, which can induce in patients, both the production of anti-ALK antibodies [94,95,96], and a T cell immune response against ALK [95,97,98,99,100]. Further validating NPM-ALK as an oncoantigen, the group of Chiarle et al. demonstrated in a mouse model the efficiency of a vaccination therapy using truncated ALK DNA [101].
- Evidence for NPM-ALK-induced escape from immune surveillance. Since the NPM-ALK oncogene-elicited immune responses failed to prevent ALK lymphomagenesis, it raised the question of the acquisition of immune escape mechanisms. Indeed, tumor cells can protect themselves from the immune system through several mechanisms, three of these being developed below because of their occurrence in ALK+ ALCL cells: (i) by the epigenetic downregulation of CD48 expression and subsequent attenuation of nature killer (NK) cell-mediated cytotoxicity against neoplastic ALK+ ALCL cells [102]; (ii) by limiting the presentation of tumor-specific antigen through the downregulation of human leucocyte antigen (HLA) molecules; a recent study reported that the inhibition of the ALK oncogene induced elevated transcript and protein expression of HLA class I, consistent with its increased representation at the cell surface [103], and thus, the ALK oncogene may allow lymphoma cells to evade the immune system by downregulating the expression of HLA class I molecules; (iii) by expressing immunosuppressive factors such as immune checkpoints, which inhibit the activity of tumor-associated T cells. In this framework, NPM-ALK was found to induce the expression of PD-L1 [104,105] through the activation of STAT3 and GRB2/SOS1 signaling networks, and the downstream involvement of the IRF4 and BATF3 transcription factors [104,106]. These important signal transduction studies highlight that NPM-ALK lymphoma can escape immune surveillance and set up the basis for clinical trials using drugs targeting the PD-1/PD-L1 axis. Two recent clinical studies reported the efficacy of anti-PD1 therapy with nivolumab in patients with ALK+ ALCL refractory to chemotherapy and ALK inhibitors [107,108]. Altogether, these fundamental and clinical findings support the development of immune-based therapies for controlling the disease.
- The role of autophagy and autophagosomes in antigen presentation and the immune control of tumors. Autophagy could likely contribute in many ways to the success of future ALK+ ALCL immunotherapies. First, with relevance to the studies showing the NPM-ALK mediated control of HLA class 1 [103] and PD-L1 cell surface expressions [104], autophagy was shown in other settings to facilitate the expression of MHC I molecules [109] and to participate in the selective degradation of the PD-L1 immune-checkpoint [110]. Second, regarding ALK tumor antigen presentation, one can hypothesize that autophagy may potentially participate in NPM-ALK epitope processing and delivery to MHC class I and II molecules. Indeed, several studies reported that the MHC class II loading compartment received cytoplasmic antigen from autophagosomes [111,112] and autophagy was shown in some cases to contribute to the loading of intracellular antigens onto MHC class I molecules [92,113]. It is thus important in this context to investigate whether NPM-ALK could be detected in autophagosomes. Moreover, such potential NPM-ALK containing autophagosomes, once released from dying ALK lymphoma cells, could be captured by dendritic cells in the tumor microenvironment and their content could therefore be redirected to MHC class I complex for processing and cross-presentation, as described previously in other cancers [114]. If so, the development of a NPM-ALK lymphoma autophagosome-based tumor vaccine warrants consideration, as proposed for other tumors [115]. Finally, since autophagosomes are known to contain ATP immunostimulatory molecules, as well as several proteins acting to alarm the immune system [116], autophagy induction in ALK+ ALCL cells and the release of their autophagosome content in the tumor microenvironment could potentially contribute to the immunogenic cell death (ICD) of the lymphoma cells. In line with this hypothesis, it is noteworthy that crizotinib treatment in EML4-ALK lung cancer cells was shown to induce the classical features of ICD [117]. Thus, whether these findings are transposable to ALK+ ALCL cells is a burning question.
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugières, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Lai, R.; Ingham, R.J. The pathobiology of the oncogenic tyrosine kinase NPM-ALK: A brief update. Ther. Adv. Hematol. 2013, 4, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-Anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef]
- Mologni, L. Inhibitors of the anaplastic lymphoma kinase. Expert Opin. Investig. Drugs 2012, 21, 985–994. [Google Scholar] [CrossRef]
- Crescenzo, R.; Inghirami, G. Anaplastic lymphoma kinase inhibitors. Curr. Opin. Pharmacol. 2015, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [PubMed]
- Larose, H.; Burke, G.A.A.; Lowe, E.J.; Turner, S.D. From bench to bedside: The past, present and future of therapy for systemic paediatric ALCL, ALK+. Br. J. Haematol. 2019, 185, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149. [Google Scholar] [CrossRef]
- Frentzel, J.; Sorrentino, D.; Giuriato, S. Targeting autophagy in ALK-associated cancers. Cancers 2017, 9, 161. [Google Scholar] [CrossRef]
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; Al Saati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL2 elevation in ALK-positive anaplastic large cell lymphoma triggers autophagy associated with cell death. Haematologica 2019, 104, 1428–1439. [Google Scholar] [CrossRef]
- Sorrentino, D.; Frentzel, J.; Mitou, G.; Blasco, R.B.; Torossian, A.; Hoareau-Aveilla, C.; Pighi, C.; Farcé, M.; Meggetto, F.; Manenti, S.; et al. High levels of mir-7-5p potentiate crizotinib-induced cytokilling and autophagic flux by targeting raf1 in npm-alk positive lymphoma cells. Cancers 2020, 12, 2951. [Google Scholar] [CrossRef]
- Shang, C.; Hassan, B.; Haque, M.; Song, Y.; Li, J.; Liu, D.; Lipke, E.; Chen, W.; Giuriato, S.; Lai, R. Crizotinib resistance mediated by autophagy is higher in the stem-like cell subset in ALK-positive anaplastic large cell lymphoma, and this effect is MYC-dependent. Cancers 2021, 13, 181. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 521–527. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 439–458. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 1–18. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Djavaheri-Mergny, M.; Pattingre, S.; Giuriato, S. The yin and the yang of autophagy in cancer cells. Medecine/Sciences 2017, 33, 328–334. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef]
- Marzec, M.; Kasprzycka, M.; Liu, X.; El-Salem, M.; Halasa, K.; Raghunath, P.N.; Bucki, R.; Wlodarski, P.; Wasik, M.A. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene 2007, 26, 5606–5614. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Mukhopadhyay, S.; Das, D.N.; Sinha, N.; Naik, P.P.; Bhutia, S.K. Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 2015, 39, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Gelebart, P.; Hegazy, S.A.; Wang, P.; Bone, K.M.; Anand, M.; Sharon, D.; Hitt, M.; Pearson, J.D.; Ingham, R.J.; Ma, Y.; et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, H.F.; Gupta, N.; Alshareef, A.; Wang, Q.; Huang, Y.H.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Lai, R. A positive feedback loop involving the Wnt/β-catenin/MYC/Sox2 axis defines a highly tumorigenic cell subpopulation in ALK-positive anaplastic large cell lymphoma. J. Hematol. Oncol. 2016, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Villalva, C.; Bougrine, F.; Delsol, G.; Brousset, P.; Diepstra, A.; Kluiver, J.; Visser, L.; Peh, S.-C.; Lim, M.; Kamps, W.A.; et al. Bcl-2 expression in anaplastic large cell lymphoma. Am. J. Pathol. 2001, 158, 1889–1890. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Sarris, A.H.; Herling, M.; Ford, R.J.; Cabanillas, F.; McDonnell, T.J.; Medeiros, L.J. Differential Expression of BCL-2 Family Proteins in ALK-Positive and ALK-Negative Anaplastic Large Cell Lymphoma of T/Null-Cell Lineage. Am. J. Pathol. 2001, 159, 527–535. [Google Scholar] [CrossRef]
- Rust, R. High expression of Mcl-1 in ALK positive and negative anaplastic large cell lymphoma. J. Clin. Pathol. 2005, 58, 520–524. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Xiao, H.L.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Decuypere, J.-P.; Parys, J.B.; Bultynck, G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Heinlein, M.; Huang, D.C.S.; Vaux, D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Vaux, D.L. BCL2 and related prosurvival proteins require BAK1 and BAX to affect autophagy. Autophagy 2014, 10, 1474–1475. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Kasprzycka, M.; Liu, X.; Raghunath, P.N.; Wlodarski, P.; Wasik, M.A. Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene 2007, 26, 813–821. [Google Scholar] [CrossRef]
- Ehrenreiter, K.; Kern, F.; Velamoor, V.; Meissl, K.; Galabova-Kovacs, G.; Sibilia, M.; Baccarini, M. Raf-1 Addiction in Ras-Induced Skin Carcinogenesis. Cancer Cell 2009, 16, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Doma, E.; Rupp, C.; Varga, A.; Kern, F.; Riegler, B.; Baccarini, M. Skin tumorigenesis stimulated by raf inhibitors relies upon raf functions that are dependent and independent of ERK. Cancer Res. 2013, 73, 6926–6937. [Google Scholar] [CrossRef]
- Ceteci, F.; Xu, J.; Ceteci, S.; Zanucco, E.; Thakur, C.; Rapp, U.R. Conditional Expression of Oncogenic C-RAF in Mouse Pulmonary Epithelial Cells Reveals Differential Tumorigenesis and Induction of Autophagy Leading to Tumor Regression. Neoplasia 2011, 13, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R.J. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef]
- Machnicki, M.M.; Stoklosa, T. BRAF—A new player in hematological neoplasms. Blood Cells Mol. Dis. 2014, 53, 77–83. [Google Scholar] [CrossRef]
- Raje, N.; Chau, I.; Hyman, D.M.; Ribrag, V.; Blay, J.-Y.; Tabernero, J.; Elez-Fernandez, M.E.; Wolf, J.S.; Sirzen, F.; Yee, A.; et al. Vemurafenib (VEM) in Relapsed Refractory Multiple Myeloma Harboring BRAFV600 Mutations (V600m): A Cohort of the Histology-Independent VE-Basket Study. Blood 2015, 126, 4263. [Google Scholar] [CrossRef]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, S.; Glimm, H.; Andrulis, M.; von Kalle, C.; Ho, A.D.; Zenz, T. BRAF Inhibition in Refractory Hairy-Cell Leukemia. N. Engl. J. Med. 2012, 366, 2038–2040. [Google Scholar] [CrossRef] [PubMed]
- Peyrade, F.; Re, D.; Ginet, C.; Gastaud, L.; Allegra, M.; Ballotti, R.; Thyss, A.; Zenz, T.; Auberger, P.; Robert, G. Low-dose vemurafenib induces complete remission in a case of hairy-cell leukemia with a V600E mutation. Haematologica 2013, 98, e20. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Quintanilla-Martinez, L.; Datta, V.; Lopategui, J.; Garshfield, G.; Nathwani, B.N. Identification of the V600D mutation in Exon 15 of the BRAF oncogene in congenital, benign langerhans cell histiocytosis. Genes Chromosom. Cancer 2013, 52, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Haroche, J.; Cohen-Aubart, F.; Emile, J.F.; Arnaud, L.; Maksud, P.; Charlotte, F.; Cluzel, P.; Drier, A.; Hervier, B.; Benameur, N.; et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013, 121, 1495–1500. [Google Scholar] [CrossRef]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tang, B.; Yu, P.W.; Tang, B.; Hao, Y.X.; Lei, X.; Luo, H.X.; Zeng, D.Z. Autophagy Protects against Oxaliplatin-Induced Cell Death via ER Stress and ROS in Caco-2 Cells. PLoS ONE 2012, 7, e51076. [Google Scholar] [CrossRef]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef]
- Tompkins, K.D.; Thorburn, A. Regulation of Apoptosis by Autophagy to Enhance Cancer Therapy. Yale J. Biol. Med. 2019, 92, 707–718. [Google Scholar]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565.e3. [Google Scholar] [CrossRef]
- Mandhair, H.K.; Arambasic, M.; Novak, U.; Radpour, R. Molecular modulation of autophagy: New venture to target resistant cancer stem cells. World J. Stem Cells 2020, 12, 303–322. [Google Scholar] [CrossRef]
- Ran, X.; Zhou, P.; Zhang, K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int. J. Oncol. 2017, 51, 644–656. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed]
- Therachiyil, L.; Haroon, J.; Sahir, F.; Siveen, K.S.; Uddin, S.; Kulinski, M.; Buddenkotte, J.; Steinhoff, M.; Krishnankutty, R. Dysregulated Phosphorylation of p53, Autophagy and Stemness Attributes the Mutant p53 Harboring Colon Cancer Cells Impaired Sensitivity to Oxaliplatin. Front. Oncol. 2020, 10, 1744. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Shen, S.; Zhang, Y.J.; Xu, C.F.; Cao, Z.T.; Wen, L.P.; Wang, J. Nanoparticle-facilitated autophagy inhibition promotes the efficacy of chemotherapeutics against breast cancer stem cells. Biomaterials 2016, 103, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Rothe, K.; Lin, H.; Lin, K.B.L.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef]
- Wu, C.; Gupta, N.; Huang, Y.H.; Zhang, H.F.; Alshareef, A.; Chow, A.; Lai, R. Oxidative stress enhances tumorigenicity and stem-like features via the activation of the Wnt/β-catenin/MYC/Sox2 axis in ALK-positive anaplastic large-cell lymphoma. BMC Cancer 2018, 18, 361. [Google Scholar] [CrossRef] [PubMed]
- Toh, P.P.C.; Luo, S.; Menzies, F.M.; Raskó, T.; Wanker, E.E.; Rubinsztein, D.C. Myc inhibition impairs autophagosome formation. Hum. Mol. Genet. 2013, 22, 5237–5248. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, J.; Zhou, L.; Han, J.; Liu, R.; Zhang, H.; Ning, T.; Gao, Z.; Liu, B.; Chen, X.; et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics 2020, 10, 1981–1996. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Bjoras, M.; Boe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.H.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef]
- Thornber, K.; Colomba, A.; Ceccato, L.; Delsol, G.; Payrastre, B.; Gaits-Iacovoni, F. Reactive oxygen species and lipoxygenases regulate the oncogenicity of NPM-ALK-positive anaplastic large cell lymphomas. Oncogene 2009, 28, 2690–2696. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.H.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, B.; Von Der Wall, E.; Hummel, M.; Dü Rkop, H. RIP1 expression is necessary for CD30-mediated cell death induction in anaplastic large-cell lymphoma cells. Lab. Investig. 2013, 93, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.-D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Laussmann, M.A.; Passante, E.; Düssmann, H.; Rauen, J.A.; Würstle, M.L.; Delgado, M.E.; Devocelle, M.; Prehn, J.H.M.; Rehm, M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011, 18, 1584–1597. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef]
- Sulkshane, P.; Teni, T. BH3 mimetic Obatoclax (GX15-070) mediates mitochondrial stress predominantly via MCL-1 inhibition and induces autophagy-dependent necroptosis in human oral cancer cells. Oncotarget 2017, 8, 60060–60079. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Baltatzis, G.; Fonseca, P.; Rodriguez, P.; Gogvadze, V.; Lennartsson, L.; Björklund, A.C.; Zhivotovsky, B.; Grandér, D.; et al. Sorafenib-induced defective autophagy promotes cell death by necroptosis. Oncotarget 2015, 6, 37066–37082. [Google Scholar] [CrossRef]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 8627, 2163–2170. [Google Scholar] [CrossRef]
- Pulford, K.; Falini, B.; Banham, A.H.; Codrington, D.; Roberton, H.; Hatton, C.; Mason, D.Y. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood 2000, 96, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Ait-Tahar, K.; Cerundolo, V.; Banham, A.H.; Hatton, C.; Blanchard, T.; Kusec, R.; Becker, M.; Smith, G.L.; Pulford, K. B and CTL responses to the ALK protein in patients with ALK-positive ALCL. Int. J. Cancer 2006, 118, 688–695. [Google Scholar] [CrossRef]
- Mussolin, L.; Bonvini, P.; Ait-Tahar, K.; Pillon, M.; Tridello, G.; Buffardi, S.; Lombardi, A.; Pulford, K.; Rosolen, A. Kinetics of humoral response to ALK and its relationship with minimal residual disease in pediatric ALCL. Leukemia 2009, 23, 400–402. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Passoni, L.; Scardino, A.; Bertazzoli, C.; Gallo, B.; Coluccia, A.M.; Lemonnier, F.A.; Kosmatopoulos, K.; Gambacorti-Passerini, C. ALK as a novel lymphoma-associated tumor antigen: Identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood 2002, 99, 2100–2106. [Google Scholar] [CrossRef]
- Passoni, L.; Gallo, B.; Biganzoli, E.; Stefanoni, R.; Massimino, M.; Di Nicola, M.; Gianni, A.M.; Gambacorti-Passerini, C. In vivo T-cell immune response against anaplastic lymphoma kinase in patients with anaplastic large cell lymphomas. Haematologica 2006, 91, 48–55. [Google Scholar]
- Ait-Tahar, K.; Barnardo, M.C.N.; Pulford, K. CD4 T-helper responses to the anaplastic lymphoma kinase (ALK) protein in patients with ALK-positive anaplastic large-cell lymphoma. Cancer Res. 2007, 67, 1898–18901. [Google Scholar] [CrossRef]
- Singh, V.K.; Werner, S.; Hackstein, H.; Lennerz, V.; Reiter, A.; Wölfel, T.; Damm-Welk, C.; Woessmann, W. Analysis of nucleophosmin–anaplastic lymphoma kinase (NPM-ALK)-reactive CD8+ T cell responses in children with NPM-ALK+ anaplastic large cell lymphoma. Clin. Exp. Immunol. 2016, 186, 96–105. [Google Scholar] [CrossRef]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef]
- Wu, R.; Ivan, E.; Sahasrabuddhe, A.A.; Shaw, T.; Mullighan, C.G.; Leventaki, V.; Elenitoba-Johnson, K.S.J.; Lim, M.S. Epigenetic Modulation of CD48 By NPM-ALK Promotes Immune Evasion in ALK+ ALCL. Blood 2019, 134, 1510. [Google Scholar] [CrossRef]
- Oh, C.Y.; Klatt, M.G.; Bourne, C.; Dao, T.; Dacek, M.M.; Brea, E.J.; Mun, S.S.; Chang, A.Y.; Korontsvit, T.; Scheinberg, D.A. ALK and RET inhibitors promote HLA Class i antigen presentation and unmask new antigens within the tumor immunopeptidome. Cancer Immunol. Res. 2019, 7, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Hong, Y.W.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Andorsky, D.J.; Yamada, R.E.; Said, J.; Pinkus, G.S.; Betting, D.J.; Timmerman, J.M. Programmed death ligand 1 is expressed by non-Hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin. Cancer Res. 2011, 17, 4232–4244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Song, Z.; Wang, H.B.; Lang, L.; Yang, Y.Z.; Xiao, W.; Webster, D.E.; Wei, W.; Barta, S.K.; Kadin, M.E.; et al. A novel model of controlling PD-L1 expression in ALK1 anaplastic large cell lymphoma revealed by CRISPR screening. Blood 2019, 134, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Hebart, H.; Lang, P.; Woessmann, W. Nivolumab for refractory anaplastic large cell lymphoma: A case report. Ann. Intern. Med. 2016, 165, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Abbou, S.; Minard-Colin, V.; Geoerger, B.; Scoazec, J.Y.; Vassal, G.; Jaff, N.; Heuberger, L.; Valteau-Couanet, D.; Brugieres, L. Efficacy of nivolumab in a patient with systemic refractory ALK+ anaplastic large cell lymphoma. Pediatr. Blood Cancer 2018, 65, e26902. [Google Scholar] [CrossRef]
- Li, B.; Lei, Z.; Lichty, B.D.; Li, D.; Zhang, G.M.; Feng, Z.H.; Wan, Y.; Huang, B. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-γ in B16 melanoma cells. Cancer Immunol. Immunother. 2010, 59, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Müller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.-X.; Yang, G.; Hao, F.; Urba, W.J.; Hu, H.-M. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008, 68, 6889–6895. [Google Scholar] [CrossRef]
- Page, D.B.; Hulett, T.W.; Hilton, T.L.; Hu, H.-M.; Urba, W.J.; Fox, B.A. Glimpse into the future: Harnessing autophagy to promote anti-tumor immunity with the DRibbles vaccine. J. Immunother. Cancer 2016, 4, 1–5. [Google Scholar] [CrossRef]
- Yi, Y.; Zhou, Z.; Shu, S.; Fang, Y.; Twitty, C.; Hilton, T.L.; Aung, S.; Urba, W.J.; Fox, B.A.; Hu, H.-M.; et al. Autophagy-assisted antigen cross-presentation: Autophagosome as the argo of shared tumor-specific antigens and DAMPs. Oncoimmunology 2012, 1, 976–978. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S.; Yamazaki, T.; Iribarren, K.; et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Younes, A. Precision therapy for lymphoma—current state and future directions. Nat. Rev. Clin. Oncol. 2014, 11, 585–596. [Google Scholar] [CrossRef]
- Moia, R.; Patriarca, A.; Schipani, M.; Ferri, V.; Favini, C.; Sagiraju, S.; Al Essa, W.; Gaidano, G. Precision medicine management of chronic lymphocytic leukemia. Cancers 2020, 12, 642. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, D.; Brimer, T.; Hussaini, M.; Sokol, L. Genomics of Peripheral T-Cell Lymphoma and Its Implications for Personalized Medicine. Front. Oncol. 2020, 10, 898. [Google Scholar] [CrossRef]
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kim, J.W.; Jung, W.J.; Koh, Y.; Yoon, S.S. Crizotinib in combination with everolimus synergistically inhibits proliferation of anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. Treat. 2018, 50, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Koh, Y.; Yoon, S.S. Synergistic effect of alectinib and everolimus on ALK-positive anaplastic large cell lymphoma growth inhibition. Anticancer Res. 2020, 40, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef] [PubMed]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Mastini, C.; Martinengo, C.; Inghirami, G.; Chiarle, R. Anaplastic lymphoma kinase: An oncogene for tumor vaccination. J. Mol. Med. 2009, 87, 669–777. [Google Scholar] [CrossRef] [PubMed]
- Stadler, S.; Singh, V.K.; Knörr, F.; Damm-Welk, C.; Woessmann, W. Immune response against ALK in children with ALK-positive anaplastic large cell lymphoma. Cancers 2018, 10, 114. [Google Scholar] [CrossRef]


{kind=link}
| Therapeutic Agent(s) | Target in the Autophagic Process | Autophagy Function | Study Model | References |
|---|---|---|---|---|
| Crizotinib + Chloroquine | Autolysosomal degradation | Pro-survival | ALK+ ALCL cells Xenografted mice ALK+ ALCL cells Stem-like ALK+ ALCL | [14,18] |
| Crizotinib + BCL2 KD | BECN1/BCL2 complex (not yet demonstrated in ALK+ ALCL) | Pro-death | ALK+ ALCL cells Xenografted mice | [16] |
| Crizotinib + RAF1 KD/KO or Crizotinib + Vemurafenib | ULK1 phosphorylation | Pro-death | ALK+ ALCL cells | [17] |
| Crizotinib + Temsirolimus | mTOR | NI | ALK+ ALCL cells Xenografted mice | [121] |
| Crizotinib + Everolimus | mTOR | NI | ALK+ ALCL cells | [122] |
| Alectinib + Everolimus | mTOR | NI | ALK+ ALCL cells | [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinos, E.; Lai, R.; Giuriato, S. The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise. Cells 2021, 10, 2517. https://doi.org/10.3390/cells10102517
Espinos E, Lai R, Giuriato S. The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise. Cells. 2021; 10(10):2517. https://doi.org/10.3390/cells10102517
Chicago/Turabian StyleEspinos, Estelle, Raymond Lai, and Sylvie Giuriato. 2021. "The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise" Cells 10, no. 10: 2517. https://doi.org/10.3390/cells10102517
APA StyleEspinos, E., Lai, R., & Giuriato, S. (2021). The Dual Role of Autophagy in Crizotinib-Treated ALK+ ALCL: From the Lymphoma Cells Drug Resistance to Their Demise. Cells, 10(10), 2517. https://doi.org/10.3390/cells10102517