
Honokiol Affects Stem Cell Viability by Suppressing Oncogenic YAP1 Function to Inhibit Colon Tumorigenesis

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Proliferation and Apoptosis Assays
2.3. Clonogenicity Assay
2.4. Cell Cycle Analyses
2.5. Real-Time Reverse-Transcription Polymerase Chain Reaction Analysis
2.6. Flow Cytometric Analyses for DCLK1
2.7. Colonosphere Assay
2.8. DCLK1 Kinase Assay
2.9. Molecular Docking
2.10. Immunoprecipitation
2.11. Western Blot Analysis
2.12. Mouse Model of Colitis-Associated Cancer
2.13. Immunohistochemistry
2.14. Statistical Analysis
3. Results
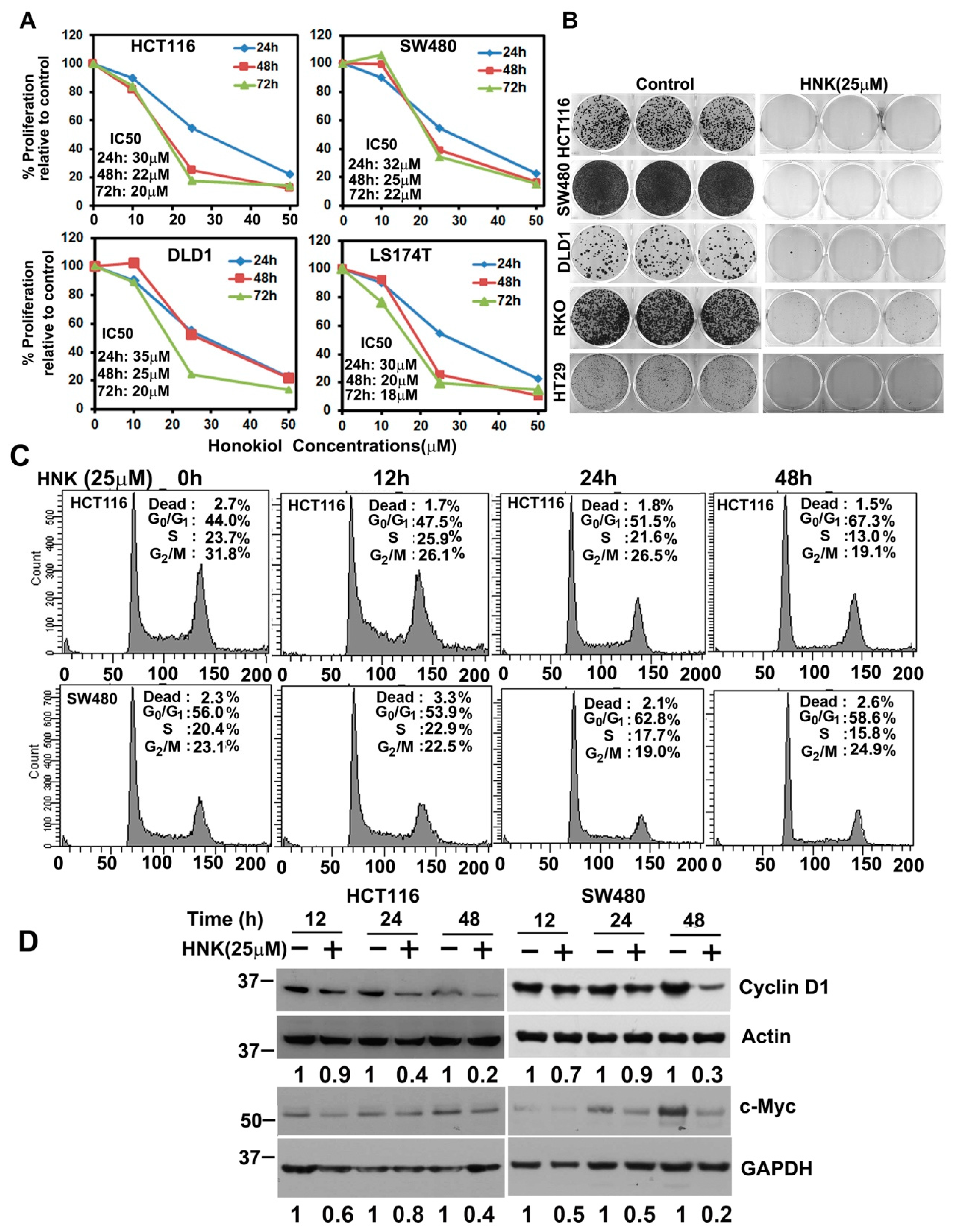
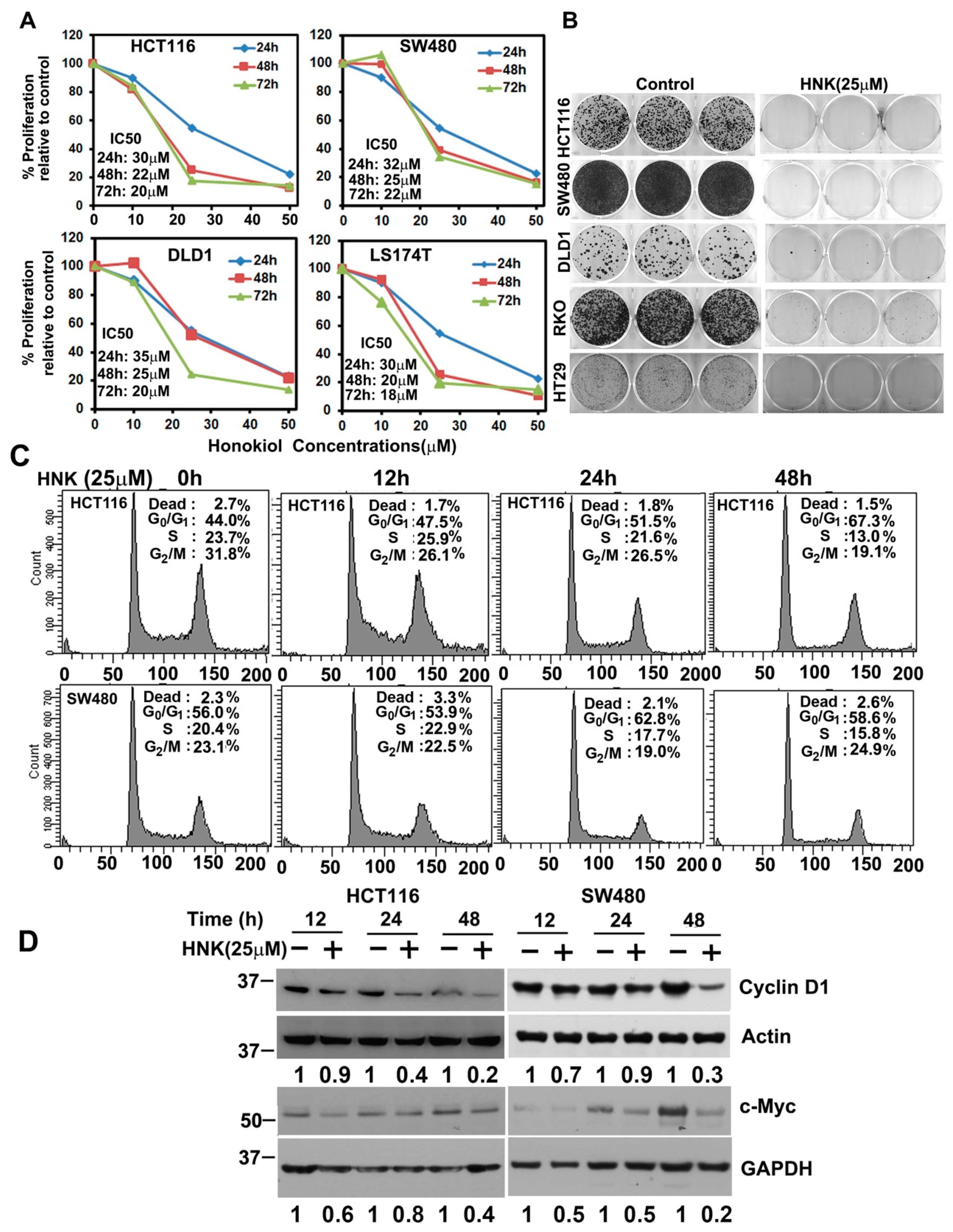
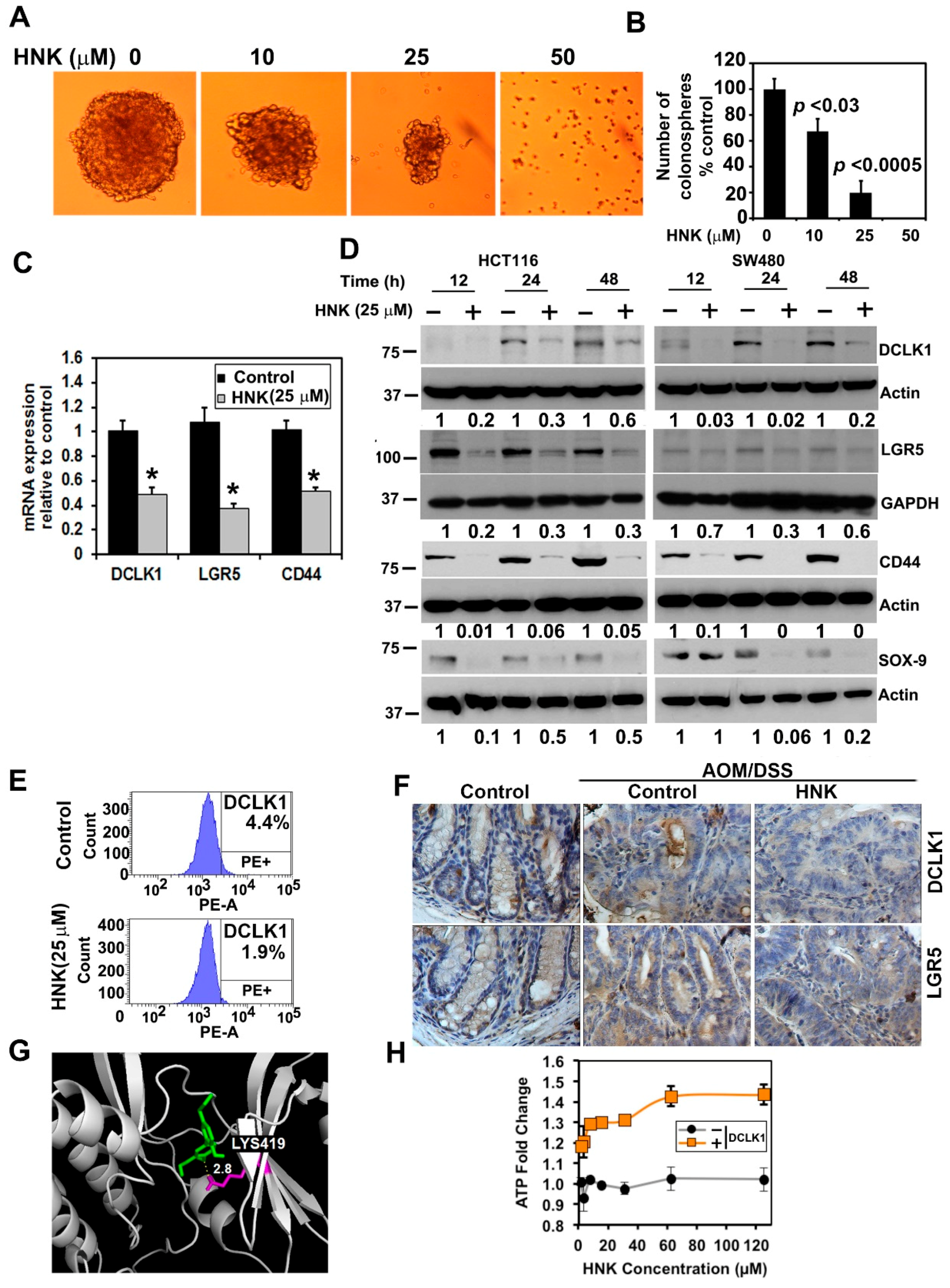
3.1. HNK Inhibits Colon Cancer Cell Proliferation and Colony Formation
3.2. HNK Prevents Colitis-Associated Cancer Growth
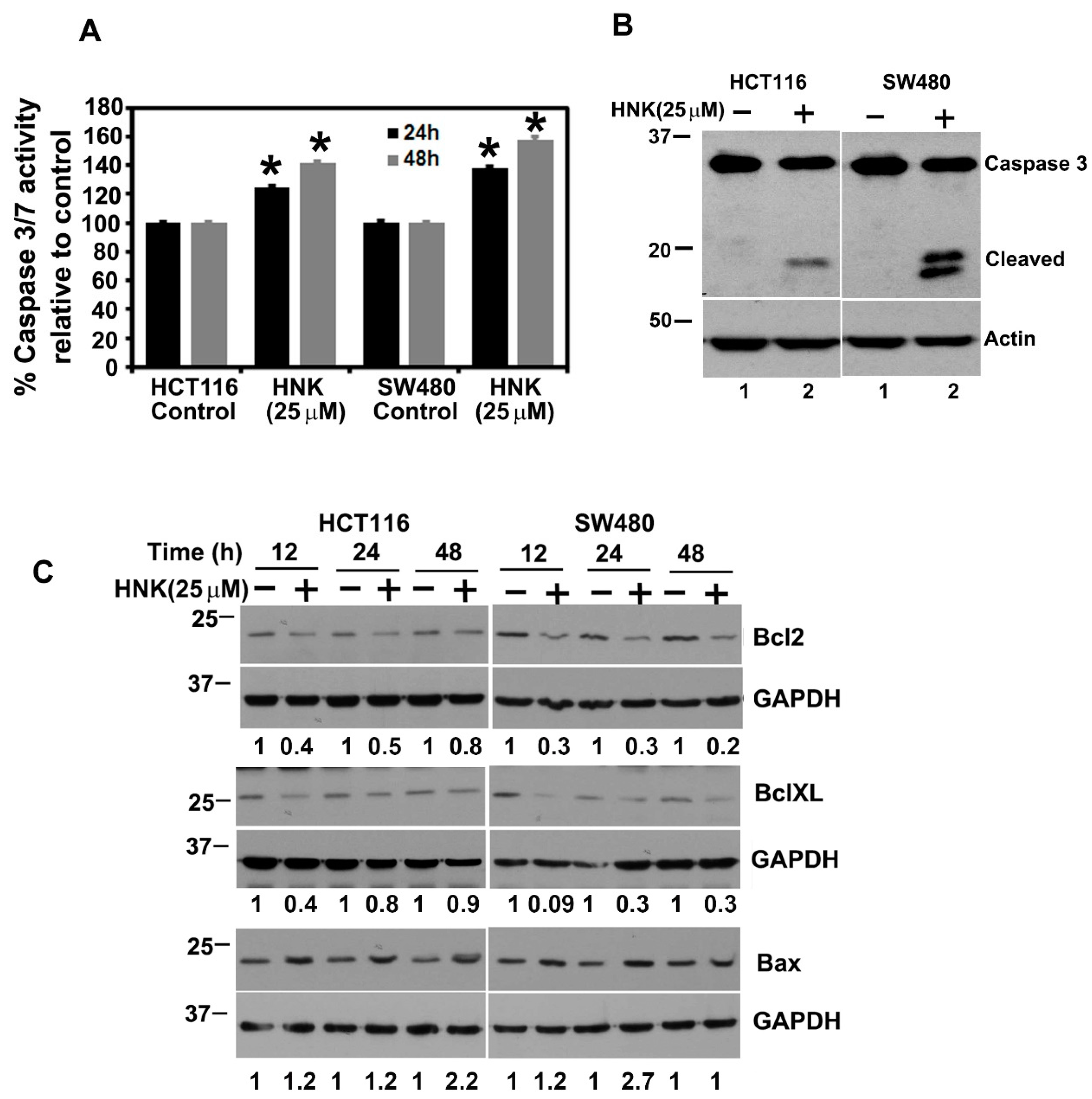
3.3. HNK Treatment Induces Apoptosis
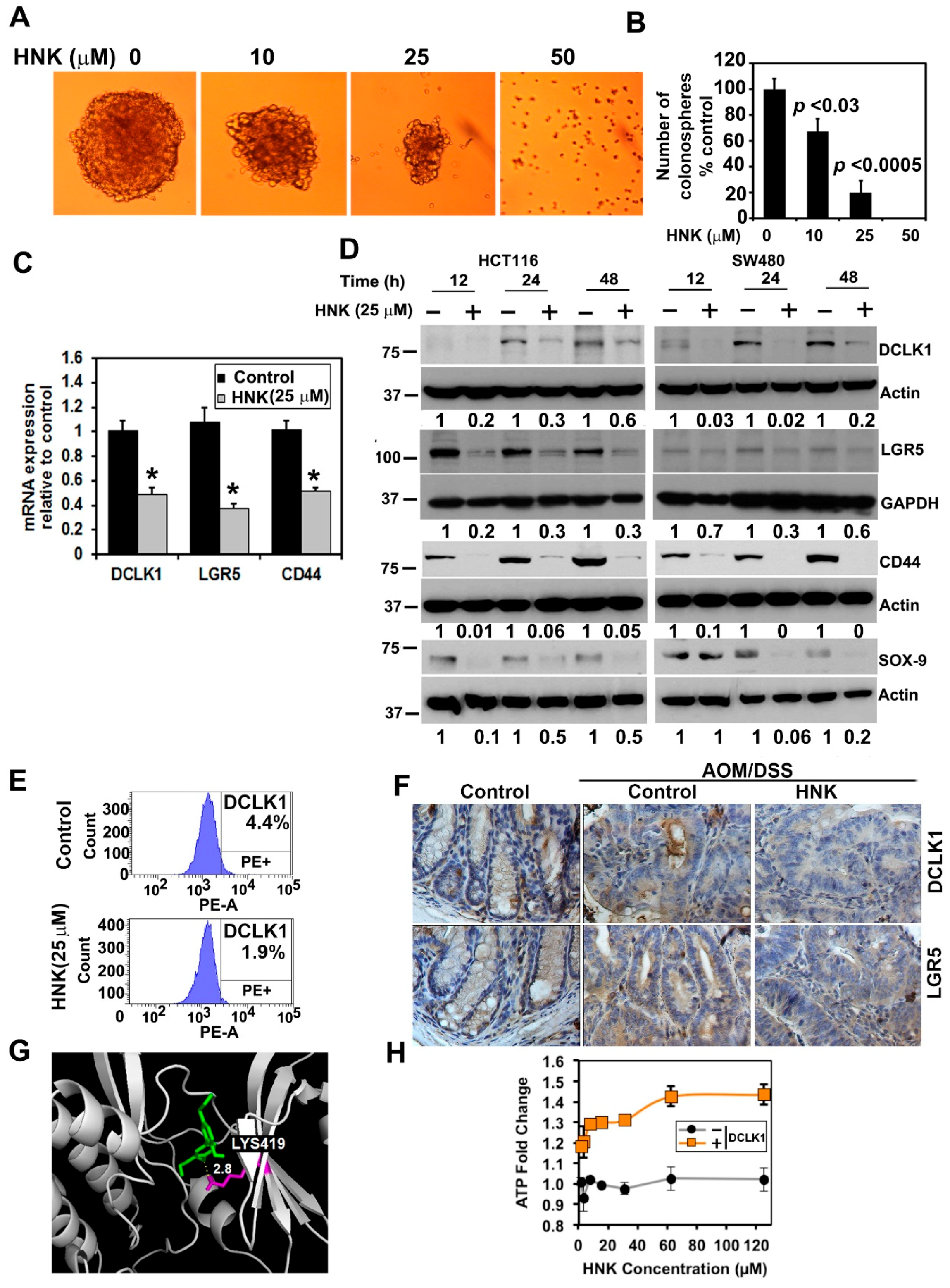
3.4. HNK Inhibits Cancer Stem Cell Marker DCLK1 Expression in Both In Vitro and Colonic Tumors
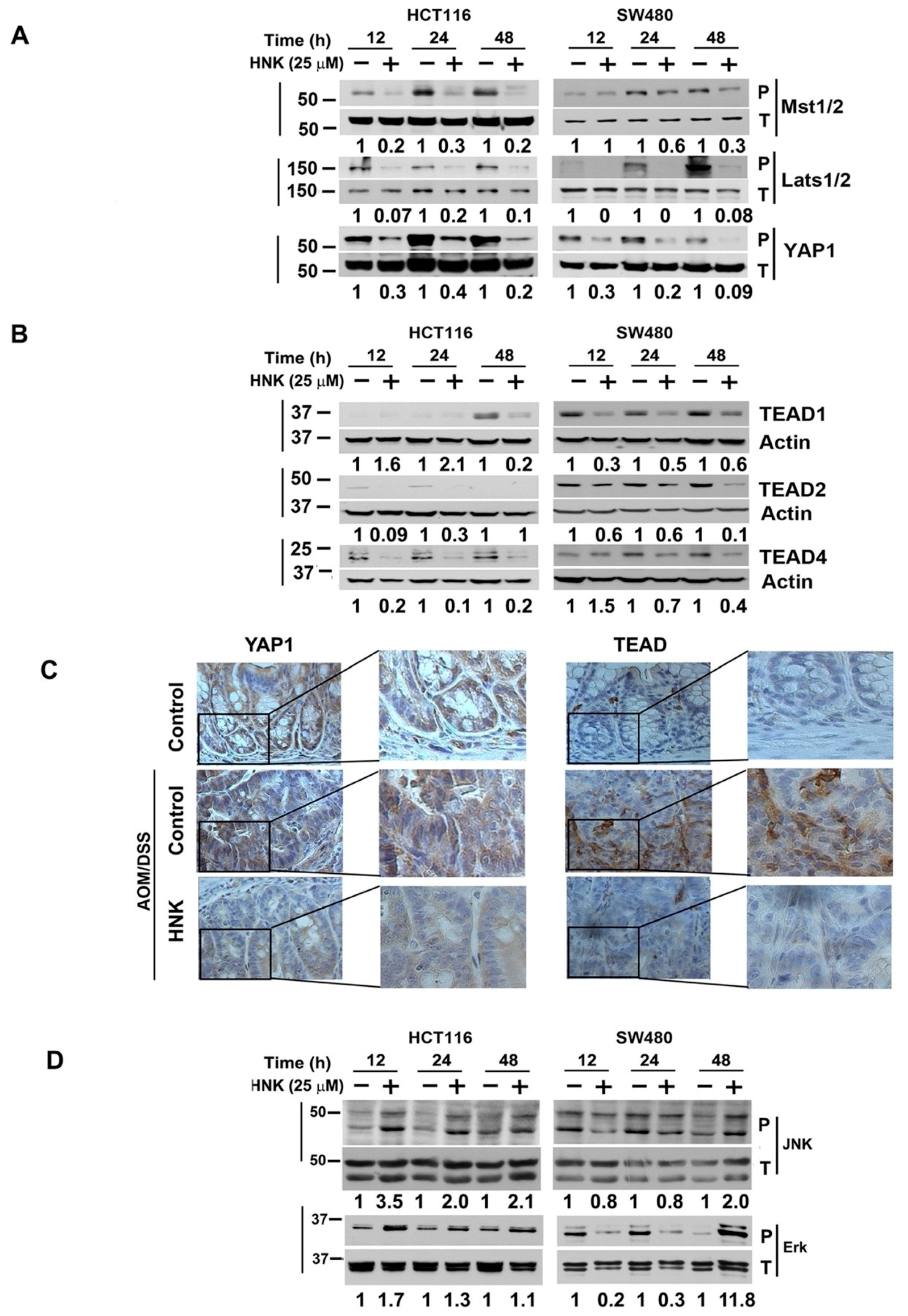
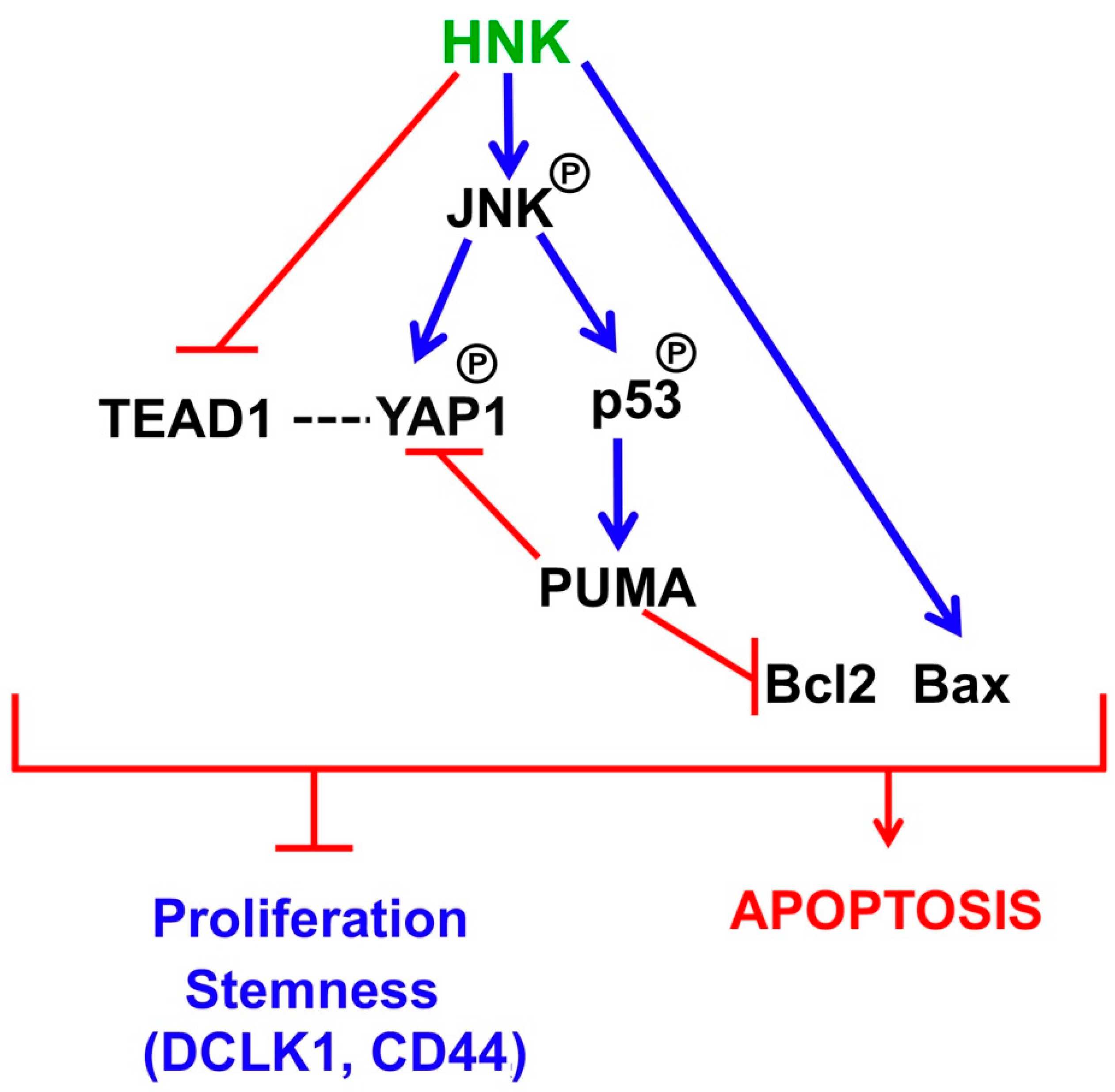
3.5. HNK Inhibits Hippo Signaling through Downregulation YAP1 in Both In Vitro and Colonic Tumors
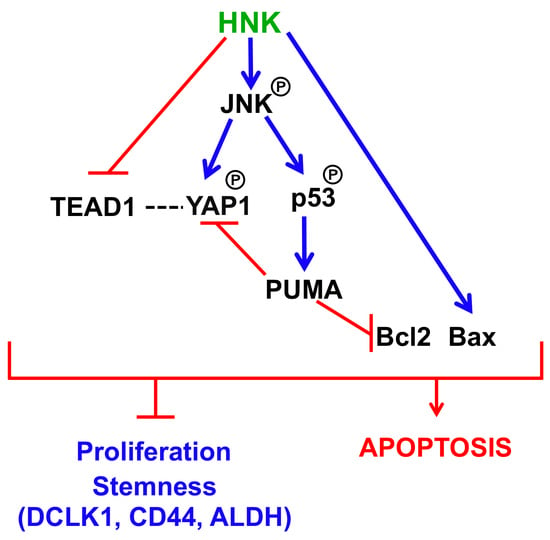
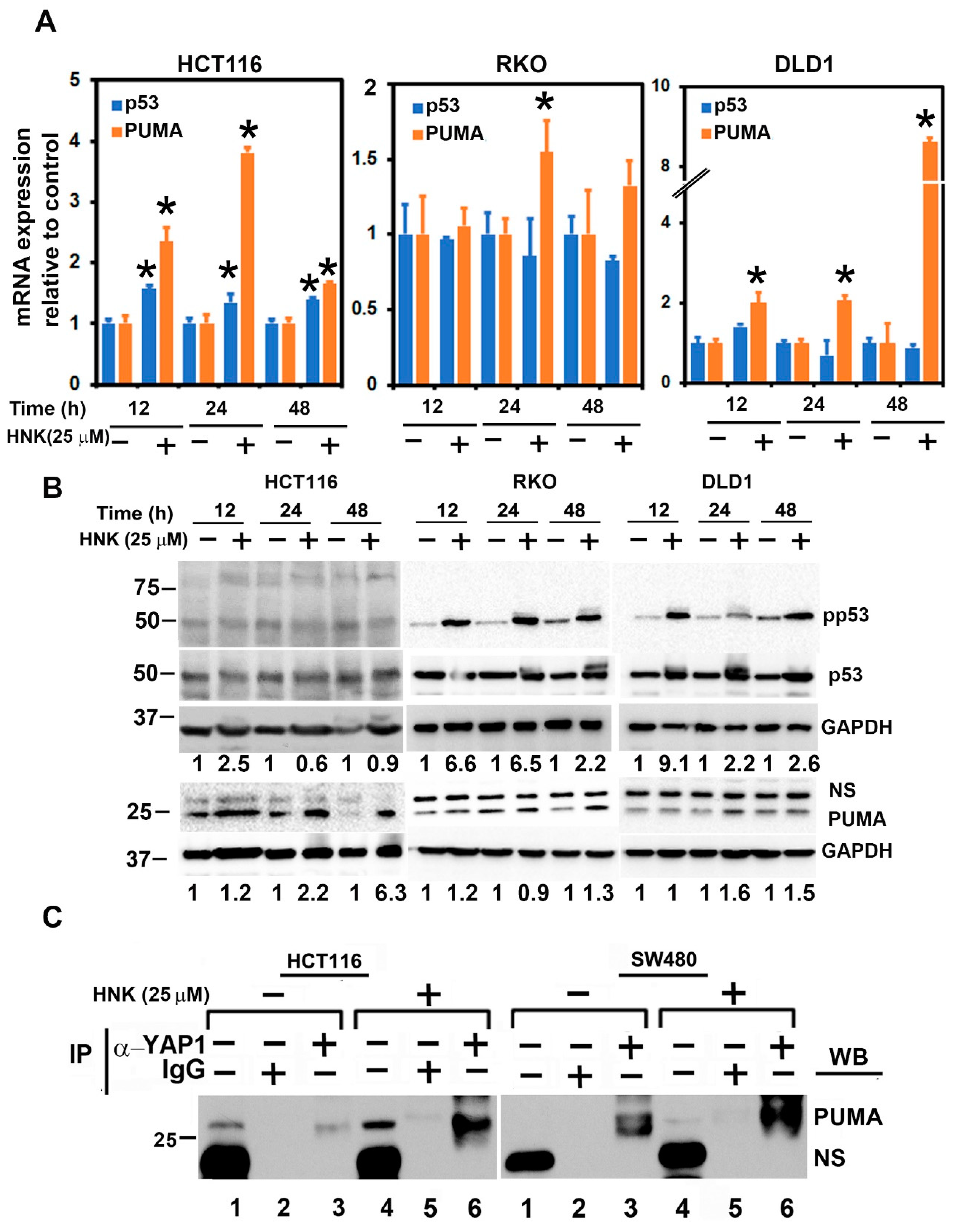
3.6. HNK Downregulates YAP1 through PUMA Mediated Apoptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HNK | Honokiol |
| CSC | Cancer stem cells |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| ERK | Extracellular signal-regulated kinase |
| JNK | Janus kinase |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Malesci, A.; Vetrano, S. Colitis-associated cancer: The dark side of inflammatory bowel disease. Gut 2011, 60, 1609–1610. [Google Scholar] [CrossRef] [PubMed]
- Tarraga Lopez, P.J.; Albero, J.S.; Rodriguez-Montes, J.A. Primary and secondary prevention of colorectal cancer. Clin. Med. Insights Gastroenterol. 2014, 7, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.M.; Chen, C.C.; Ko, F.N.; Lee, L.G.; Huang, T.F.; Chen, Y.P.; Hsu, H.Y. Two antiplatelet agents from Magnolia officinalis. Thromb. Res. 1988, 50, 757–765. [Google Scholar] [CrossRef]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Banik, K.; Ranaware, A.M.; Deshpande, V.; Nalawade, S.P.; Padmavathi, G.; Bordoloi, D.; Sailo, B.L.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; et al. Honokiol for cancer therapeutics: A traditional medicine that can modulate multiple oncogenic targets. Pharmacol. Res. 2019, 144, 192–209. [Google Scholar] [CrossRef]
- Ponnurangam, S.; Mammen, J.M.; Ramalingam, S.; He, Z.; Zhang, Y.; Umar, S.; Subramaniam, D.; Anant, S. Honokiol in combination with radiation targets notch signaling to inhibit colon cancer stem cells. Mol. Cancer Ther. 2012, 11, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.J.; Lai, G.M.; Yeh, C.T.; Lai, M.T.; Shih, P.H.; Chao, W.J.; Whang-Peng, J.; Chuang, S.E.; Lai, T.Y. Honokiol Eliminates Human Oral Cancer Stem-Like Cells Accompanied with Suppression of Wnt/beta-Catenin Signaling and Apoptosis Induction. Evid. Based Complement. Altern. Med. 2013, 2013, 146136. [Google Scholar] [CrossRef] [Green Version]
- Lai, I.C.; Shih, P.H.; Yao, C.J.; Yeh, C.T.; Wang-Peng, J.; Lui, T.N.; Chuang, S.E.; Hu, T.S.; Lai, T.Y.; Lai, G.M. Elimination of cancer stem-like cells and potentiation of temozolomide sensitivity by Honokiol in glioblastoma multiforme cells. PLoS ONE 2015, 10, e0114830. [Google Scholar] [CrossRef] [Green Version]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Mol. Oncol. 2014, 8, 565–580. [Google Scholar] [CrossRef]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol activates LKB1-miR-34a axis and antagonizes the oncogenic actions of leptin in breast cancer. Oncotarget 2015, 6, 29947–29962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef]
- May, R.; Sureban, S.M.; Hoang, N.; Riehl, T.E.; Lightfoot, S.A.; Ramanujam, R.; Wyche, J.H.; Anant, S.; Houchen, C.W. Doublecortin and CaM kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells 2009, 27, 2571–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, R.; Riehl, T.E.; Hunt, C.; Sureban, S.M.; Anant, S.; Houchen, C.W. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells 2008, 26, 630–637. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Ramalingam, S.; Subramaniam, D.; Natarajan, G.; Anant, S.; Houchen, C.W. Selective blockade of DCAMKL-1 results in tumor growth arrest by a Let-7a MicroRNA-dependent mechanism. Gastroenterology 2009, 137, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M.; Wong, K.F.; Jiang, X.; Qiao, Y.; Luk, J.M. Regulators of mammalian Hippo pathway in cancer. Biochim. Biophys. Acta 2012, 1826, 357–364. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, D.; Chi, F.; Zhao, B. The Hippo pathway regulates stem cell proliferation, self-renewal, and differentiation. Protein Cell 2012, 3, 291–304. [Google Scholar] [CrossRef]
- Stanger, B.Z. Quit your YAPing: A new target for cancer therapy. Genes Dev. 2012, 26, 1263–1267. [Google Scholar] [CrossRef] [Green Version]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.; Schiripo, T.A.; Haber, D.; Hariharan, I.K. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.P.; Zhu, J.S.; Zhang, Q.; Wang, X.Y. A breakdown of the Hippo pathway in gastric cancer. Hepato-Gastroenterology 2011, 58, 1611–1617. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. Expression of Yes-associated protein in common solid tumors. Hum. Pathol. 2008, 39, 1582–1589. [Google Scholar] [CrossRef] [Green Version]
- Avruch, J.; Zhou, D.; Fitamant, J.; Bardeesy, N.; Mou, F.; Barrufet, L.R. Protein kinases of the Hippo pathway: Regulation and substrates. Semin. Cell Dev. Biol. 2012, 23, 770–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xing, D.; Liu, L. PUMA promotes Bax translocation by both directly interacting with Bax and by competitive binding to Bcl-X L during UV-induced apoptosis. Mol. Biol. Cell 2009, 20, 3077–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J. PUMA Kills Stem Cells to Stall Cancer? Mol. Cell. Pharm. 2009, 1, 112–118. [Google Scholar] [CrossRef]
- Landegren, U. Measurement of cell numbers by means of the endogenous enzyme hexosaminidase. Applications to detection of lymphokines and cell surface antigens. J. Immunol. Methods 1984, 67, 379–388. [Google Scholar] [CrossRef]
- Subramaniam, D.; May, R.; Sureban, S.M.; Lee, K.B.; George, R.; Kuppusamy, P.; Ramanujam, R.P.; Hideg, K.; Dieckgraefe, B.K.; Houchen, C.W.; et al. Diphenyl difluoroketone: A curcumin derivative with potent in vivo anticancer activity. Cancer Res. 2008, 68, 1962–1969. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, N.; Woetzel, N.; Meiler, J. Bcl: Cluster: A method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System. IEEE Int. Conf. Comput. Adv. Bio. Med. Sci. 2011, 2011, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Thaker, A.I.; Shaker, A.; Rao, M.S.; Ciorba, M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). JoVE J. 2012. [Google Scholar] [CrossRef] [Green Version]
- Mateyak, M.K.; Obaya, A.J.; Sedivy, J.M. C-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell Biol. 1999, 19, 4672–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boatright, K.M.; Salvesen, G.S. Caspase activation. Biochem. Soc. Symp. 2003, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.B.; Auerbach, W.; Lee, J.S.; Stephen, D.; Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002, 129, 4753–4761. [Google Scholar] [CrossRef]
- Patel, O.; Dai, W.; Mentzel, M.; Griffin, M.D.; Serindoux, J.; Gay, Y.; Fischer, S.; Sterle, S.; Kropp, A.; Burns, C.J.; et al. Biochemical and Structural Insights into Doublecortin-like Kinase Domain 1. Structure 2016, 24, 1550–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Castellan, M.; Battilana, G.; Zanconato, F.; Azzolin, L.; Giulitti, S.; Cordenonsi, M.; Piccolo, S. YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat. Commun. 2017, 8, 15206. [Google Scholar] [CrossRef]
- Corvaisier, M.; Bauzone, M.; Corfiotti, F.; Renaud, F.; El Amrani, M.; Monte, D.; Truant, S.; Leteurtre, E.; Formstecher, P.; Van Seuningen, I.; et al. Regulation of cellular quiescence by YAP/TAZ and Cyclin E1 in colon cancer cells: Implication in chemoresistance and cancer relapse. Oncotarget 2016, 7, 56699–56712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Chen, Y.; Zhang, R.; Wu, Y.; Ma, Y.; Fang, X.; Shen, S. Honokiol induces apoptosis and autophagy via the ROS/ERK1/2 signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Pincus, M.R.; Ronai, Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA 1998, 95, 10541–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [Green Version]
- Ambroise, G.; Portier, A.; Roders, N.; Arnoult, D.; Vazquez, A. Subcellular localization of PUMA regulates its pro-apoptotic activity in Burkitts lymphoma B cells. Oncotarget 2015, 6, 38181–38194. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Subramaniam, D.; Ramalingam, S.; Dhar, A.; Postier, R.G.; Umar, S.; Zhang, Y.; Anant, S. Honokiol radiosensitizes colorectal cancer cells: Enhanced activity in cells with mismatch repair defects. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G929–G937. [Google Scholar] [CrossRef]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Milas, L.; Raju, U.; Liao, Z.; Ajani, J. Targeting molecular determinants of tumor chemo-radioresistance. Semin. Oncol. 2005, 32, S78–S81. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Ramalingam, S.; Houchen, C.W.; Anant, S. Cancer stem cells: A novel paradigm for cancer prevention and treatment. Mini Rev. Med. Chem. 2010, 10, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Nautiyal, J.; Kanwar, S.S.; Yu, Y.; Majumdar, A.P. Combination of dasatinib and curcumin eliminates chemo-resistant colon cancer cells. J. Mol. Signal 2011, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5 (+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Landeghem, L.; Santoro, M.A.; Krebs, A.E.; Mah, A.T.; Dehmer, J.J.; Gracz, A.D.; Scull, B.P.; McNaughton, K.; Magness, S.T.; Lund, P.K. Activation of two distinct Sox9-EGFP-expressing intestinal stem cell populations during crypt regeneration after irradiation. Am. J. Physiol. Gastrointes. Liver Physiol. 2012, 302, G1111–G1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, J.; Tang, J.; Liu, X.; Zhong, Q.; Wang, F.; Hu, W.; Yuan, Z.; Nie, C.; Wei, Y. JNK- and Akt-mediated Puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem. J. 2012, 444, 291–301. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, D.; Ponnurangam, S.; Ramalingam, S.; Kwatra, D.; Dandawate, P.; Weir, S.J.; Umar, S.; Jensen, R.A.; Anant, S. Honokiol Affects Stem Cell Viability by Suppressing Oncogenic YAP1 Function to Inhibit Colon Tumorigenesis. Cells 2021, 10, 1607. https://doi.org/10.3390/cells10071607
Subramaniam D, Ponnurangam S, Ramalingam S, Kwatra D, Dandawate P, Weir SJ, Umar S, Jensen RA, Anant S. Honokiol Affects Stem Cell Viability by Suppressing Oncogenic YAP1 Function to Inhibit Colon Tumorigenesis. Cells. 2021; 10(7):1607. https://doi.org/10.3390/cells10071607
Chicago/Turabian StyleSubramaniam, Dharmalingam, Sivapriya Ponnurangam, Satish Ramalingam, Deep Kwatra, Prasad Dandawate, Scott J. Weir, Shahid Umar, Roy A. Jensen, and Shrikant Anant. 2021. "Honokiol Affects Stem Cell Viability by Suppressing Oncogenic YAP1 Function to Inhibit Colon Tumorigenesis" Cells 10, no. 7: 1607. https://doi.org/10.3390/cells10071607
APA StyleSubramaniam, D., Ponnurangam, S., Ramalingam, S., Kwatra, D., Dandawate, P., Weir, S. J., Umar, S., Jensen, R. A., & Anant, S. (2021). Honokiol Affects Stem Cell Viability by Suppressing Oncogenic YAP1 Function to Inhibit Colon Tumorigenesis. Cells, 10(7), 1607. https://doi.org/10.3390/cells10071607