
A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients

 , ,
, ,  , , , ,
, , , , 

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Generation of Chemoresistant Cell Lines
2.3. Colorectal Cancer Tumor Samples
2.4. Xenograft Tumor Samples
2.5. Determining IC50
2.6. Real Time PCR
2.7. Western Blotting
2.8. Sphere Formation Assay
2.9. Nanog Reporter Assay
2.10. Immunofluorescence
2.11. Validation of the Genes in the Patient Datasets
2.12. Statistical Analysis
3. Results
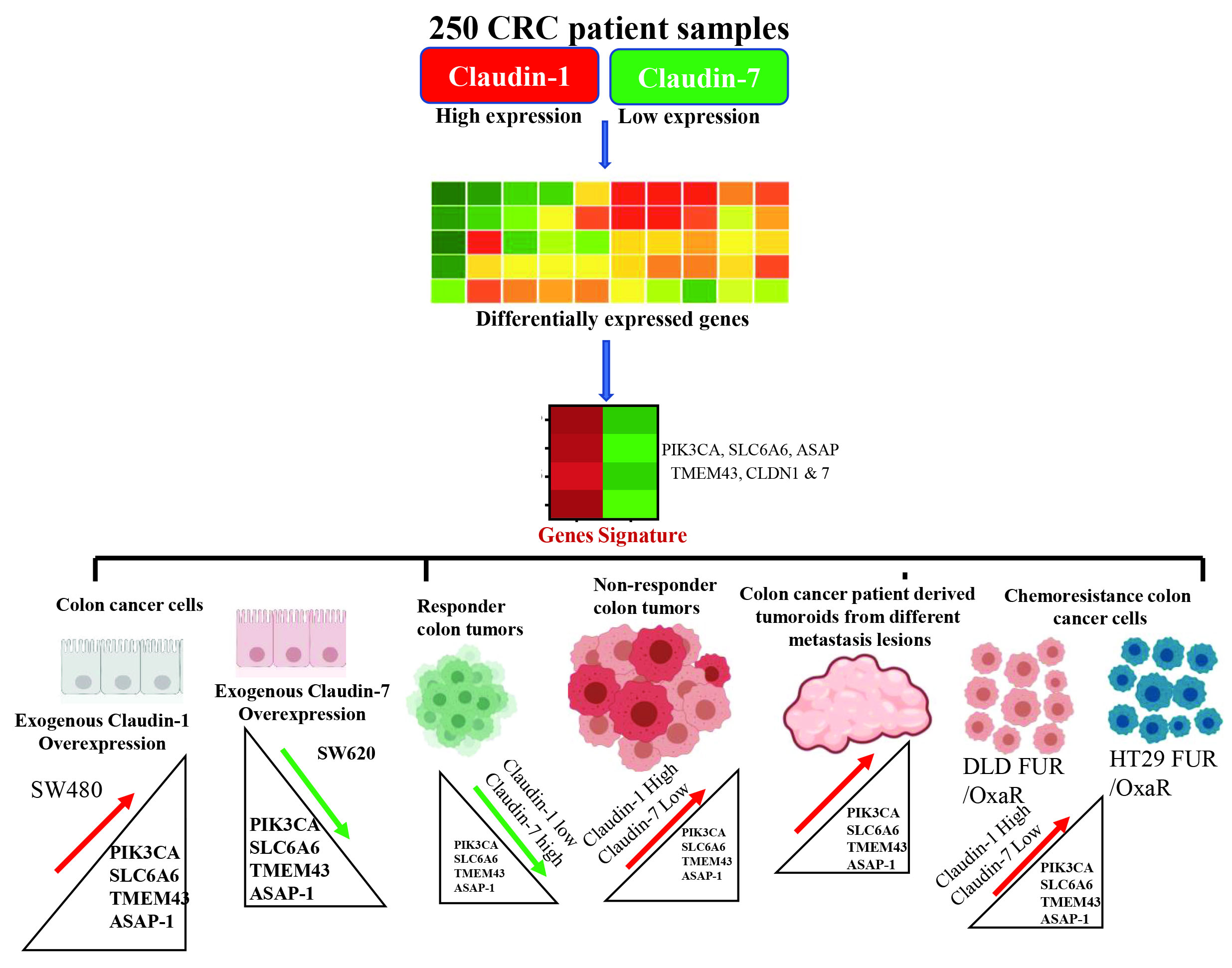
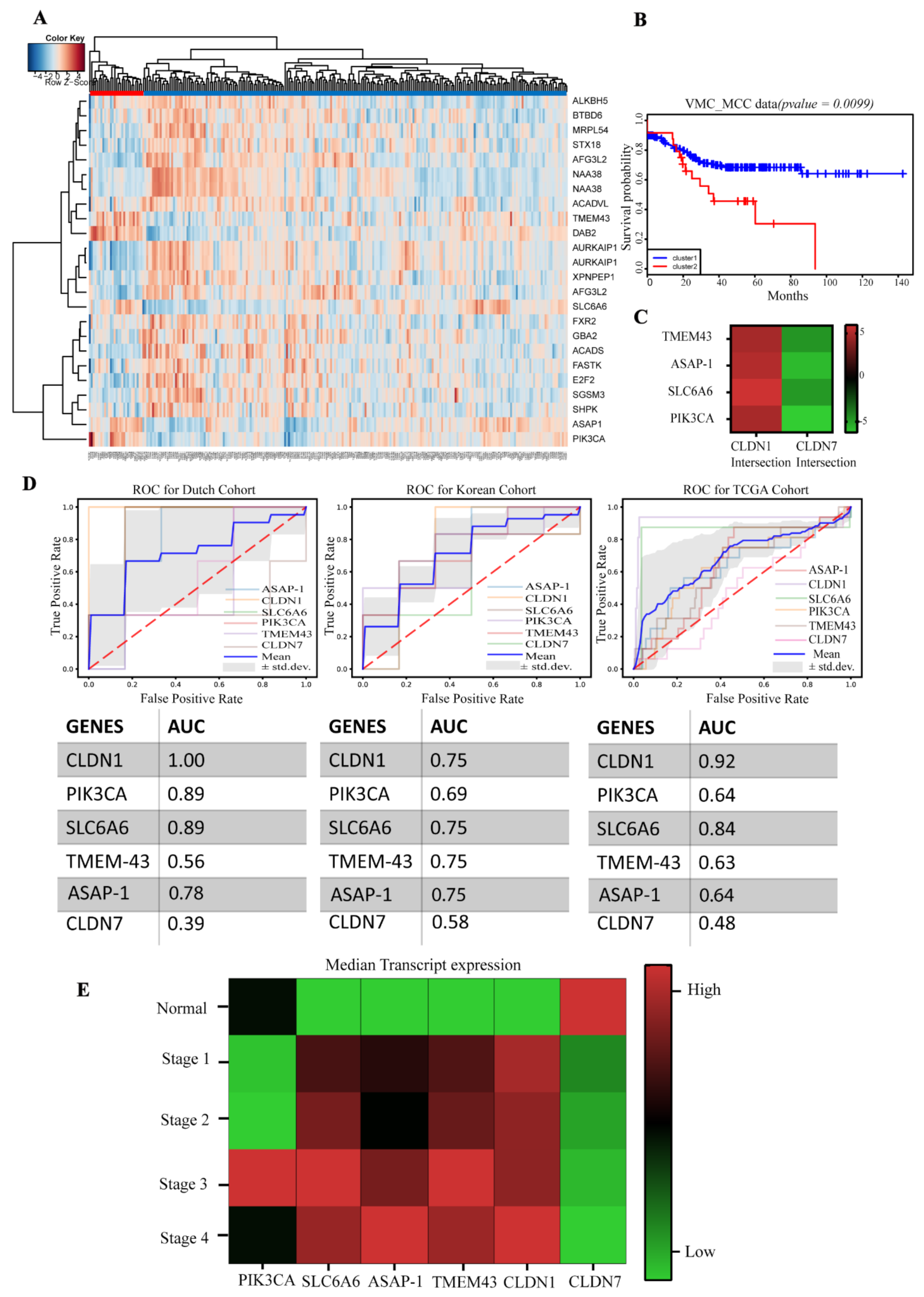
3.1. Identification of a Claudin-Associated Molecular Signature
3.2. Validation of the Gene Signature in the TCGA Dataset
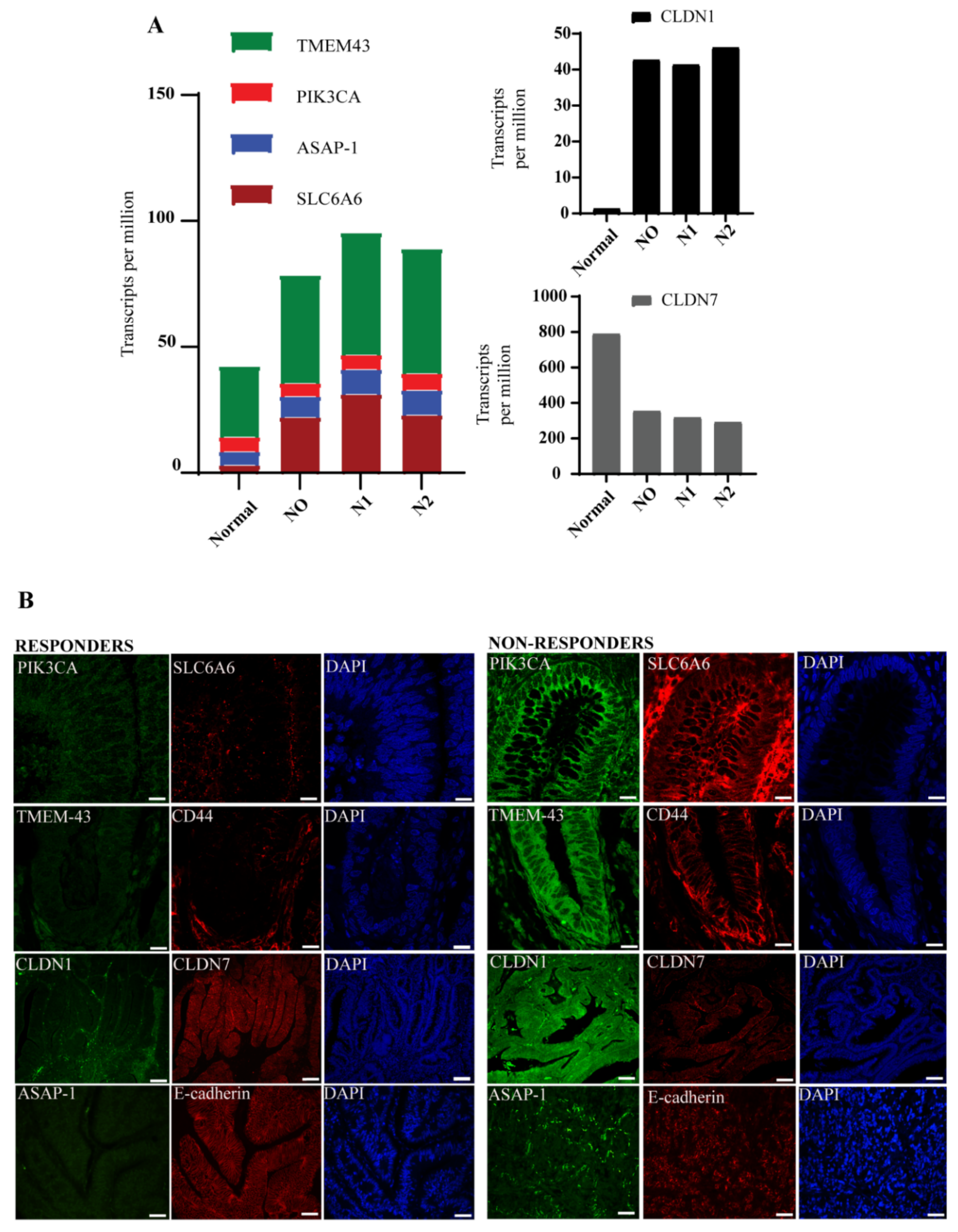
3.3. Molecular Signature Distinguishes Responders from Non-Responder Patients
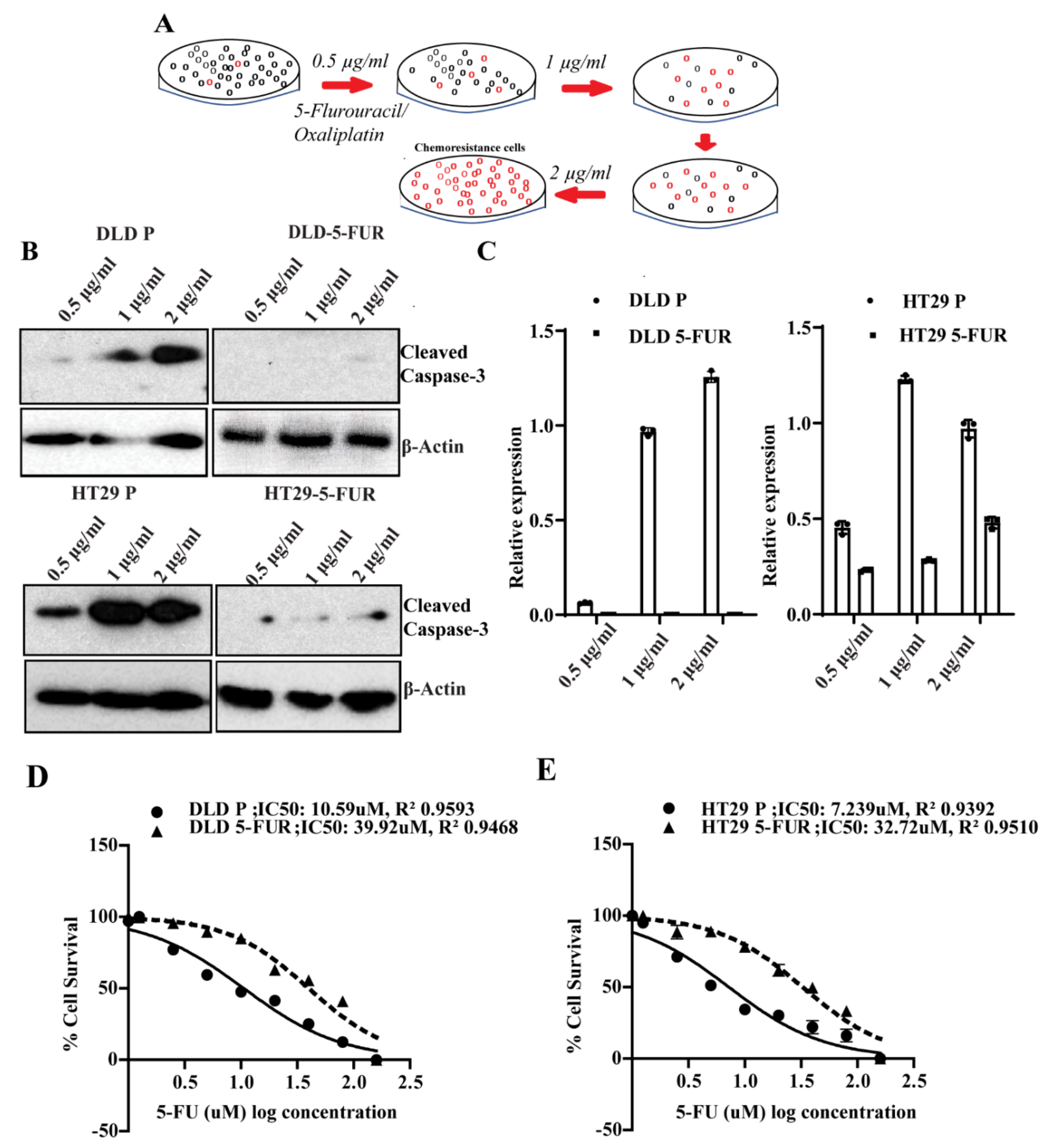
3.4. CLDN1 and CLDN7 Affect Sensitivity of CRC Cells to 5-FU
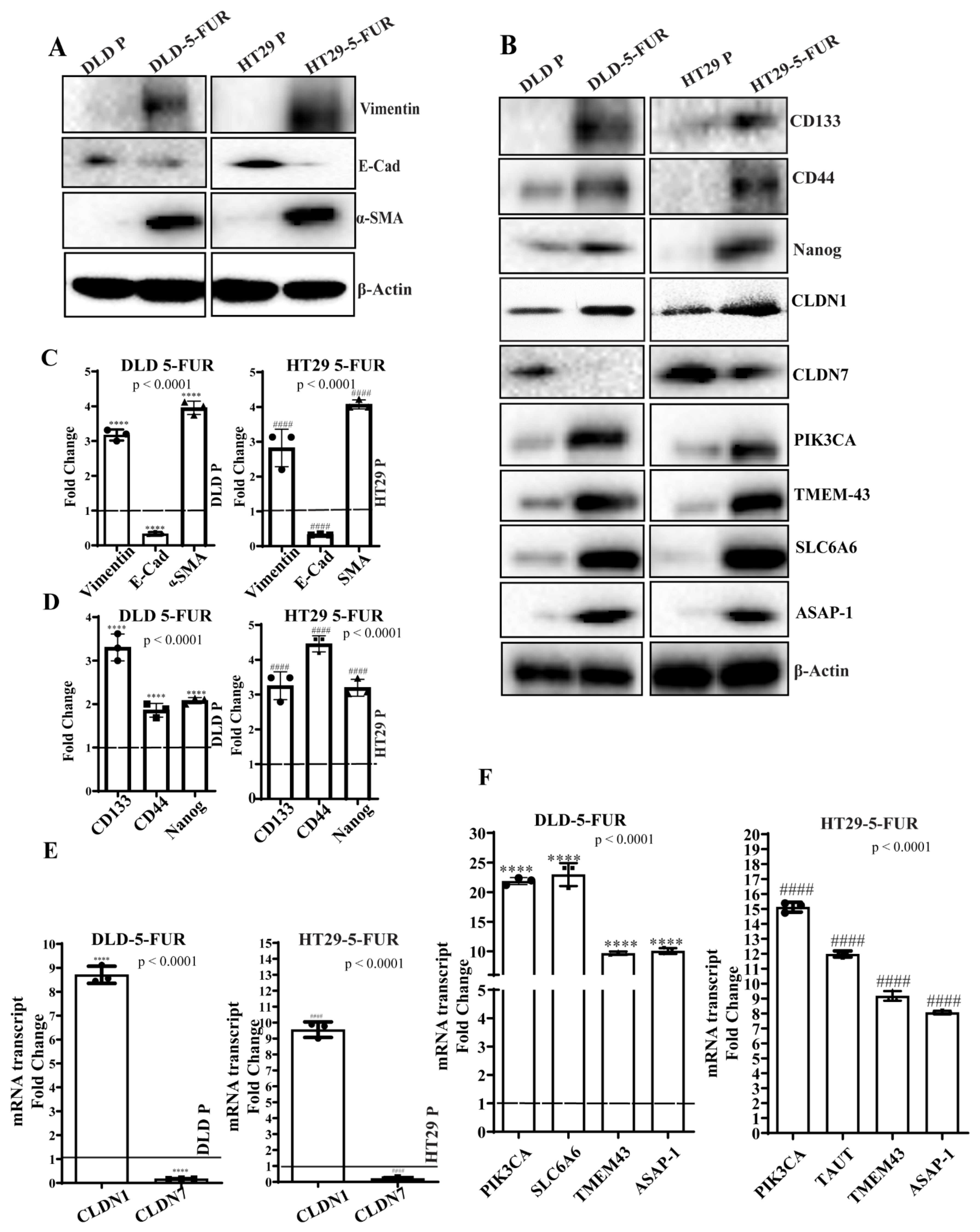
3.5. Validation of the Claudin-Associated Molecular Signature in CRC Cell Lines
3.6. Signature Gene Expression in Chemoresistant Cell Line Variants
3.7. Knockdown of PIK3CA and SLC6A6 Suppresses Stemness and EMT Markers and Enhances Chemosensitivity in DLD/HT29 5-FUR Cells
3.8. Metastatic and Recurrent Colonic Tumoroids Display Increased Claudin-Associated Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Lugli, A. Prognostic and predictive factors in colorectal cancer. J. Clin. Pathol. 2008, 61, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, J.; Li, S.; Lv, M.; Chen, Z.; Li, E.; Yi, M.; Yang, J. Tumor location as a novel high risk parameter for stage II colorectal cancers. PLoS ONE 2017, 12, e0179910. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Goldberg, R.M.; Lenz, H.J.; Shields, A.F.; Gibney, G.T.; Tan, A.R.; Brown, J.; Eisenberg, B.; Heath, E.I.; Phuphanich, S.; et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA Cancer J. Clin. 2019, 69, 305–343. [Google Scholar] [CrossRef] [PubMed]
- Mousa, L.; Salem, M.E.; Mikhail, S. Biomarkers of Angiogenesis in Colorectal Cancer. Biomark. Cancer 2015, 7, 13–19. [Google Scholar] [CrossRef]
- Ward, T.H.; Cummings, J.; Dean, E.; Greystoke, A.; Hou, J.M.; Backen, A.; Ranson, M.; Dive, C. Biomarkers of apoptosis. Br. J. Cancer 2008, 99, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Drucker, E.; Krapfenbauer, K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Gowrikumar, S.; Singh, A.B.; Dhawan, P. Role of Claudin Proteins in Regulating Cancer Stem Cells and Chemoresistance-Potential Implication in Disease Prognosis and Therapy. Int. J. Mol. Sci. 2019, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 2016, 1, e85902. [Google Scholar] [CrossRef] [PubMed]
- Ouban, A. Claudin-1 role in colon cancer: An update and a review. Histol. Histopathol. 2018, 33, 1013–1019. [Google Scholar] [PubMed]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Pope, J.L.; Smith, J.J.; Ahmad, R.; Chen, X.; Washington, M.K.; Beauchamp, R.D.; Singh, A.B.; Dhawan, P. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene 2015, 34, 4570–4580. [Google Scholar] [CrossRef]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauve, C.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Bose, D.; Zimmerman, L.J.; Pierobon, M.; Petricoin, E.; Tozzi, F.; Parikh, A.; Fan, F.; Dallas, N.; Xia, L.; Gaur, P.; et al. Chemoresistant colorectal cancer cells and cancer stem cells mediate growth and survival of bystander cells. Br. J. Cancer 2011, 105, 1759–1767. [Google Scholar] [CrossRef]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G., 2nd; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef]
- Smith, J.J.; Deane, N.G.; Wu, F.; Merchant, N.B.; Zhang, B.; Jiang, A.; Lu, P.; Johnson, J.C.; Schmidt, C.; Bailey, C.E.; et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 2010, 138, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Shen, L.; Yu, M.; Ni, J.; Dong, X.; Zhou, Y.; Wu, S. Colon cancer cells treated with 5fluorouracil exhibit changes in polylactosaminetype Nglycans. Mol. Med. Rep. 2014, 9, 1697–1702. [Google Scholar] [CrossRef]
- Shaheen, S.; Ahmed, M.; Lorenzi, F.; Nateri, S.A. Spheroid-Formation (Colonosphere) Assay for In Vitro Assessment and Expansion of Stem Cells in Colon Cancer. Stem Cell Rev. Rep. 2016, 12, 492–499. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Lawler, M.; Alsina, D.; Adams, R.A.; Anderson, A.S.; Brown, G.; Fearnhead, N.S.; Fenwick, S.W.; Halloran, S.P.; Hochhauser, D.; Hull, M.A.; et al. Critical research gaps and recommendations to inform research prioritisation for more effective prevention and improved outcomes in colorectal cancer. Gut 2018, 67, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Tibiche, C.; Zou, J.; Zaman, N.; Trifiro, M.; O’Connor-McCourt, M.; Wang, E. Identification and Construction of Combinatory Cancer Hallmark-Based Gene Signature Sets to Predict Recurrence and Chemotherapy Benefit in Stage II Colorectal Cancer. JAMA Oncol. 2016, 2, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Chen, J.; Liu, L.; Zhao, G.; Dong, P.; Wu, B.; Wang, J.; Dong, L. Establishment of a 12-gene expression signature to predict colon cancer prognosis. PeerJ 2018, 6, e4942. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J. Emerging roles of claudins in human cancer. Int. J. Mol. Sci. 2013, 14, 18148–18180. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.H.; Hwang, D.; An, J.; Chung, H.S.; Yang, E.G.; Kim, S.Y. Extracellular pyruvate kinase M2 facilitates cell migration by upregulating claudin-1 expression in colon cancer cells. Biochem. Cell Biol. 2020, 98, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Stromberg, V.; Askari, S.; Ahmad, A.; Befekadu, R.; Nilsson, T.K. Expression of claudin 1, claudin 4, and claudin 7 in colorectal cancer and its relation with CLDN DNA methylation patterns. Tumour Biol. 2017, 39, 1010428317697569. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Park, M.Y.; Sung, M.K. 5-Fluorouracil-induced changes of intestinal integrity biomarkers in BALB/c mice. J. Cancer Prev. 2013, 18, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, R.; Maruhashi, R.; Eguchi, H.; Kitabatake, K.; Tsukimoto, M.; Furuta, T.; Matsunaga, T.; Endo, S.; Ikari, A. Decrease in paracellular permeability and chemosensitivity to doxorubicin by claudin-1 in spheroid culture models of human lung adenocarcinoma A549 cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 769–780. [Google Scholar] [CrossRef]
- Hoggard, J.; Fan, J.; Lu, Z.; Lu, Q.; Sutton, L.; Chen, Y.H. Claudin-7 increases chemosensitivity to cisplatin through the upregulation of caspase pathway in human NCI-H522 lung cancer cells. Cancer Sci. 2013, 104, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Morikawa, T.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Yamauchi, M.; Nosho, K.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; et al. Prognostic role of PIK3CA mutation in colorectal cancer: Cohort study and literature review. Clin. Cancer Res. 2012, 18, 2257–2268. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.N.; Rajput, A.; Rose, R.; Hauser, J.; Beko, A.; Kuropatwinski, K.; LeVea, C.; Hoffman, R.M.; Brattain, M.G.; Wang, J. Mutant PIK3CA-bearing colon cancer cells display increased metastasis in an orthotopic model. Cancer Res. 2007, 67, 5851–5858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yasunaga, M.; Matsumura, Y. Role of SLC6A6 in promoting the survival and multidrug resistance of colorectal cancer. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef]
- Lin, D.; Watahiki, A.; Bayani, J.; Zhang, F.; Liu, L.; Ling, V.; Sadar, M.D.; English, J.; Fazli, L.; So, A.; et al. ASAP1, a gene at 8q24, is associated with prostate cancer metastasis. Cancer Res. 2008, 68, 4352–4359. [Google Scholar] [CrossRef]
- Muller, T.; Stein, U.; Poletti, A.; Garzia, L.; Rothley, M.; Plaumann, D.; Thiele, W.; Bauer, M.; Galasso, A.; Schlag, P.; et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 2010, 29, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Schmit, K.; Michiels, C. TMEM Proteins in Cancer: A Review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Wrzesinski, T.; Szelag, M.; Cieslikowski, W.A.; Ida, A.; Giles, R.; Zodro, E.; Szumska, J.; Pozniak, J.; Kwias, Z.; Bluyssen, H.A.; et al. Expression of pre-selected TMEMs with predicted ER localization as potential classifiers of ccRCC tumors. BMC Cancer 2015, 15, 518. [Google Scholar] [CrossRef]
- McAuley, J.R.; Freeman, T.J.; Ekambaram, P.; Lucas, P.C.; McAllister-Lucas, L.M. CARMA3 Is a Critical Mediator of G Protein-Coupled Receptor and Receptor Tyrosine Kinase-Driven Solid Tumor Pathogenesis. Front. Immunol. 2018, 9, 1887. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhu, Y.; Zhou, Z.; Gumin, J.; Bengtsson, L.; Wu, W.; Songyang, Z.; Lang, F.F.; Lin, X. TMEM43/LUMA is a key signaling component mediating EGFR-induced NF-kappaB activation and tumor progression. Oncogene 2017, 36, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef]
- Loboda, A.; Nebozhyn, M.V.; Watters, J.W.; Buser, C.A.; Shaw, P.M.; Huang, P.S.; Van’t Veer, L.; Tollenaar, R.A.; Jackson, D.B.; Agrawal, D.; et al. EMT is the dominant program in human colon cancer. BMC Med. Genomics 2011, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Toledo-Guzman, M.E.; Bigoni-Ordonez, G.D.; Ibanez Hernandez, M.; Ortiz-Sanchez, E. Cancer stem cell impact on clinical oncology. World J. Stem Cells 2018, 10, 183–195. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Cai, H.; Du, B.; Zhang, L.; Ma, W.; Hu, Y.; Feng, S.; Miao, G. MACC1 facilitates chemoresistance and cancer stem cell-like properties of colon cancer cells through the PI3K/AKT signaling pathway. Mol. Med. Rep. 2017, 6, 8747–8754. [Google Scholar] [CrossRef]
- Garcia-Carracedo, D.; Cai, Y.; Qiu, W.; Saeki, K.; Friedman, R.A.; Lee, A.; Li, Y.; Goldberg, E.M.; Stratikopoulos, E.E.; Parsons, R.; et al. PIK3CA and p53 Mutations Promote 4NQO-Initated Head and Neck Tumor Progression and Metastasis in Mice. Mol. Cancer Res. 2020, 18, 822–834. [Google Scholar] [CrossRef]
- Dazhi, W.; Jing, D.; Chunling, R.; Mi, Z.; Zhixuan, X. Elevated SLC6A6 expression drives tumorigenesis and affects clinical outcomes in gastric cancer. Biomark. Med. 2019, 13, 95–104. [Google Scholar] [CrossRef]
- Hsu, H.H.; Chen, M.C.; Baskaran, R.; Lin, Y.M.; Day, C.H.; Lin, Y.J.; Tu, C.C.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef]
- Singh, A.B.; Sharma, A.; Dhawan, P. Claudin-1 expression confers resistance to anoikis in colon cancer cells in a Src-dependent manner. Carcinogenesis 2012, 33, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Gowrikumar, S.; Ahmad, R.; Uppada, S.B.; Washington, M.K.; Shi, C.; Singh, A.B.; Dhawan, P. Upregulated claudin-1 expression promotes colitis-associated cancer by promoting beta-catenin phosphorylation and activation in Notch/p-AKT-dependent manner. Oncogene 2019, 38, 5321–5337. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Natl. Acad. Sci. USA 2013, 110, 21124–21129. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Zhou, Y.Y.; Chen, F.; Wang, B.; Li, J.; Deng, Y.W.; Liu, W.D.; Wang, Z.G.; Li, Y.W.; Li, D.Z.; et al. Taurine inhibits serum deprivation-induced osteoblast apoptosis via the taurine transporter/ERK signaling pathway. Braz. J. Med. Biol. Res. 2011, 44, 618–623. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gowrikumar, S.; Primeaux, M.; Pravoverov, K.; Wu, C.; Szeglin, B.C.; Sauvé, C.-E.G.; Thapa, I.; Bastola, D.; Chen, X.S.; Smith, J.J.; et al. A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients. Cells 2021, 10, 2211. https://doi.org/10.3390/cells10092211
Gowrikumar S, Primeaux M, Pravoverov K, Wu C, Szeglin BC, Sauvé C-EG, Thapa I, Bastola D, Chen XS, Smith JJ, et al. A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients. Cells. 2021; 10(9):2211. https://doi.org/10.3390/cells10092211
Chicago/Turabian StyleGowrikumar, Saiprasad, Mark Primeaux, Kristina Pravoverov, Chao Wu, Bryan C. Szeglin, Charles-Etienne Gabriel Sauvé, Ishwor Thapa, Dhundy Bastola, Xi Steven Chen, J. Joshua Smith, and et al. 2021. "A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients" Cells 10, no. 9: 2211. https://doi.org/10.3390/cells10092211
APA StyleGowrikumar, S., Primeaux, M., Pravoverov, K., Wu, C., Szeglin, B. C., Sauvé, C.-E. G., Thapa, I., Bastola, D., Chen, X. S., Smith, J. J., Singh, A. B., & Dhawan, P. (2021). A Claudin-Based Molecular Signature Identifies High-Risk, Chemoresistant Colorectal Cancer Patients. Cells, 10(9), 2211. https://doi.org/10.3390/cells10092211