
B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
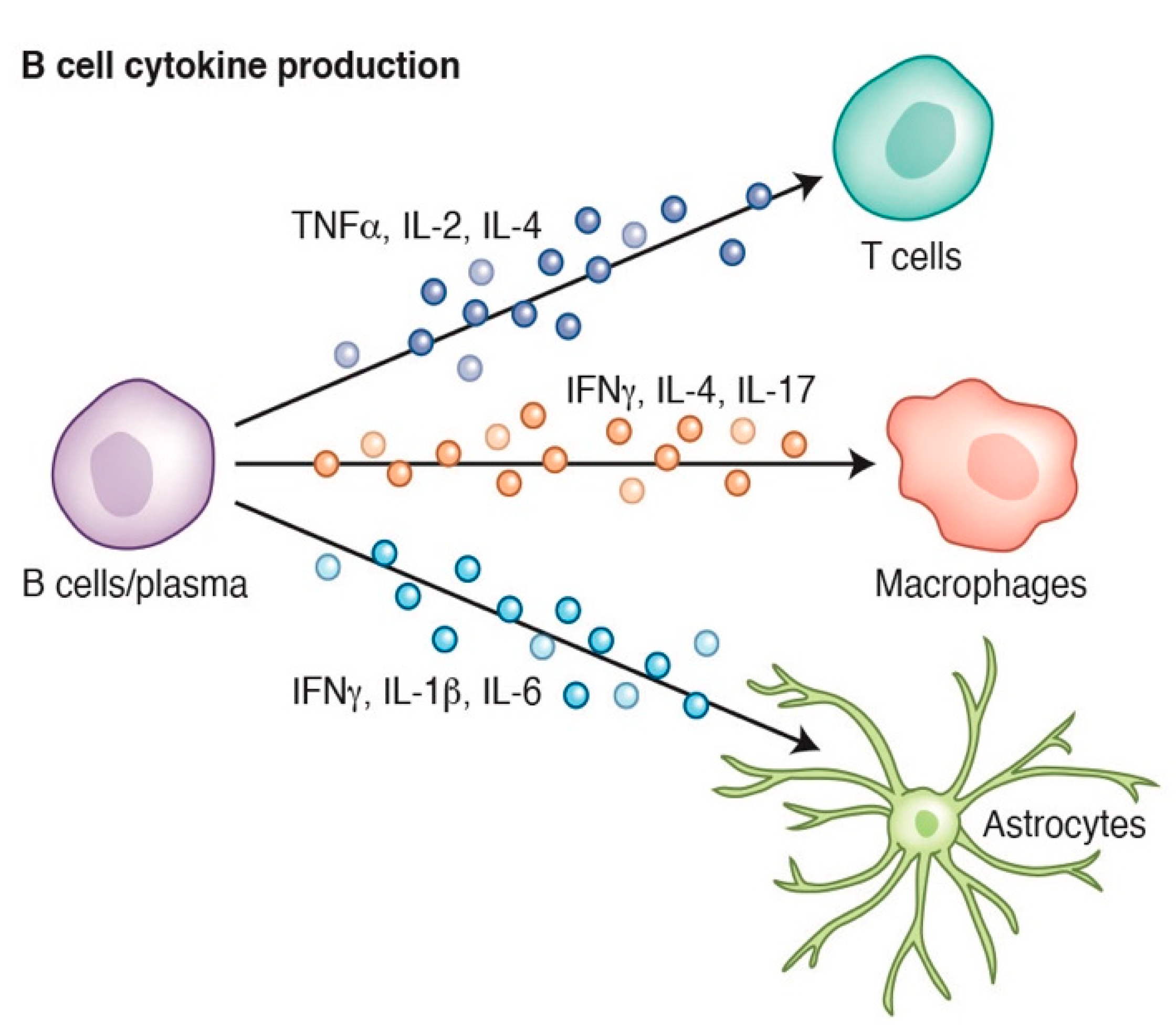
2. B Cell Biology
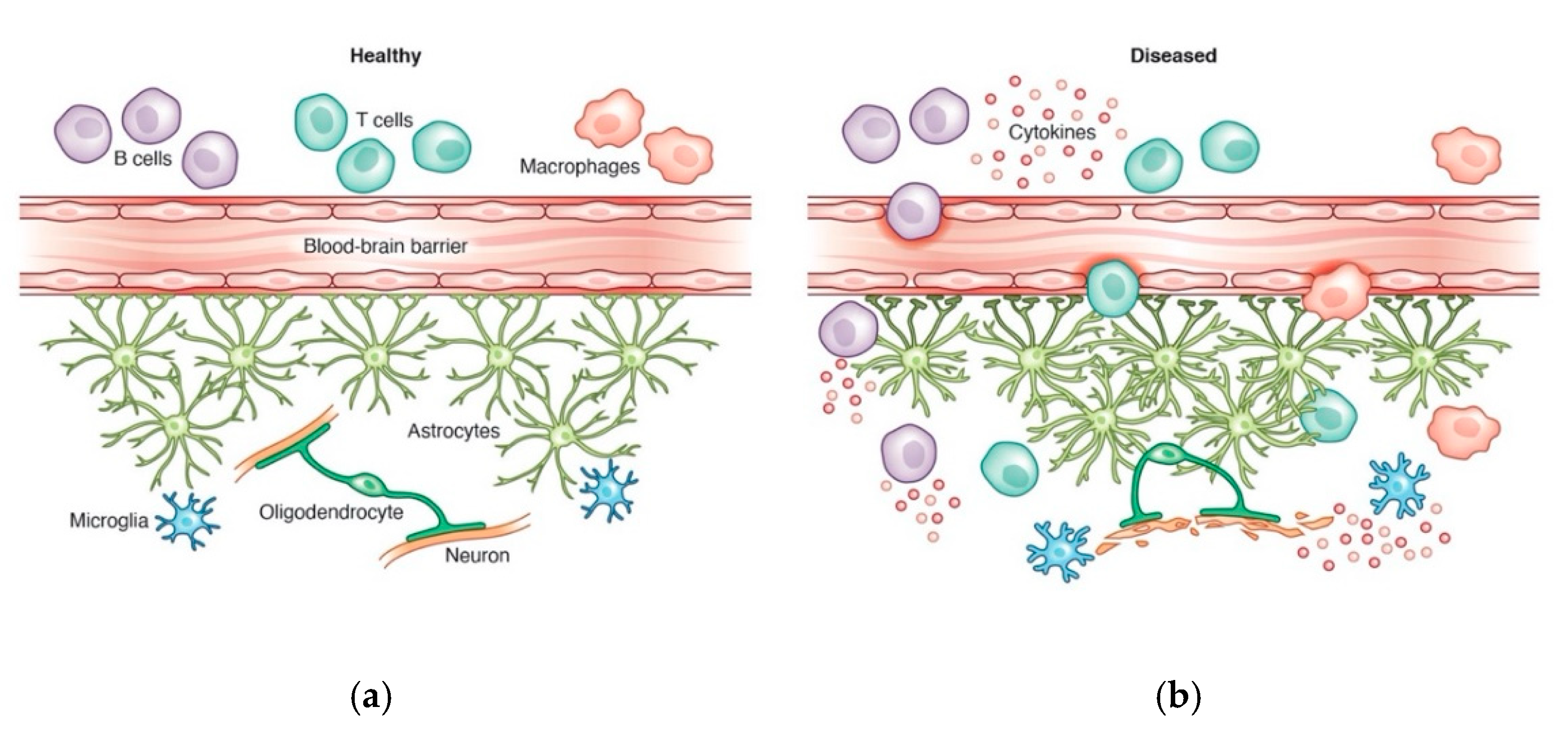
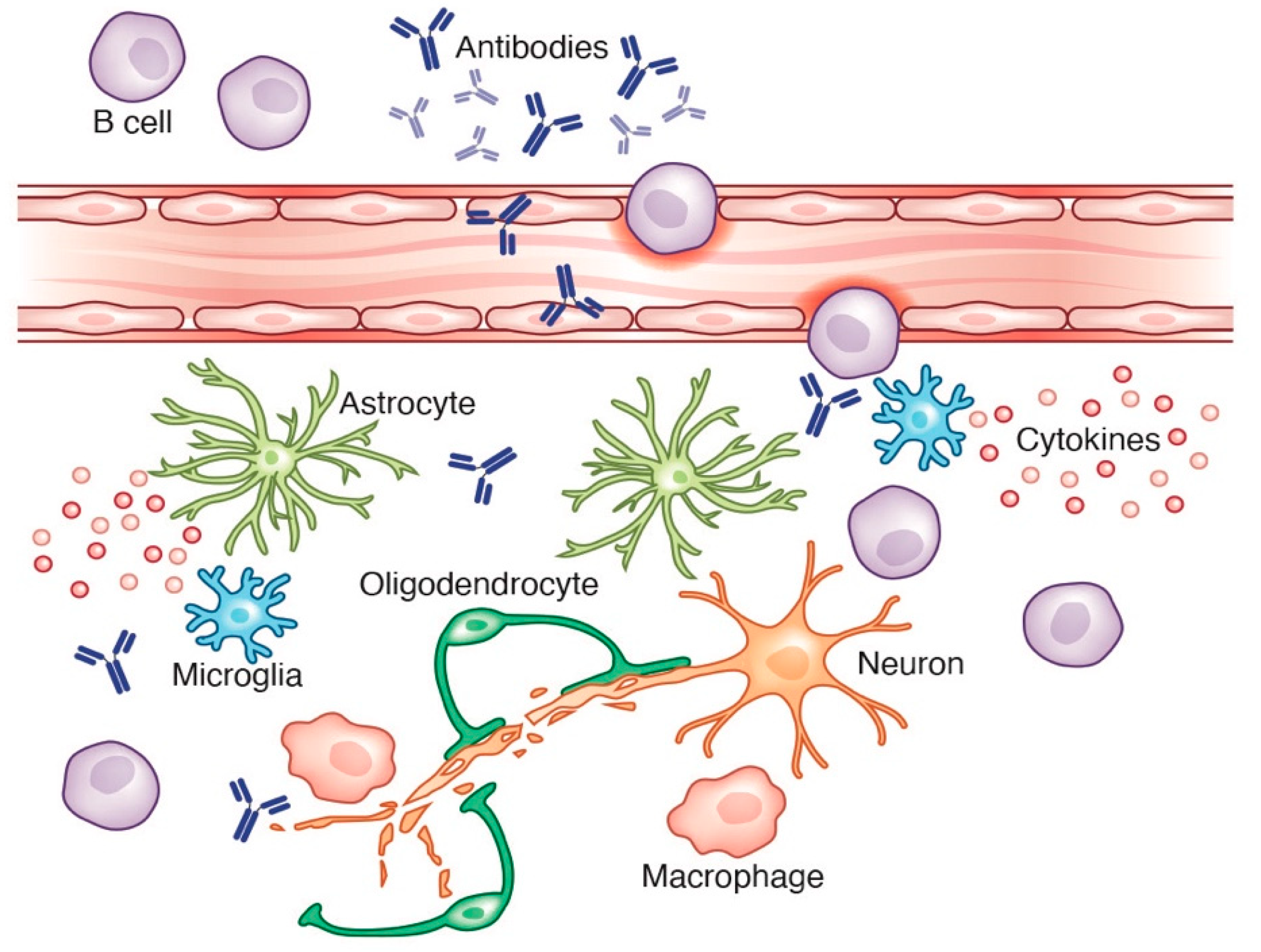
3. CNS Immune Regulation
4. B Cells in Neurological Diseases
4.1. Multiple Sclerosis
4.2. Parkinson’s Disease
4.3. Alzheimer’s Disease
5. Further Consideration and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J.; Patrick, D.J.; Hui, E.K.-W.; Lu, J.Z. Blood-Brain Barrier Transport, Plasma Pharmacokinetics, and Neuropathology Following Chronic Treatment of the Rhesus Monkey with a Brain Penetrating Humanized Monoclonal Antibody Against the Human Transferrin Receptor. Mol. Pharm. 2018, 15, 5207–5216. [Google Scholar] [CrossRef]
- Pinheiro, M.A.L.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.; Rouhani, S.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nat. Cell Biol. 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Harling-Berg, C.; Knopf, P.M.; Merriam, J.; Cserr, H.F. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J. Neuroimmunol. 1989, 25, 185–193. [Google Scholar] [CrossRef]
- Widner, H.; Möller, G.; Johansson, B.B. Immune Response in Deep Cervical Lymph Nodes and Spleen in the Mouse after Antigen Deposition in Different Intracerebral Sites. Scand. J. Immunol. 1988, 28, 563–571. [Google Scholar] [CrossRef]
- Harling-Berg, C.J.; Park, J.T.; Knopf, P.M. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J. Neuroimmunol. 1999, 101, 111–127. [Google Scholar] [CrossRef]
- Stern, J.N.; Yaari, G.; Heiden, J.V.; Church, G.; Donahue, W.F.; Hintzen, R.; Huttner, A.J.; Laman, J.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef]
- Palanichamy, A.; Apeltsin, L.; Kuo, T.C.; Sirota, M.; Wang, S.; Pitts, S.J.; Sundar, P.D.; Telman, D.; Zhao, L.Z.; Derstine, M.; et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6, 248ra106. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Grummel, V.; Wemlinger, S.; Buck, D.; Weber, M.S.; Berthele, A.; Hemmer, B. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J. Neurol. 2014, 261, 130–143. [Google Scholar] [CrossRef]
- De Graaf, M.T.; Smitt, P.A.E.S.; Luitwieler, R.L.; Van Velzen, C.; Broek, P.D.M.V.D.; Kraan, J.; Gratama, J.W. Central memory CD4+ T cells dominate the normal cerebrospinal fluid. Cytom. Part B Clin. Cytom. 2010, 80, 43–50. [Google Scholar] [CrossRef]
- Wagnon, I.; Hélie, P.; Bardou, I.; Regnauld, C.; Lesec, L.; Leprince, J.; Naveau, M.; Delaunay, B.; Toutirais, O.; Lemauff, B.; et al. Autoimmune encephalitis mediated by B-cell response against N-methyl-d-aspartate receptor. Brain 2020, 143, 2957–2972. [Google Scholar] [CrossRef] [PubMed]
- Pellkofer, H.L.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Gerdes, L.A.; Havla, J.; Bittner, R.; Canis, M.; Meinl, E.; Hohlfeld, R.; et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011, 76, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; DuPree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic Intestinal Inflammatory Condition Generates IL-10-Producing Regulatory B Cell Subset Characterized by CD1d Upregulation. Immunology 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Li, H.; Gommerman, J.; Prat, A.; Bar-Or, A. Antibody-Independent Function of Human B Cells Contributes to Antifungal T Cell Responses. J. Immunol. 2017, 198, 3245–3254. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Vinuesa, C.; Randall, K.L.; Mackay, F.; Brink, R. Control systems and decision making for antibody production. Nat. Immunol. 2010, 11, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Odendahl, M.; Mei, H.; Hoyer, B.F.; Jacobi, A.M.; Hansen, A.; Muehlinghaus, G.; Berek, C.; Hiepe, F.; Manz, R.; Radbruch, A.; et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 2005, 105, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Arce, S.; Luger, E.; Muehlinghaus, G.; Cassese, G.; Hauser, A.; Horst, A.; Lehnert, K.; Odendahl, M.; Hönemann, D.; Heller, K.-D.; et al. CD38 low IgG-secreting cells are precursors of various CD38 high-expressing plasma cell populations. J. Leukoc. Biol. 2004, 75, 1022–1028. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; De Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Bove, S. Specific B lymphocytes efficiently pick up, process and present antigen to T cells. Behring Inst. Mitt. 1985, 77, 82–87. [Google Scholar]
- Von Bergwelt-Baildon, M.S.; Vonderheide, R.H.; Maecker, B.; Hirano, N.; Anderson, K.S.; Butler, M.O.; Xia, Z.; Zeng, W.Y.; Wucherpfennig, K.W.; Nadler, L.M.; et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: Potential for clinical application. Blood 2002, 99, 3319–3325. [Google Scholar] [CrossRef] [PubMed]
- Moutai, T.; Yamana, H.; Nojima, T.; Kitamura, D. A Novel and Effective Cancer Immunotherapy Mouse Model Using Antigen-Specific B Cells Selected In Vitro. PLoS ONE 2014, 9, e92732. [Google Scholar] [CrossRef]
- Rossetti, R.A.M.; Lorenzi, N.P.C.; Yokochi, K.; Rosa, M.B.S.D.F.; Benevides, L.; Margarido, P.F.R.; Baracat, E.C.; Carvalho, J.P.; Villa, L.L.; Lepique, A.P. B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 2018, 13, e0199034. [Google Scholar] [CrossRef]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct Effector Cytokine Profiles of Memory and Naive Human B Cell Subsets and Implication in Multiple Sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef]
- Duddy, M.E.; Alter, A.; Bar-Or, A. Distinct Profiles of Human B Cell Effector Cytokines: A Role in Immune Regulation? J. Immunol. 2004, 172, 3422–3427. [Google Scholar] [CrossRef]
- Barr, T.A.; Brown, S.; Mastroeni, P.; Gray, D. TLR and B Cell Receptor Signals to B Cells Differentially Program Primary and Memory Th1 Responses toSalmonella enterica. J. Immunol. 2010, 185, 2783–2789. [Google Scholar] [CrossRef]
- Menard, L.C.; Minns, L.A.; Darche, S.; Mielcarz, D.W.; Foureau, D.M.; Roos, D.S.; Dzierszinski, F.; Kasper, L.H.; Buzoni-Gatel, D. B Cells Amplify IFN-γ Production By T Cells via a TNF-α-Mediated Mechanism. J. Immunol. 2007, 179, 4857–4866. [Google Scholar] [CrossRef]
- Randall, T.D.; Carragher, D.M.; Rangel-Moreno, J. Development of Secondary Lymphoid Organs. Annu. Rev. Immunol. 2008, 26, 627–650. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Duan, H.; Zhang, T.; Pan, Y.; Kou, Z.; Zhang, X.; Lu, Y.; Wang, S.; Yang, Z. IL-17A promotes microglial activation and neuroinflammation in mouse models of intracerebral haemorrhage. Mol. Immunol. 2016, 73, 151–157. [Google Scholar] [CrossRef]
- Suzumura, A.; Sawada, M.; Itoh, Y.; Marunouchi, T. Interleukin-4 induces proliferation and activation of microglia but suppresses their induction of class II major histocompatibility complex antigen expression. J. Neuroimmunol. 1994, 53, 209–218. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Hyvärinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-stimulation with IL-1β and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Savarin, C.; Hinton, D.R.; Valentin-Torres, A.; Chen, Z.; Trapp, B.D.; Bergmann, C.C.; Stohlman, S. Astrocyte response to IFN-γ limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J. Neuroinflamm. 2015, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Putatunda, R.; Zhang, Y.; Soni, P.V.; Li, F.; Zhang, T.; Xin, M.; Luo, J.J.; Bethea, J.R.; Cheng, Y.; et al. Lymphotoxin β receptor-mediated NFκB signaling promotes glial lineage differentiation and inhibits neuronal lineage differentiation in mouse brain neural stem/progenitor cells. J. Neuroinflamm. 2018, 15, 1–14. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF–producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef]
- Stone, S.L.; Peel, J.N.; Scharer, C.; Risley, C.A.; Chisolm, D.A.; Schultz, M.D.; Yu, B.; Ballesteros-Tato, A.; Wojciechowski, W.; Mousseau, B.; et al. T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-γ on B Cells. Immunology 2019. [Google Scholar] [CrossRef]
- Smolders, J.; Remmerswaal, E.; Schuurman, K.G.; Melief, J.; Van Eden, C.G.; van Lier, R.; Huitinga, I.; Hamann, J. Characteristics of differentiated CD8+ and CD4+ T cells present in the human brain. Acta Neuropathol. 2013, 126, 525–535. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [PubMed]
- Shelestak, J.; Singhal, N.; Frankle, L.; Tomor, R.; Sternbach, S.; McDonough, J.; Freeman, E.; Clements, R. Increased blood-brain barrier hyperpermeability coincides with mast cell activation early under cuprizone administration. PLoS ONE 2020, 15, e0234001. [Google Scholar] [CrossRef] [PubMed]
- Lepennetier, G.; Hracsko, Z.; Unger, M.; Van Griensven, M.; Grummel, V.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Kowarik, M.C. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J. Neuroinflamm. 2019, 16. [Google Scholar] [CrossRef]
- Blumenfeld-Kan, S.; Staun-Ram, E.; Miller, A. Fingolimod reduces CXCR4-mediated B cell migration and induces regulatory B cells-mediated anti-inflammatory immune repertoire. Mult. Scler. Relat. Disord. 2019, 34, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Thrash, J.C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef]
- Yoshizaki, S.; Tamaru, T.; Hara, M.; Kijima, K.; Tanaka, M.; Konno, D.-J.; Matsumoto, Y.; Nakashima, Y.; Okada, S. Microglial inflammation after chronic spinal cord injury is enhanced by reactive astrocytes via the fibronectin/β1 integrin pathway. J. Neuroinflamm. 2021, 18, 1–15. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Liberto, C.M.; Albrecht, P.J.; Herx, L.M.; Yong, V.W.; Levison, S. Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 2004, 89, 1092–1100. [Google Scholar] [CrossRef]
- Van Der Poel, M.; Ulas, T.; Mizee, M.R.; Hsiao, C.-C.; Miedema, S.; Adelia; Schuurman, K.G.; Helder, B.; Tas, S.W.; Schultze, J.L.; et al. Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.A.S.; Grandbarbe, L.; Buckley, N.J.; Niclou, S.P.; Michelucci, A. Transcriptional and epigenetic mechanisms underlying astrocyte identity. Prog. Neurobiol. 2019, 174, 36–52. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.; Chakraborty, C.; Münch, A.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Nicholas, R.; Cruciani, C.; Castellaro, M.; Romualdi, C.; Rossi, S.; Pitteri, M.; Benedetti, M.D.; Gajofatto, A.; et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann. Neurol. 2018, 83, 739–755. [Google Scholar] [CrossRef]
- Inglese, M.; Oesingmann, N.; Casaccia, P.; Fleysher, L. Progressive Multiple Sclerosis and Gray Matter Pathology: An MRI Perspective. Mt. Sinai J. Med. 2011, 78, 258–267. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Baroncini, D.; Ghezzi, A.; Annovazzi, P.; Colombo, B.; Martinelli, V.; Minonzio, G.; Moiola, L.; Rodegher, M.; Zaffaroni, M.; Comi, G. Natalizumab versus fingolimod in patients with relapsing-remitting multiple sclerosis non-responding to first-line injectable therapies. Mult. Scler. J. 2016, 22, 1315–1326. [Google Scholar] [CrossRef]
- Frischer, J.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2006, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Wei, W.; Belachew, S.; Arnold, D.L. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult. Scler. J. 2018, 25, 1915–1925. [Google Scholar] [CrossRef]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Hussman, J.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.; Haines, J.L.; Pericak-Vance, M. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016, 17, 305–312. [Google Scholar] [CrossRef]
- Weber, M.S.; Prod’Homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.E.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Hart, B.A.; Dunham, J.; Faber, B.W.; Laman, J.D.; Van Horssen, J.; Bauer, J.; Kap, Y.S. A B Cell-Driven Autoimmune Pathway Leading to Pathological Hallmarks of Progressive Multiple Sclerosis in the Marmoset Experimental Autoimmune Encephalomyelitis Model. Front. Immunol. 2017, 8, 804. [Google Scholar] [CrossRef]
- Häusler, D.; Häusser-Kinzel, S.; Feldmann, L.; Torke, S.; Lepennetier, G.; Bernard, C.C.A.; Zamvil, S.S.; Brück, W.; Lehmann-Horn, K.; Weber, M.S. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc. Natl. Acad. Sci. USA 2018, 115, 9773–9778. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Berge, I.J.M.T.; Van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.C.; Cambrook, H.E.; O’Connor, R.A.; Anderton, S.M. Induction of Passive EAE Using Myelin-Reactive CD4+ T Cells. Methods Mol. Biol. 2014, 1193, 187–198. [Google Scholar] [CrossRef]
- Maimaitijiang, G.; Watanabe, M.; Shinoda, K.; Isobe, N.; Nakamura, Y.; Masaki, K.; Matsushita, T.; Yoshikai, Y.; Kira, J.-I. Long-term use of interferon-β in multiple sclerosis increases Vδ1−Vδ2−Vγ9− γδ T cells that are associated with a better outcome. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef]
- Scheu, S.; Ali, S.; Mann-Nüttel, R.; Richter, L.; Arolt, V.; Dannlowski, U.; Kuhlmann, T.; Klotz, L.; Alferink, J. Interferon β-Mediated Protective Functions of Microglia in Central Nervous System Autoimmunity. Int. J. Mol. Sci. 2019, 20, 190. [Google Scholar] [CrossRef]
- Kap, Y.S.; Bauer, J.; Van Driel, N.; Bleeker, W.K.; Parren, P.W.; Kooi, E.-J.; Geurts, J.J.; Laman, J.D.; Craigen, J.L.; Blezer, E.; et al. B-Cell Depletion Attenuates White and Gray Matter Pathology in Marmoset Experimental Autoimmune Encephalomyelitis. J. Neuropathol. Exp. Neurol. 2011, 70, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Kap, Y.S.; Van Driel, N.; Blezer, E.; Parren, P.W.H.I.; Bleeker, W.K.; Laman, J.D.; Craigen, J.L.; Hart, B.A. ’T Late B Cell Depletion with a Human Anti-Human CD20 IgG1κ Monoclonal Antibody Halts the Development of Experimental Autoimmune Encephalomyelitis in Marmosets. J. Immunol. 2010, 185, 3990–4003. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kümpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef]
- Barnett, M.H.; Parratt, J.D.E.; Cho, E.-S.; Prineas, J.W. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 2009, 65, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Brändle, S.M.; Obermeier, B.; Senel, M.; Bruder, J.; Mentele, R.; Khademi, M.; Olsson, T.; Tumani, H.; Kristoferitsch, W.; Lottspeich, F.; et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 7864–7869. [Google Scholar] [CrossRef]
- Petzold, A. Intrathecal oligoclonal IgG synthesis in multiple sclerosis. J. Neuroimmunol. 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S.; Thompson, E.J.; Deisenhammer, F.; Giovannoni, G.; Grimsley, G.; Keir, G.; Öhman, S.; Racke, M.K.; Sharief, M.; Sindic, C.J.M.; et al. Recommended Standard of Cerebrospinal Fluid Analysis in the Diagnosis of Multiple Sclerosis: A consensus statement. Arch. Neurol. 2005, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Shu, Y.; Dai, Y.; Liu, X.; Chang, Y.; Huang, Q.; Kermode, A.G.; Qiu, W. B cell depleting therapy for multiple sclerosis overlapping with neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2018, 22, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Ramaglia, V.; Sheikh-Mohamed, S.; Legg, K.; Park, C.; Rojas, O.L.; Zandee, S.; Fu, F.; Ornatsky, O.; Swanson, E.C.; Pitt, D.; et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Eggers, E.L.; Michel, B.A.; Wu, H.; Wang, S.-Z.; Bevan, C.J.; Abounasr, A.; Pierson, N.S.; Bischof, A.; Kazer, M.; Leitner, E.; et al. Clonal relationships of CSF B cells in treatment-naive multiple sclerosis patients. JCI Insight 2017, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Zinnhardt, B.; Belloy, M.; Fricke, I.; Orije, J.; Guglielmetti, C.; Hermann, S.; Wagner, S.; Schäfers, M.; Van Der Linden, A.; Jacobs, A.H. Molecular Imaging of Immune Cell Dynamics During De- and Remyelination in the Cuprizone Model of Multiple Sclerosis by [18F]DPA-714 PET and MRI. Theranostics 2019, 9, 1523–1537. [Google Scholar] [CrossRef]
- Chu, T.; Zhang, Y.P.; Tian, Z.; Ye, C.; Zhu, M.; Shields, L.B.E.; Kong, M.; Barnes, G.N.; Shields, C.B.; Cai, J. Dynamic response of microglia/macrophage polarization following demyelination in mice. J. Neuroinflamm. 2019, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.J.; Tiwari-Woodruff, S.; Sofroniew, M.V. Reactive Astrocytes Form Scar-Like Perivascular Barriers to Leukocytes during Adaptive Immune Inflammation of the CNS. J. Neurosci. 2009, 29, 11511–11522. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 898–915. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s Disease: Substantia Nigra Regional Selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Papachroni, K.K.; Ninkina, N.; Papapanagiotou, A.; Hadjigeorgiou, G.M.; Xiromerisiou, G.; Papadimitriou, A.; Kalofoutis, A.; Buchman, V.L. Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J. Neurochem. 2006, 101, 749–756. [Google Scholar] [CrossRef]
- Bas, J.; Calopa, M.; Mestre, M.; Molleví, D.G.; Cutillas, B.; Ambrosio, S. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 2001, 113, 146–152. [Google Scholar] [CrossRef]
- Orr, C.; Rowe, D.; Mizuno, Y.; Mori, H.; Halliday, G.M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 2005, 128, 2665–2674. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal Blood–Brain Barrier Permeability in Parkinson’S Disease. Br. J. Pharmacol. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Grove, J.; Grandinetti, A.; Curb, J.; Yee, M.; Blanchette, P.; Ross, G.; Rodriguez, B. Association of Fibrinogen with Parkinson Disease in Elderly Japanese-American Men: A Prospective Study. Neuroepidemiology 2010, 34, 50–54. [Google Scholar] [CrossRef]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2008, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Besong-Agbo, D.; Wolf, E.; Jessen, F.; Oechsner, M.; Hametner, E.; Poewe, W.; Reindl, M.; Oertel, W.H.; Noelker, C.; Bacher, M.; et al. Naturally occurring -synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology 2012, 80, 169–175. [Google Scholar] [CrossRef]
- Han, M.; Nagele, E.; DeMarshall, C.; Acharya, N.; Nagele, R. Diagnosis of Parkinson’s Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS ONE 2012, 7, e32383. [Google Scholar] [CrossRef]
- He, Y.; Le, W.-D.; Appel, S.H. Role of Fcγ Receptors in Nigral Cell Injury Induced by Parkinson Disease Immunoglobulin Injection into Mouse Substantia Nigra. Exp. Neurol. 2002, 176, 322–327. [Google Scholar] [CrossRef]
- Stevens, C.; Rowe, D.; Morel-Kopp, M.-C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Souto-Carneiro, M.M.; Mahadevan, V.; Takada, K.; Fritsch-Stork, R.; Nanki, T.; Brown, M.; Fleisher, T.; Wilson, M.; Goldbach-Mansky, R.; Lipsky, P. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res. Ther. 2009, 11, R84. [Google Scholar] [CrossRef]
- Hansen, A.; Odendahl, M.; Reiter, K.; Jacobi, A.M.; Feist, E.; Scholze, J.; Burmester, G.R.; Lipsky, P.E.; Dorner, T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 2002, 46, 2160–2171. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Normal and Abnormal Biology of the β-Amyloid Precursor Protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Knezevic, D.; Mizrahi, R. Molecular imaging of neuroinflammation in Alzheimer’s disease and mild cognitive impairment. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 80, 123–131. [Google Scholar] [CrossRef]
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J. Neuroinflamm. 2014, 11, 120. [Google Scholar] [CrossRef]
- Lagarde, J.; Sarazin, M.; Bottlaender, M. In vivo PET imaging of neuroinflammation in Alzheimer’s disease. J. Neural Transm. 2017, 125, 847–867. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.D.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood-Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood–Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; Van Der Vies, S.M.; Rozemuller, A.J.; Van Horssen, J.; De Vries, H.E. Amyloid Beta Induces Oxidative Stress-Mediated Blood–Brain Barrier Changes in Capillary Amyloid Angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Alkaslasi, M.; Cho, N.; Dhillon, N.; Shelest, O.; Haro-Lopez, P.; Linaval, N.; Ghoulian, J.; Yang, A.; Vit, J.-P.; Avalos, P.; et al. Poor Corticospinal Motor Neuron Health Is Associated with Increased Symptom Severity in the Acute Phase Following Repetitive Mild TBI and Predicts Early ALS Onset in Genetically Predisposed Rodents. Brain Sci. 2021, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Montrasio, F.; Pattamatta, A.; Tusi, S.K.; Bardhi, O.; Meyer, K.D.; Hayes, L.; Nakamura, K.; Banez-Coronel, M.; Coyne, A.; et al. Antibody Therapy Targeting RAN Proteins Rescues C9 ALS/FTD Phenotypes in C9orf72 Mouse Model. Neuron 2020, 105, 645–662.e11. [Google Scholar] [CrossRef] [PubMed]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Niebroj-Dobosz, I. Auto-antibodies against proteins of spinal cord cells in cerebrospinal fluid of patients with amyotrophic lateral sclerosis (ALS). Folia Neuropathol. 2006, 44, 191–196. [Google Scholar]
- Naor, S.; Keren, Z.; Bronshtein, T.; Goren, E.; Machluf, M.; Melamed, D. Development of ALS-like disease in SOD-1 mice deficient of B lymphocytes. J. Neurol. 2009, 256, 1228–1235. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.J.; Abu-Rub, M.; Miller, R.H. B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells 2021, 10, 1605. https://doi.org/10.3390/cells10071605
Ahn JJ, Abu-Rub M, Miller RH. B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells. 2021; 10(7):1605. https://doi.org/10.3390/cells10071605
Chicago/Turabian StyleAhn, Julie J., Mohammad Abu-Rub, and Robert H. Miller. 2021. "B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights" Cells 10, no. 7: 1605. https://doi.org/10.3390/cells10071605
APA StyleAhn, J. J., Abu-Rub, M., & Miller, R. H. (2021). B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells, 10(7), 1605. https://doi.org/10.3390/cells10071605