
RelB and Neuroinflammation

Abstract
1. Introduction
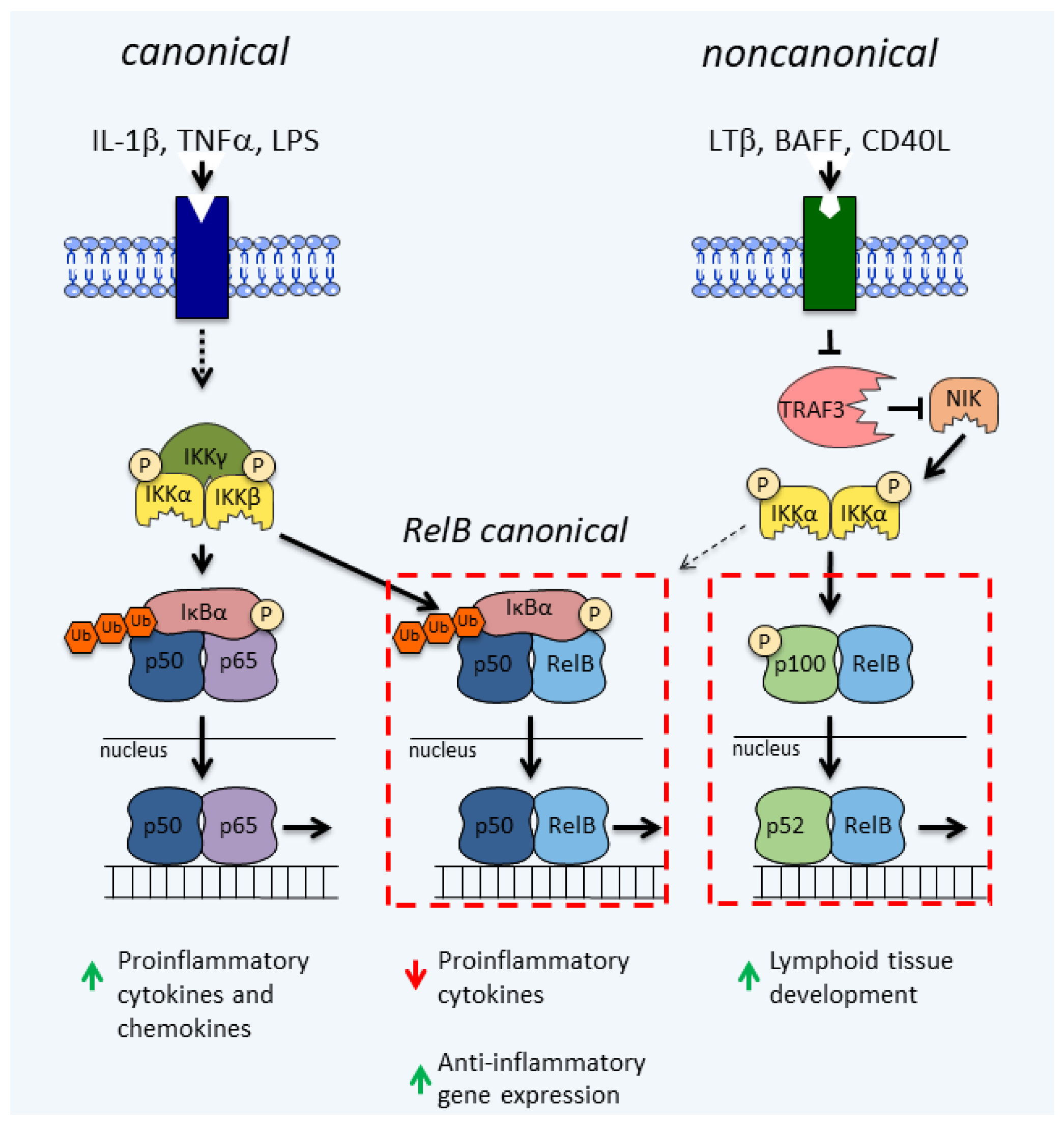
2. The NF-κB Signaling Pathways
3. RelB
3.1. RelB in the CNS
3.2. RelB in Microglia
3.3. RelB in Astrocytes
3.4. RelB in Oligodendrocytes
3.5. RelB in Neurons
3.6. RelB in Other Cells of the CNS
4. The Immunosuppressive Role of RelB in GBM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, A.M.; Williams, G.P.; Wallen, Z.D.; Standaert, D.G.; Harms, A.S. Innate and Adaptive Immune Responses in Parkinson’s Disease, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 252, ISBN 9780444642608. [Google Scholar]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, 427. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate immunity and neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Falcão, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.S.; Floriddia, E.M.; Vanichkina, D.P.; French-Constant, C. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Parsadaniantz, S.M.; Franklin, R.J.M.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef]
- Zeis, T.; Enz, L.; Schaeren-Wiemers, N. The immunomodulatory oligodendrocyte. Brain Res. 2016, 1641, 139–148. [Google Scholar] [CrossRef]
- Zaman, V.; Shields, D.C.; Shams, R.; Drasites, K.P.; Matzelle, D.; Haque, A.; Banik, N.L. Cellular and molecular pathophysiology in the progression of Parkinson’s disease. Metab. Brain Dis. 2021, 3, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, 169–176. [Google Scholar] [CrossRef]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Redmann, M.; Darley-Usmar, V.; Zhang, J. The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: Implications for neurodegenerative diseases. Aging Dis. 2016, 7, 150–162. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Jin, W.; Qazi, T.J.; Quan, Z.; Li, N.; Qing, H. Dysregulation of Transcription Factors: A Key Culprit Behind Neurodegenerative Disorders. Neuroscientist 2019, 25, 548–565. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor κB (NF-κB)–Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Yang, M.G.; Sun, L.; Han, J.; Zheng, C.; Liang, H.; Zhu, J.; Jin, T. Biological characteristics of transcription factor RelB in different immune cell types: Implications for the treatment of multiple sclerosis. Mol. Brain 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Shih, V.F.S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Körner, M.; Baud, V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation. Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK Complex, a Central Regulator of NF- B Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.B.; Weih, D.S.; Sivakumar, V.; Weih, F. RelB is required for Peyer’s patch development: Differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Coope, H.J.; Atkinson, P.G.P.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.J.; Klaus, G.G.B.; Johnston, L.H.; Ley, S.C. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef]
- Saitoh, T.; Nakayama, M.; Nakano, H.; Yagita, H.; Yamamoto, N.; Yamaoka, S. TWEAK Induces NF-κB2 p100 processing and long lasting nf-κB Activation. J. Biol. Chem. 2003, 278, 36005–36012. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to Induce TNFα-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Gardam, S.; Sierro, F.; Basten, A.; Mackay, F.; Brink, R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B Cells by the BAFF Receptor. Immunity 2008, 28, 391–401. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.Z.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-κB-Inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 77, 401–409. [Google Scholar] [CrossRef]
- Fong, A.; Sun, S.C. Genetic evidence for the essential role of β-transducin repeat-containing protein in the inducible processing of NF-κB2/p100. J. Biol. Chem. 2002, 277, 22111–22114. [Google Scholar] [CrossRef]
- Millet, P.; McCall, C.; Yoza, B. RelB: An outlier in leukocyte biology. J. Leukoc. Biol. 2013, 94, 941–951. [Google Scholar] [CrossRef]
- Gasparini, C.; Foxwell, B.M.J.; Feldmann, M. RelB/p50 regulates CCL19 production, but fails to promote human DC maturation. Eur. J. Immunol. 2009, 39, 2215–2223. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Yester, J.W.; Singh, S.K.; Biswas, D.D.; Surace, M.J.; Waters, M.R.; Hauser, K.F.; Yao, Z.; Boyce, B.F.; Kordula, T. RelB/p50 Complexes Regulate Cytokine-Induced YKL-40 Expression. J. Immunol. 2015, 194, 2862–2870. [Google Scholar] [CrossRef]
- Shih, V.F.S.; Davis-Turak, J.; MacAl, M.; Huang, J.Q.; Ponomarenko, J.; Kearns, J.D.; Yu, T.; Fagerlund, R.; Asagiri, M.; Zuniga, E.I.; et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat. Immunol. 2012, 13, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Solan, N.J.; Miyoshi, H.; Carmona, E.M.; Bren, G.D.; Paya, C.V. RelB cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem. 2002, 277, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.J.; Savinova, O.V.; Talwar, R.; Kearns, J.D.; Hoffmann, A.; Ghosh, G. Stabilization of RelB requires multidomain interactions with p100/p52. J. Biol. Chem. 2008, 283, 12324–12332. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.; Ryseck, R.P.; Bravo, R. Expression of relB transcripts during lymphoid organ development: Specific expression in dendritic antigen-presenting cells. Development 1993, 118, 1221–1231. [Google Scholar] [CrossRef]
- Gupta, A.S.; Waters, M.R.; Biswas, D.D.; Brown, L.N.; Surace, M.J.; Floros, C.; Siebenlist, U.; Kordula, T. RelB controls adaptive responses of astrocytes during sterile inflammation. Glia 2019, 67, 1449–1461. [Google Scholar] [CrossRef]
- Chen, X.; Yoza, B.K.; El Gazzar, M.; Hu, J.Y.Q.; Cousart, S.L.; McCall, C.E. RelB Sustains IκBα Expression during Endotoxin Tolerance. Clin. Vaccine Immunol. 2009, 16, 104–110. [Google Scholar] [CrossRef]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef]
- Marienfeld, R.; May, M.J.; Berberich, I.; Serfling, E.; Ghosh, S.; Neumann, M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003, 278, 19852–19860. [Google Scholar] [CrossRef]
- RELB RELB proto-oncogene, NF-kB subunit [ Homo sapiens (human). Available online: https://www.ncbi.nlm.nih.gov/gene/5971 (accessed on 18 May 2021).
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the RelB gene is regulated by NF-κB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef]
- Lernbecher, T.; Kistler, B.; Wirth, T. Two distinct mechanisms contribute to the constitutive activation of RelB in lymphoid cells. EMBO J. 1994, 13, 4060–4069. [Google Scholar] [CrossRef] [PubMed]
- Ammon, C.; Mondal, K.; Andreesen, R.; Krause, S.W. Differential expression of the transcription factor NF-κB during human mononuclear phagocyte differentiation to macrophages and dendritic cells. Biochem. Biophys. Res. Commun. 2000, 268, 99–105. [Google Scholar] [CrossRef]
- Ruben, S.M.; Klement, J.F.; Coleman, T.A.; Maher, M.; Chen, C.H.; Rosen, C.A. I-Rel: A novel rel-related protein that inhibits NF-κB transcriptional activity. Genes Dev. 1992, 6, 745–760. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cramer, P.; Müller, C.W. Engineering of diffraction-quality crystals of the NF-κB p52 homodimer:DNA complex. FEBS Lett. 1997, 405, 373–377. [Google Scholar] [CrossRef]
- Ghosh, G.; Van Duyne, G.; Ghosh, S.; Sigler, P.B. Structure of nf-κb p50 homodimer bound to a κb site. Nature 1995, 373, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.W.; Rey, F.A.; Sodeoka, M.; Verdine, G.L.; Harrison, S.C. Structure of the nf-κb p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κb bound to DNA. Nature 1998, 391, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Q.; Ghosh, S.; Ghosh, G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol. 1998, 5, 67–73. [Google Scholar] [CrossRef]
- Huang, D.B.; Chen, Y.Q.; Ruetsche, M.; Phelps, C.B.; Ghosh, G. X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure 2001, 9, 669–678. [Google Scholar] [CrossRef]
- Ryseck, R.P.; Novotny, J.; Bravo, R. Characterization of elements determining the dimerization properties of RelB and p50. Mol. Cell. Biol. 1995, 15, 3100–3109. [Google Scholar] [CrossRef]
- Maier, H.J.; Marienfeld, R.; Wirth, T.; Baumann, B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J. Biol. Chem. 2003, 278, 39242–39250. [Google Scholar] [CrossRef] [PubMed]
- Ryseck, R.P.; Bull, P.; Takamiya, M.; Bours, V.; Siebenlist, U.; Dobrzanski, P.; Bravo, R. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol. Cell. Biol. 1992, 12, 674–684. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; Ghosh, S. Rel/NF-κB and IκB proteins: An overview. Semin. Cancer Biol. 1997, 8, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, A.K.; Huang, D.B.; Wang, V.Y.F.; Vu, D.; Ghosh, G. X-ray Structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB Sites. J. Mol. Biol. 2007, 373, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Dobrzanski, P.; Ryseck, R.P.; Bravo, R. Both N- and C-terminal domains of RelB are required for full transactivation: Role of the N-terminal leucine zipper-like motif. Mol. Cell. Biol. 1993, 13, 1572–1582. [Google Scholar] [CrossRef]
- Baud, V.; Collares, D. Post-translational modifications of RelB NF-κB subunit and associated functions. Cells 2016, 5, 22. [Google Scholar] [CrossRef]
- Marienfeld, R.; Berberich-Siebelt, F.; Berberich, I.; Denk, A.; Serfling, E.; Neumann, M. Signal-specific and phosphorylation-dependent RelB degradation: A potential mechanism of NF-κB control. Oncogene 2001, 20, 8142–8147. [Google Scholar] [CrossRef]
- Neumann, M.; Klar, S.; Wilisch-Neumann, A.; Hollenbach, E.; Kavuri, S.; Leverkus, M.; Kandolf, R.; Brunner-Weinzierl, M.C.; Klingel, K. Glycogen synthase kinase-3Β is a crucial mediator of signal-induced RelB degradation. Oncogene 2011, 30, 2485–2492. [Google Scholar] [CrossRef]
- Authier, H.; Billot, K.; Derudder, E.; Bordereaux, D.; Riviere, P.; Rodrigues-Ferreira, S.; Nahmias, C.; Baud, V. IKK phosphorylates RelB to modulate its promoter specificity and promote fibroblast migration downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 14794–14799. [Google Scholar] [CrossRef]
- Weih, D.S.; Yilmaz, Z.B.; Weih, F. Essential Role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol. 2001, 167, 1909–1919. [Google Scholar] [CrossRef]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-κB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef]
- Burkly, L.; Hession, C.; Ogata, L.; Reilly, C.; Marconl, L.A.; Olson, D.; Tizard, R.; Gate, R.; Lo, D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature 1995, 373, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Vaira, S.; Johnson, T.; Hirbe, A.C.; Alhawagri, M.; Anwisye, I.; Sammut, B.; O’Neal, J.; Zou, W.; Weilbaecher, K.N.; Faccio, R.; et al. RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Weih, F.; Durham, S.K.; Barton, D.S.; Sha, W.C.; Baltimore, D.; Bravo, R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J. Immunol. 1996, 157, 3974–3979. [Google Scholar] [PubMed]
- Yoza, B.K.; Hu, J.Y.-Q.; Cousart, S.L.; Forrest, L.M.; McCall, C.E. Induction of RelB participates in endotoxin tolerance. J. Immunol. 2006, 177, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Strong, R.; Zhang, J.; Grotta, J.C.; Aronowski, J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-κB subunit, iNOS, and COX-2 expression. J. Neurochem. 2007, 101, 652–663. [Google Scholar] [CrossRef]
- Richa, R.; Yadawa, A.K.; Chaturvedi, C.M. Hyperglycemia and high nitric oxide level induced oxidative stress in the brain and molecular alteration in the neurons and glial cells of laboratory mouse, Mus musculus. Neurochem. Int. 2017, 104, 64–79. [Google Scholar] [CrossRef]
- Mitra, S.; Ghosh, N.; Sinha, P.; Chakrabarti, N.; Bhattacharyya, A. Alteration of nuclear factor-kappaB pathway promote neuroinflammation depending on the functions of estrogen receptors in substantia nigra after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment. Neurosci. Lett. 2016, 616, 86–92. [Google Scholar] [CrossRef]
- Riphagen, J.M.; Ramakers, I.H.G.M.; Freeze, W.M.; Pagen, L.H.G.; Hanseeuw, B.J.; Verbeek, M.M.; Verhey, F.R.J.; Jacobs, H.I.L. Linking APOE-ε4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol. Aging 2020, 85, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Nho, K.; Kim, S.; Horgusluoglu, E.; Risacher, S.L.; Shen, L.; Kim, D.; Lee, S.; Foroud, T.; Shaw, L.M.; Trojanowski, J.Q.; et al. Association analysis of rare variants near the APOE region with CSF and neuroimaging biomarkers of Alzheimer’s disease. BMC Med. Genom. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, E.; Chen, Q.; Goldman, A.L.; Tan, H.Y.; Healy, K.; Zoltick, B.; Das, S.; Kolachana, B.; Callicott, J.H.; Dickinson, D.; et al. Late-Onset Alzheimer’s Disease polygenic risk profile score predicts hippocampal function. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 673–679. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J. Immunol. Res. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mosser, C.A.; Baptista, S.; Arnoux, I.; Audinat, E. Microglia in CNS development: Shaping the brain for the future. Prog. Neurobiol. 2017, 149, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, M.Z.; Yang, Z.Y.; Jin, W.L. Microglia in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 270–280. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the central nervous system. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Schaafsma, W.; Zhang, X.; van Zomeren, K.C.; Jacobs, S.; Georgieva, P.B.; Wolf, S.A.; Kettenmann, H.; Janova, H.; Saiepour, N.; Hanisch, U.K.; et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain. Behav. Immun. 2015, 48, 205–221. [Google Scholar] [CrossRef]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar, S.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef]
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar] [CrossRef]
- Kiebala, M.; Polesskaya, O.; Yao, Z.; Perry, S.W.; Maggirwa, S.B. Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Laspia, M.F.; Rice, A.P.; Mathews, M.B. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 1989, 59, 283–292. [Google Scholar] [CrossRef]
- Gupta, A.S.; Biswas, D.D.; Brown, L.S.N.; Mockenhaupt, K.; Marone, M.; Hoskins, A.; Siebenlist, U.; Kordula, T. A detrimental role of RelB in mature oligodendrocytes during experimental acute encephalomyelitis. J. Neuroinflammation 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, J.; Chen, Z.; Wang, H.; Xue, H.; Yang, C.; Guo, Q.; Qi, Y.; Guo, X.; Qian, M.; et al. Transfer of MicroRNA via macrophage-derived extracellular vesicles promotes proneural-to-mesenchymal transition in glioma stem cells. Cancer Immunol. Res. 2020, 8, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.M.; Lee, D.W.; Jung, J.-U.; Sitcheran, R. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-κB-inducing kinase (NIK) and noncanonical NF-κB signaling. Mol. Cancer 2015, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, N.; Nakatani, Y.; Yamashita, D.; Ohue, S.; Ohnishi, T.; Kondo, T. Eva1 maintains the stem-like character of glioblastoma-initiating cells by activating the noncanonical NF-κB signaling pathway. Cancer Res. 2016, 76, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.R.; Gupta, A.S.; Mockenhaupt, K.; Brown, L.S.N.; Biswas, D.D.; Kordula, T. RelB acts as a molecular switch driving chronic inflammation in glioblastoma multiforme. Oncogenesis 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Luo, L.; Sun, B.; Sun, D. Roles of glial ion transporters in brain diseases. Glia 2020, 68, 472–494. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef]
- Zhang, Y.; Barres, B.A. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 2010, 20, 588–594. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef]
- Farmer, W.T.; Murai, K. Resolving Astrocyte Heterogeneity in the CNS. Front. Cell. Neurosci. 2017, 11, 1–7. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Jang, E.; Kim, J.-H.; Lee, S.; Kim, J.-H.; Seo, J.-W.; Jin, M.; Lee, M.-G.; Jang, I.-S.; Lee, W.-H.; Suk, K. Phenotypic Polarization of Activated Astrocytes: The Critical Role of Lipocalin-2 in the Classical Inflammatory Activation of Astrocytes. J. Immunol. 2013, 191, 5204–5219. [Google Scholar] [CrossRef]
- Beurel, E. HDAC6 regulates LPS-tolerance in astrocytes. PLoS ONE 2011, 6, e25804. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. Modulation of NF-κB activity by exchange of dimers. Mol. Cell 2003, 11, 1563–1574. [Google Scholar] [CrossRef]
- Shih, V.F.S.; Kearns, J.D.; Basak, S.; Savinova, O.V.; Ghosh, G.; Hoffmann, A. Kinetic control of negative feedback regulators of NF-κB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc. Natl. Acad. Sci. USA 2009, 106, 9619–9624. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Bojanowski, K.; Chaudhuri, R.K. A novel fumarate, isosorbide di-(methyl fumarate) (IDMF), replicates astrocyte transcriptome responses to dimethyl fumarate (DMF) but specifically down-regulates genes linked to a reactive phenotype. Biochem. Biophys. Res. Commun. 2020, 532, 475–481. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An epigenetic journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 480–481. [Google Scholar] [CrossRef]
- Dowling, P.; Husar, W.; Menonna, J.; Donnenfeld, H.; Cook, S.; Sidhu, M. Cell death and birth in multiple sclerosis brain. J. Neurol. Sci. 1997, 149, 1–11. [Google Scholar] [CrossRef]
- Pender, M.P.; Nguyen, K.B.; McCombe, P.A.; Kerr, J.F.R. Apoptosis in the nervous system in experimental allergic encephalomyelitis. J. Neurol. Sci. 1991, 104, 81–87. [Google Scholar] [CrossRef]
- Hisahara, S.; Okano, H.; Miura, M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci. Res. 2003, 46, 387–397. [Google Scholar] [CrossRef]
- Schmitz, T.; Chew, L.J. Cytokines and myelination in the central nervous system. Sci. World J. 2008, 8, 1119–1147. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, R.S.T.J.; Wing, M.G.; Compston, A. Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-κB. Eur. J. Neurosci. 2001, 13, 959–967. [Google Scholar] [CrossRef]
- Hamanoue, M.; Yoshioka, A.; Ohashi, T.; Eto, Y.; Takamatsu, K. NF-kappaB prevents TNF-alpha-induced apoptosis in an oligodendrocyte cell line. Neurochem. Res. 2004, 29, 1571–1576. [Google Scholar] [CrossRef]
- Stone, S.; Jamison, S.; Yue, Y.; Durose, W.; Schmidt-Ullrich, R.; Lin, W. NF-κB activation protects oligodendrocytes against inflammation. J. Neurosci. 2017, 37, 9332–9344. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Yue, Y.; Stone, S.; Wu, S.; Lin, W. NF-κB activation accounts for the cytoprotective effects of PERK activation on oligodendrocytes during EAE. J. Neurosci. 2020, 40, 6444–6456. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.-L.; McKinsey, T.A.; Liu, L.; Gentry, J.J.; Malim, M.H.; Ballard, D.W. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF- B control. Proc. Natl. Acad. Sci. USA 1997, 94, 10057–10062. [Google Scholar] [CrossRef]
- Tsunemoto, R.; Lee, S.; Szűcs, A.; Chubukov, P.; Sokolova, I.; Blanchard, J.W.; Eade, K.T.; Bruggemann, J.; Wu, C.; Torkamani, A.; et al. Diverse reprogramming codes for neuronal identity. Nature 2018, 557, 375–380. [Google Scholar] [CrossRef]
- Zhang, R.S.; Liakath-Ali, K.; Südhof, T.C. Latrophilin-2 and latrophilin-3 are redundantly essential for parallel-fiber synapse function in cerebellum. Elife 2020, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hassan, B.A.; Hiesinger, P.R. Beyond Molecular Codes: Simple Rules to Wire Complex Brains. Cell 2015, 163, 285–291. [Google Scholar] [CrossRef]
- Yogev, S.; Shen, K. Cellular and Molecular Mechanisms of Synaptic Specificity. Annu. Rev. Cell Dev. Biol. 2014, 30, 417–437. [Google Scholar] [CrossRef]
- Li, H.; Shuster, S.A.; Li, J.; Luo, L. Linking neuronal lineage and wiring specificity. Neural Dev. 2018, 13, 1–19. [Google Scholar] [CrossRef]
- Engelmann, C.; Haenold, R. Transcriptional Control of Synaptic Plasticity by Transcription Factor NF-κB. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Meffert, M.K. Cellular specificity of NF-κB function in the nervous system. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Riemann, M.; Carlstedt, S.; Grimlowski, R.; Andreas, N.; Koliesnik, I.; Meier, E.; Austerfield, P.; Haenold, R. Identification of undescribed Relb expression domains in the murine brain by new Relb:cre-katushka reporter mice. Dev. Dyn. 2020, 249, 983–997. [Google Scholar] [CrossRef]
- Schmeisser, M.J.; Baumann, B.; Johannsen, S.; Vindedal, G.F.; Jensen, V.; Hvalby, O.C.; Sprengel, R.; Seither, J.; Maqbool, A.; Magnutzki, A.; et al. IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J. Neurosci. 2012, 32, 5688–5703. [Google Scholar] [CrossRef] [PubMed]
- Bhakar, A.L.; Tannis, L.L.; Zeindler, C.; Russo, M.P.; Jobin, C.; Park, D.S.; MacPherson, S.; Barker, P.A. Constitutive nuclear factor-κB activity is required for central neuron survival. J. Neurosci. 2002, 22, 8466–8475. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, X.; Liu, M.; Cao, J.; Chen, J.; Wang, H.; Niu, H.; Yu, Z.; Yu, J.; Wang, T.; et al. A new alternative NF-ΚB Pathway mediated the neuroprotection of GDNF on 6-OHDA-induced da neurons neurotoxicity. Brain Res. 2012, 1437, 38–49. [Google Scholar] [CrossRef]
- Xiao, X.; Putatunda, R.; Zhang, Y.; Soni, P.V.; Li, F.; Zhang, T.; Xin, M.; Luo, J.J.; Bethea, J.R.; Cheng, Y.; et al. Lymphotoxin β receptor-mediated NFκB signaling promotes glial lineage differentiation and inhibits neuronal lineage differentiation in mouse brain neural stem/progenitor cells. J. Neuroinflammation 2018, 15, 1–14. [Google Scholar] [CrossRef]
- Calandria, J.M.; Asatryan, A.; Balaszczuk, V.; Knott, E.J.; Jun, B.K.; Mukherjee, P.K.; Belayev, L.; Bazan, N.G. NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neural cell survival. Cell Death Differ. 2015, 22, 1363–1377. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.H.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflammation 2018, 15, 1–18. [Google Scholar] [CrossRef]
- Rudziak, P.; Ellis, C.G.; Kowalewska, P.M. Role and Molecular Mechanisms of Pericytes in Regulation of Leukocyte Diapedesis in Inflamed Tissues. Mediators Inflamm. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Qin, W.; Li, J.; Zhu, R.; Gao, S.; Fan, J.; Xia, M.; Zhao, R.C.; Zhang, J. Melatonin protects blood-brain barrier integrity and permeability by inhibiting matrix metalloproteinase-9 via the NOTCH3/NF-kB pathway. Aging 2019, 11, 11391–11415. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.-H. Interleukin-1β induces pericyte apoptosis via the NF-κB pathway in diabetic retinopathy. Biochem. Biophys. Res. Commun. 2021, 546, 46–53. [Google Scholar] [CrossRef]
- Liang, Q.; Zhang, L.; Wood, R.W.; Ji, R.C.; Boyce, B.F.; Schwarz, E.M.; Wang, Y.; Xing, L. Avian reticuloendotheliosis viral oncogene related B regulates lymphatic endothelial cells during vessel maturation and is required for lymphatic vessel function in adult mice. Am. J. Pathol. 2019, 189, 2516–2530. [Google Scholar] [CrossRef]
- Andreas, N.; Potthast, M.; Geiselhöringer, A.-L.; Garg, G.; de Jong, R.; Riewaldt, J.; Russkamp, D.; Riemann, M.; Girard, J.-P.; Blank, S.; et al. RelB deficiency in dendritic cells protects from autoimmune inflammation due to spontaneous accumulation of Tissue T regulatory cells. J. Immunol. 2019, 203, 2602–2613. [Google Scholar] [CrossRef] [PubMed]
- Obara-Michlewska, M.; Szeliga, M. Targeting glutamine addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Caniglia, J.L.; Jalasutram, A.; Asuthkar, S.; Sahagun, J.; Park, S.; Ravindra, A.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Beyond glucose: Alternative sources of energy in glioblastoma. Theranostics 2021, 11, 2048–2057. [Google Scholar] [CrossRef]
- Russo, C.; Lisi, L.; Tentori, L.; Navarra, P.; Graziani, G.; Combs, C. Exploiting microglial functions for the treatment of glioblastoma. Curr. Cancer Drug Targets 2017, 17, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. J. Am. Med. Assoc. 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Tanaka, S.; Louis, D.N.; Curry, W.T.; Batchelor, T.T.; Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat. Rev. Clin. Oncol. 2013, 10, 14–26. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Behnan, J.; Stangeland, B.; Hosainey, S.A.M.; Joel, M.; Olsen, T.K.; Micci, F.; Glover, J.C.; Isakson, P.; Brinchmann, J.E. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene 2017, 36, 570–584. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Cheng, W.; Ren, X.; Zhang, C.; Cai, J.; Liu, Y.; Han, S.; Wu, A. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology 2016, 86, 2226–2234. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef]
- Verhaak, R.G.W. Moving the needle: Optimizing classification for glioma. Sci. Transl. Med. 2016, 8, 350fs14. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.I.; Petritsch, C.; Swartling, F.J.; Itsara, M.; Sim, F.J.; Auvergne, R.; Goldenberg, D.D.; Vandenberg, S.R.; Nguyen, K.N.; Yakovenko, S.; et al. Non-stem cell origin for oligodendroglioma. Cancer Cell 2010, 18, 669–682. [Google Scholar] [CrossRef]
- Ledur, P.F.; Liu, C.; He, H.; Harris, A.R.; Minussi, D.C.; Zhou, H.Y.; Shaffrey, M.E.; Asthagiri, A.; Lopes, M.B.S.; Schiff, D.; et al. Culture conditions tailored to the cell of origin are critical for maintaining native properties and tumorigenicity of glioma cells. Neuro. Oncol. 2016, 18, 1413–1424. [Google Scholar] [CrossRef]
- Sutcliffe, M.D.; Galvao, R.P.; Wang, L.; Kim, J.; Rosenfeld, L.K.; Singh, S.; Zong, H.; Janes, K.A. Premalignant oligodendrocyte precursor cells stall in a heterogeneous state of replication stress prior to gliomagenesis. Cancer Res. 2021, 81, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 1–12. [Google Scholar] [CrossRef]
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Deumelandt, K.; van der Vliet, H.J.; Wesseling, P.; de Gruijl, T.D. Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: How to break a vicious cycle. Biochim. Biophys. Acta 2014, 1846, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef]
- Osterberg, N.; Ferrara, N.; Vacher, J.; Gaedicke, S.; Niedermann, G.; Weyerbrock, A.; Doostkam, S.; Schaefer, H.E.; Plate, K.H.; Machein, M.R. Decrease of VEGF-A in myeloid cells attenuates glioma progression and prolongs survival in an experimental glioma model. Neuro. Oncol. 2016, 18, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Ku, M.C.; Markovic, D.; Dzaye, O.D.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 2014, 135, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Langenfurth, A.; Rinnenthal, J.L.; Vinnakota, K.; Prinz, V.; Carlo, A.S.; Stadelmann, C.; Siffrin, V.; Peaschke, S.; Endres, M.; Heppner, F.; et al. Membrane-type 1 metalloproteinase is upregulated in microglia/brain macrophages in neurodegenerative and neuroinflammatory diseases. J. Neurosci. Res. 2014, 92, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H. Microglia in the tumor microenvironment: Taking their TOLL on glioma biology. Neuro. Oncol. 2015, 17, 171–173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, J.; Chen, X.; Chu, J.; Xu, B.; Meisen, W.H.; Chen, L.; Zhang, L.; Zhang, J.; He, X.; Wang, Q.E.; et al. TGFβ treatment enhances glioblastoma virotherapy by inhibiting the innate immune response. Cancer Res. 2015, 75, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased Regulatory T-Cell Fraction Amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients with Malignant Glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro. Oncol. 2006, 8, 234–243. [Google Scholar] [CrossRef]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef]
- Jahani-As, A.; Yin, H.; Soleimani, V.D.; Haque, T.; Luchman, H.A.; Chang, N.C.; Sincennes, M.C.; Puram, S.V.; Scott, A.M.; Lorimer, I.A.J.; et al. Control of glioblastoma tumorigenesis by feed-forward cytokine signaling. Nat. Neurosci. 2016, 19, 798–806. [Google Scholar] [CrossRef]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Cajal, S.R.Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis article. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef]
- Priego, N.; Valiente, M. The potential of astrocytes as immune modulators in brain tumors. Front. Immunol. 2019, 10, 1314. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.C.; Maier, L.M.; D’Amico, R.; Mandigo, C.E.; Fontana, E.J.; Waziri, A.; Assanah, M.C.; Canoll, P.; Anderson, R.C.E.; Anderson, D.E.; et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol. 2009, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Long, W.; Xing, C.; Chu, J.; Luo, M.; Wang, H.Y.; Liu, Q.; Wang, R.F. Cancer stem cells and immunosuppressive microenvironment in glioma. Front. Immunol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Hong, J.-H.; Hsueh, C.; Chiang, C.-S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Gonçalves, R.M.; Klafke, K.; de Souza, P.O.; Dillenburg, F.C.; Carro, L.; Gelain, D.P.; Moreira, J.C.F. Inflammatory landscape of human brain tumors reveals an NFκB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017, 390, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Wang, K.; Huang, R.; Liu, Y.; Zhang, Y.; Hu, H. RELB: A novel prognostic marker for glioblastoma as identified by population-based analysis. Oncol. Lett. 2019, 18, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Ramakrishnan, D.; Valenta, J.; Parney, I.F.; Bayless, K.J.; Sitcheran, R. The NF-κB RelB Protein Is an Oncogenic Driver of Mesenchymal Glioma. PLoS ONE 2013, 8, e57489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, L.; Yin, L. Silencing LncRNA LOXL1-AS1 attenuates mesenchymal characteristics of glioblastoma via NF-κB pathway. Biochem. Biophys. Res. Commun. 2018, 500, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-mesenchymal transition: Phenotypic plasticity to acquire multitherapy resistance in glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Walker, K.; Ayokanmbi, A.; Gordon, E.R.; Whetsel, J.; Smith, C.M.; Sanchez, R.G.; Lubin, F.D.; Chakraborty, A.; Tran, A.N.; et al. Chromodomain helicase DNA-Binding Protein 7 is suppressed in the perinecrotic/ischemic microenvironment and is a novel regulator of glioblastoma angiogenesis. Stem Cells 2019, 37, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Hummelink, K.; Hollingsworth, F.; Wani, K.; James, J.D.; Goodman, L.D.; Conroy, S.; Long, L.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 1–22. [Google Scholar] [CrossRef]
- Singh, S.K.; Bhardwaj, R.; Wilczynska, K.M.; Dumur, C.I.; Kordula, T. A complex of nuclear factor I-X3 and STAT3 regulates astrocyte and glioma migration through the secreted glycoprotein YKL-40. J. Biol. Chem. 2011, 286, 29893–29903. [Google Scholar] [CrossRef]
- Pflug, K.M. Sitcheran Targeting NF-κB-Inducing Kinase (NIK) in immunity, inflammation, and cancer. Int. J. Mol. Sci. 2020, 21, 8470. [Google Scholar] [CrossRef]
- Jung, J.U.; Ravi, S.; Lee, D.W.; McFadden, K.; Kamradt, M.L.; Toussaint, L.G.; Sitcheran, R. NIK/MAP3K14 regulates mitochondrial dynamics and trafficking to promote cell invasion. Curr. Biol. 2016, 26, 3288–3302. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.L.; Lee, D.W.; Jung, J.-U.; Ravi, S.; Pogue, C.B.; Toussaint, L.G.; Bayless, K.J.; Sitcheran, R. NIK regulates MT1-MMP activity and promotes glioma cell invasion independently of the canonical NF-κB pathway. Oncogenesis 2016, 5, e231. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Jennewein, C.; Eckhardt, I.; Rajalingam, K.; Fulda, S. Identification of non-canonical NF-kB signaling as a critical mediator of smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Didonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Tchoghandjian, A.; Jennewein, C.; Eckhardt, I.; Momma, S.; Figarella-Branger, D.; Fulda, S. Smac mimetic promotes glioblastoma cancer stem-like cell differentiation by activating NF-κB. Cell Death Differ. 2014, 21, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, I.; Roesler, S.; Fulda, S. Identification of DR5 as a critical, NF-κB-regulated mediator of Smac-induced apoptosis. Cell Death Dis. 2013, 4, 1–14. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Perrot-Applanat, M.; Vacher, S.; Toullec, A.; Pelaez, I.; Velasco, G.; Cormier, F.; Saad, H.E.S.; Lidereau, R.; Baud, V.; Bièche, I. Similar NF-κB Gene Signatures in TNF-α treated human endothelial cells and breast tumor biopsies. PLoS ONE 2011, 6, e21589. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
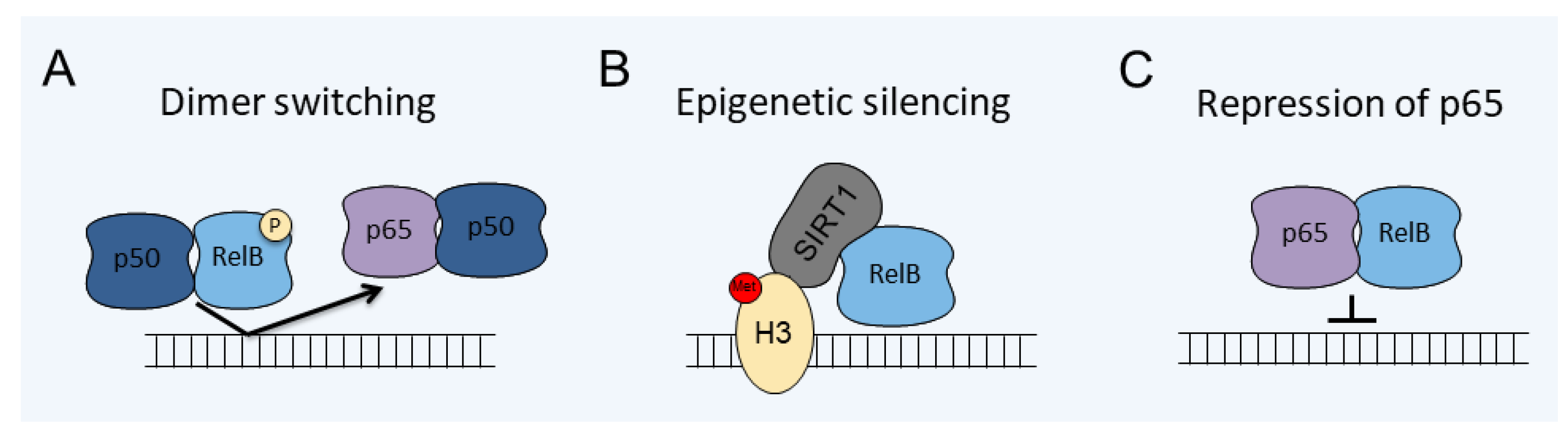
| Model | Mechanism | Cell Type | Reference |
|---|---|---|---|
| LPS induced tolerance | Epigenetic silencing | Mouse microglia | [99] |
| HIV/Tat induction | Repression of p65 | Mouse microglia | [102] |
| IL-1β induced tolerance | Dimer switching/phosphorylation | Human astrocytes | [47] |
| EAE | Repression of p65 | Mouse oligodendrocytes | [104] |
| miRNA inhibition of CDH7 | Activation of RelB/p50 | Human glioma stem cells | [105] |
| TWEAK induction | Activation of noncanonical pathway (RelB/p52) | Human glioma cells | [106] |
| Overexpression of Eva1 | Activation of noncanonical pathway (RelB/p52) | Human glioma-initiating cells | [107] |
| IL-1 β and OSM stimulation | RelB/p50/YY1 complex formation | Human GBM cells | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mockenhaupt, K.; Gonsiewski, A.; Kordula, T. RelB and Neuroinflammation. Cells 2021, 10, 1609. https://doi.org/10.3390/cells10071609
Mockenhaupt K, Gonsiewski A, Kordula T. RelB and Neuroinflammation. Cells. 2021; 10(7):1609. https://doi.org/10.3390/cells10071609
Chicago/Turabian StyleMockenhaupt, Karli, Alexandra Gonsiewski, and Tomasz Kordula. 2021. "RelB and Neuroinflammation" Cells 10, no. 7: 1609. https://doi.org/10.3390/cells10071609
APA StyleMockenhaupt, K., Gonsiewski, A., & Kordula, T. (2021). RelB and Neuroinflammation. Cells, 10(7), 1609. https://doi.org/10.3390/cells10071609