
Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications

 ,
, 

Abstract
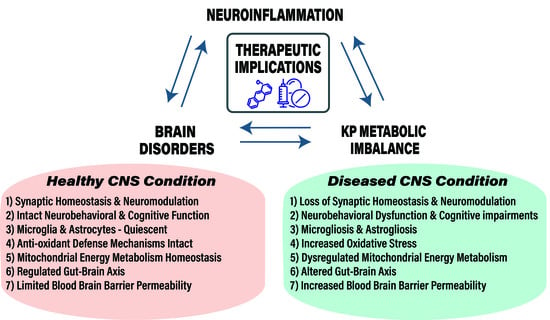
Highlights
- -
- Disturbances in KP metabolism and its regulation affect CNS disease progression and associated pathology with notable changes in KP metabolite levels, enzyme function and gene induction.
- -
- KP of tryptophan metabolism is ubiquitous in eukaryotic cells and regulates several key biological systems including oxidative stress, energy metabolism, immune function, gut-microbiota functions and neurotransmitter functions.
- -
- Aging, genetic and environmental risk factors ensue a feedforward loop between neuroinflammation and KP metabolic imbalance during CNS disease.
- -
- Broadly targeting neuroinflammation in CNS disorders with currently available anti-inflammatory pharmacotherapy is inefficacious for a lack of specificity.
- -
- Therapeutic targeting of KP for CNS diseases requires a better understanding of KP metabolite functions, cellular and molecular events affected by KP and neuroinflammation in effector cells like microglia and astrocytes.
- -
- In this review, we describe and discuss evidence from the field that elaborates on KP metabolite production and function, alterations in KP metabolic imbalance arising during CNS diseases and potential targets tested in pre-clinical or clinical stages.
1. Introduction
2. Neuroinflammation
3. The Kynurenine Pathway
4. KP Metabolism, Immune Cell Trafficking and Neuroimmune Signaling
5. The Gut-Microbiota-Brain Axis and KP
6. Brain Regional Heterogeneity in KP Metabolism
7. KP Metabolites and Molecular Mechanisms
7.1. Kynurenine
7.2. 3-Hydroxykynurenine (3-HK)
7.3. 3-Hydroxyanthranillic Acid (3-HANA)
7.4. Quinolinic Acid (QA)
7.5. Kynurenic Acid (KA)
7.6. Xanthurenic Acid (XA)
7.7. Cinnabarinic Acid
7.8. Picolinic Acid (PA)
8. Translational and Therapeutic Considerations
9. Single Nucleotide Polymorphisms (SNP’s) in The Kynurenine Pathway of Tryptophan Metabolism
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-MT | 1-methyltryptophan |
| 3-HAAO | 3-hydoxyanthranillic acid dioxygenase |
| 3-HK | 3-Hydroxykynurenine |
| α7nAChR | Alpha-7-nicotinic acetylcholine receptor |
| AβPP/PS | double transgenic mice expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9) |
| ACMSD | 2-Amino-3-carboxymuconic-6-semiladehyde decarboxylase |
| AD | Alzheimer’s disease |
| AhR | Aryl hydrocarbon receptor |
| BBB | blood–brain barrier |
| CA | Cinnabarinic acid |
| CA1 | Cornu Ammonis 1 |
| CNS | Central nervous system |
| COX2 | Cyclooxygenase 2 |
| CSF | Cerebrospinal fluid |
| FCMTE | Familial cortical myoclonic tremor with epilepsy |
| GlyB | Glycine B Site |
| GPR35 | G-protein couples receptor 35 |
| GWAS | Genome Wide Association Studies |
| HD | Huntington’s disease |
| HIV | Human immunodeficiency virus |
| ICV | Intracerebroventricular |
| IDO | Indoleamine-2,3-dioxygenase |
| KAT | Kynurenine aminotransferase |
| KA | Kynurenic acid |
| KAT | Kynurenine aminotransferase |
| Kyn | Kynurenine |
| KMO | Kynurenine-3-monooxygenase |
| KP | kynurenine Pathway |
| LAT1 | Large amino acid transporter 1 |
| LPS | Lipopolysaccharide |
| LTP | Long term potentiation |
| MDD | Major depressive disorder |
| mGLUR | Metabotropic glutamate receptors |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NAD | Nicotinamide adenine dinucleotide |
| NMDAR | N-methyl-D-aspartate receptor |
| PD | Parkinson’s disease |
| PA | Picolinic acid |
| PDD | Postpartum depression |
| QA | Quinolinic acid |
| SBI | Surgical brain injury |
| SNI | Spared nerve injury |
| SNP | Single nucleotide polymorphism |
| SNRI | Selective norepinephrine reuptake inhibitor |
| SSRI | Selective serotonin reuptake inhibitor |
| TCA | Tricyclic antidepressants |
| TDO | Tryptophan-2,3-dioxygnease |
| TRD | Treatment resistant depression |
| TRP | Tryptophan |
| VGLUT | Vesicle glutamate transporter |
References
- Feigin, V.L.; Krishnamurthi, R.V.; Theadom, A.M.; Abajobir, A.A.; Mishra, S.R.; Ahmed, M.B.; Abate, K.H.; Mengistie, M.A.; Wakayo, T.; Abd-Allah, F.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Gooch, C.L.; Pracht, E.; Borenstein, A.R. The burden of neurological disease in the United States: A summary report and call to action. Ann. Neurol. 2017, 81, 479–484. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflammation 2019, 16, 1–24. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Fourrier, C.; Singhal, G.; Baune, B.T. Neuroinflammation and cognition across psychiatric conditions. CNS Spectr. 2019, 24, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Vogels, T.; Murgoci, A.N.; Hromádka, T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun. 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Pieber, M.; Han, J.; Blomgren, K.; Zhang, X.M.; Harris, R.A.; Lund, H. Absence of microglia or presence of peripherally-derived macrophages does not affect tau pathology in young or old hTau mice. Glia 2020, 68, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. Series Editors : Marco Colonna and David Holtzmann CNS inflammation and neurodegeneration. J. Clin. Invest. 2017, 127, 1–11. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Solano Fonseca, R.; Mahesula, S.; Apple, D.M.; Raghunathan, R.; Dugan, A.; Cardona, A.; O’Connor, J.; Kokovay, E. Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev. 2016, 25, 542–555. [Google Scholar] [CrossRef]
- Jurgens, H.A.; Amancherla, K.; Johnson, R.W. Influenza Infection Induces Neuroinflammation, Alters Hippocampal Neuron Morphology, and Impairs Cognition in Adult Mice. J. Neurosci. 2012, 32, 3958–3968. [Google Scholar] [CrossRef]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef]
- Singhal, G.; Baune, B.T. Inflammatory Abnormalities in Major Depressive Disorder. In Major Depressive Disorder; Elsevier: Amsterdam, The Netherlands, 2020; pp. 75–89. [Google Scholar]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral effects of interferon-α in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Larkin, P.B.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Funakoshi, H.; Nakamura, T.; Schwarcz, R.; Muchowski, P.J. Tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase 1 make separate, tissue-specific contributions to basal and inflammation-induced kynurenine pathway metabolism in mice. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2345–2354. [Google Scholar] [CrossRef]
- Laugeray, A.; Launay, J.M.; Callebert, J.; Mutlu, O.; Guillemin, G.J.; Belzung, C.; Barone, P.R. Chronic treatment with the IDO1 inhibitor 1-methyl-D-tryptophan minimizes the behavioural and biochemical abnormalities induced by unpredictable chronic mild stress in mice—Comparison with fluoxetine. PLoS ONE 2016, 11, e0164337. [Google Scholar] [CrossRef]
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019, 92, E2070–E2080. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of Rofecoxib or Naproxen vs Placebo on Alzheimer Disease Progression: A Randomized Controlled Trial. J. Am. Med. Assoc. 2003, 289, 2819–2826. [Google Scholar] [CrossRef]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef]
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A.; McIntyre, R.S. Novel Therapeutic Targets for Major Depressive Disorder. In Neurobiology of Depression; Elsevier: Amsterdam, The Netherlands, 2019; pp. 383–400. [Google Scholar]
- Nettis, M.A.; Lombardo, G.; Hastings, C.; Zajkowska, Z.; Mariani, N.; Nikkheslat, N.; Worrell, C.; Enache, D.; McLaughlin, A.; Kose, M.; et al. Augmentation therapy with minocycline in treatment-resistant depression patients with low-grade peripheral inflammation: Results from a double-blind randomised clinical trial. Neuropsychopharmacology 2021, 46, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.I.; Cullen, C.; Umer, M.; Carvalho, A.F.; Kloiber, S.; Meyer, J.H.; Ortiz, A.; Knyahnytska, Y.; Husain, M.O.; Giddens, J.; et al. Minocycline as adjunctive treatment for treatment-resistant depression: Study protocol for a double blind, placebo-controlled, randomized trial (MINDEP2). BMC Psychiatry 2020, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rosenblat, J.D.; McIntyre, R.S. Efficacy and tolerability of minocycline for depression: A systematic review and meta-analysis of clinical trials. J. Affect. Disord. 2018, 227, 219–225. [Google Scholar] [CrossRef]
- Verdonk, F.; Petit, A.C.; Abdel-Ahad, P.; Vinckier, F.; Jouvion, G.; de Maricourt, P.; De Medeiros, G.F.; Danckaert, A.; Van Steenwinckel, J.; Blatzer, M.; et al. Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain. Behav. Immun. 2019, 81, 361–373. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Lord, B.; Grigoleit, J.S.; He, Y.; Fraser, I.; Campbell, S.N.; Taylor, N.; Aluisio, L.; O’Connor, J.C.; Papp, M.; et al. Neuropsychopharmacology of JNJ-55308942: Evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacology 2018, 43, 2586–2596. [Google Scholar] [CrossRef]
- Mallah, K.; Couch, C.; Borucki, D.M.; Toutonji, A.; Alshareef, M.; Tomlinson, S. Anti-inflammatory and Neuroprotective Agents in Clinical Trials for CNS Disease and Injury: Where Do We Go From Here? Front. Immunol. 2020, 11, 1–26. [Google Scholar] [CrossRef]
- Colpo, G.D.; Venna, V.R.; McCullough, L.D.; Teixeira, A.L. Systematic review on the involvement of the kynurenine pathway in stroke: Pre-clinical and Clinical Evidence. Front. Neurol. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients with Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Parashos, S.A.; Luo, S.; Biglan, K.M.; Bodis-Wollner, I.; Liang, G.S.; Ross, G.W.; Tilley, B.C.; Shulman, L.M. Measuring disease progression in early parkinson disease the national institutes of health exploratory trials in parkinson disease (NET-PD) experience. JAMA Neurol. 2014, 71, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J.; Ritzel, R.M.; Barrett, J.P.; Doran, S.J.; Jiao, Y.; Leach, J.B.; Szeto, G.L.; Wu, J.; Stoica, B.A.; Faden, A.I.; et al. Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J. Neurosci. 2020, 40, 2960–2974. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Redus, L.; Chen, C.; Martinez, P.A.; Strong, R.; Li, S.; O’Connor, J.C. Cognitive Dysfunction Precedes the Onset of Motor Symptoms in the MitoPark Mouse Model of Parkinson’s Disease. PLoS ONE 2013, 8, e71341. [Google Scholar] [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Stone, T.W.; Forrest, C.M.; Darlington, L.G. Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J. 2012, 279, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.J.; Yuasa, H.J.; Austin, C.J.D.; Weiser, S.; Hunt, N.H. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int. J. Biochem. Cell Biol. 2009, 41, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Li, J. Biochemical identification and crystal structure of kynurenine formamidase from Drosophila melanogaster. Biochem. J. 2012, 446, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitron, R.; Ugalde Muniz, P.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez-De La Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Baumgartner, R.; Forteza, M.J.; Ketelhuth, D.F.J. The interplay between cytokines and the Kynurenine pathway in inflammation and atherosclerosis. Cytokine 2019, 122, 154148. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Möller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 1–22. [Google Scholar] [CrossRef]
- Lawson, M.A.; Parrott, J.M.; McCusker, R.H.; Dantzer, R.; Kelley, K.W.; O’Connor, J.C. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J. Neuroinflammation 2013, 10, 1. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Briley, E.M.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Herkenham, M.; Dantzer, R.; Kelley, K.W. Induction of IDO by Bacille Calmette-Guérin Is Responsible for Development of Murine Depressive-Like Behavior. J. Immunol. 2009, 182, 3202–3212. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2008, 14, 511. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320, 595–597. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Schwarcz, R.; Whetsell, W.; Mangano, R. Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brain. Science 1983, 219, 316–318. [Google Scholar] [CrossRef]
- Foster, A.C.; White, R.J.; Schwarcz, R. Synthesis of Quinolinic Acid by 3-Hydroxyanthranilic Acid Oxygenase in Rat Brain Tissue In Vitro. J. Neurochem. 1986, 47, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.C.; Zinkand, W.C.; Schwarcz, R. Quinolinic Acid Phosphoribosyltransferase in Rat Brain. J. Neurochem. 1985, 44, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Perozzi, S.; Cimadamore, F.; Orsomando, G.; Raffaelli, N. Tissue expression and biochemical characterization of human 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase, a key enzyme in tryptophan catabolism. FEBS J. 2007, 274, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Hoffman, G.E.; Melendez-Ferro, M.; Albuquerque, E.X.; Schwarcz, R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia 2007, 55, 78–92. [Google Scholar] [CrossRef]
- Baran, H.; Schwarcz, R. Presence of 3-Hydroxyanthranilic Acid in Rat Tissues and Evidence for Its Production from Anthranilic Acid in the Brain. J. Neurochem. 1990, 55, 738–744. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Cavallari, M.; Zappulla, C.; Ulivieri, M.; Napoletano, F.; Capi, M.; Corigliano, V.; et al. Xanthurenic Acid Activates mGlu2/3 metabotropic glutamate receptors and is a potential trait marker for schizophrenia. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Sathyasaikumar, K.V.; Tararina, M.; Wu, H.-Q.; Neale, S.A.; Weisz, F.; Salt, T.E.; Schwarcz, R. Xanthurenic Acid Formation from 3-Hydroxykynurenine in the Mammalian Brain: Neurochemical Characterization and Physiological Effects. Neuroscience 2017, 367, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharmacol. 2012, 81, 643–656. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood–Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef] [PubMed]
- Strasser, B.; Becker, K.; Fuchs, D.; Gostner, J.M. Kynurenine pathway metabolism and immune activation: Peripheral measurements in psychiatric and co-morbid conditions. Neuropharmacology 2017, 112, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.L.; Case, J.A.C.; Khan, O.; Ricart, T.; Hanna, A.; Alonso, C.M.; Gabbay, V. The role of the kynurenine pathway in suicidality in adolescent major depressive disorder. Psychiatry Res. 2015, 227, 206–212. [Google Scholar] [CrossRef]
- Yoshida, R.; Imanishi, J.; Oku, T.; Kishida, T.; Hayaishi, O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc. Natl. Acad. Sci. USA 1981, 78, 129–132. [Google Scholar] [CrossRef]
- Musso, T.; Gusella, G.L.; Brooks, A.; Longo, D.L.; Varesio, L. Interleukin-4 inhibits indoleamine 2,3-dioxygenase expression in human monocytes. Blood 1994, 83, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.C.O.; Andre, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-y and Tumor Necrosis Factor-Mediate the Upregulation of Indoleamine 2,3-Dioxygenase and the Induction of Depressive-Like Behavior in Mice in Response to Bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gál, E.M.; Sherman, A.D. L-kynurenine: Its synthesis and possible regulatory function in brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol.-Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-monooxygenase: An influential mediator of neuropathology. Front. Psychiatry 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Farzi, A.; Reichmann, F.; Meinitzer, A.; Mayerhofer, R.; Jain, P.; Hassan, A.M.; Fröhlich, E.E.; Wagner, K.; Painsipp, E.; Rinner, B.; et al. Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain. Behav. Immun. 2015, 44, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 2013, 33, 13820–13833. [Google Scholar] [CrossRef]
- Majerova, P.; Michalicova, A.; Cente, M.; Hanes, J.; Vegh, J.; Kittel, A.; Kosikova, N.; Cigankova, V.; Mihaljevic, S.; Jadhav, S.; et al. Trafficking of immune cells across the bloodbrain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS ONE 2019, 14, e0217216. [Google Scholar] [CrossRef]
- Owe-Young, R.; Webster, N.L.; Mukhtar, M.; Pomerantz, R.J.; Smythe, G.; Walker, D.; Armati, P.J.; Crowe, S.M.; Brew, B.J. Kynurenine pathway metabolism in human blood-brain-barrier cells: Implications for immune tolerance & neurotoxicity. J. Neurochem. 2008, 105, 1346–1357. [Google Scholar] [CrossRef]
- Däubener, W.; Spors, B.; Hucke, C.; Adam, R.; Stins, M.; Kim, K.S.; Schroten, H. Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect. Immun. 2001, 69, 6527–6531. [Google Scholar] [CrossRef]
- Adam, R.; Russing, D.; Adams, O.; Ailyati, A.; Sik Kim, K.; Schroten, H.; Daubener, W. Role of human brain microvascular endothelial cells during central nervous system infection. Significance of indoleamine 2,3-dioxygenase in antimicrobial defence and immunoregulation. Thromb. Haemost. 2005, 94, 341–346. [Google Scholar] [CrossRef]
- Liu, H.; Liu, L.; Liu, K.; Bizargity, P.; Hancock, W.W.; Visner, G.A. Reduced Cytotoxic Function of Effector CD8+ T Cells Is Responsible for Indoleamine 2,3-Dioxygenase-Dependent Immune Suppression. J. Immunol. 2009, 183, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Donley, D.W.; Olson, A.R.; Raisbeck, M.F.; Fox, J.H.; Gigley, J.P. Huntingtons disease mice infected with toxoplasma gondii demonstrate early kynurenine pathway activation, altered CD8+ T-Cell responses, and premature mortality. PLoS ONE 2016, 11, e0162404. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Zheng, X.; Hou, Y.; Hu, M.; Wang, H.; Bao, X.; Zhou, F.; Wang, G.; Hao, H. Regulation of proinflammatory monocyte activation by the kynurenine-AhR axis underlies immunometabolic control of depressive behavior in mice. FASEB J. 2018, 32. [Google Scholar] [CrossRef]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, A.S.; Borba, L.A.; Zugno, A.I.; Quevedo, J.; Réus, G.Z. Gut microbiota–brain axis in depression: The role of neuroinflammation. Eur. J. Neurosci. 2019, 1–14. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.; Farzi, A.; Zhu, W. Tryptophan Metabolism: A Link Between the Gut Microbiota and Brain. Adv. Nutr. 2019, 1–15. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12. [Google Scholar] [CrossRef]
- Luczynski, P.; Whelan, S.O.; O’Sullivan, C.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Adult microbiota-deficient mice have distinct dendritic morphological changes: Differential effects in the amygdala and hippocampus. Eur. J. Neurosci. 2016, 44, 2654–2666. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 1–22. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; MacRi, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology 2010, 139, 2102–2112. [Google Scholar] [CrossRef]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain. Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; Redus, L.; Santana-Coelho, D.; Morales, J.; Gao, X.; O’Connor, J.C. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl. Psychiatry 2016, 6, e918. [Google Scholar] [CrossRef]
- Parrott, J.M.; Redus, L.; Connor, J.C.O. Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J. Neuroinflammation 2016, 1–15. [Google Scholar] [CrossRef]
- Heyes, M.P.; Nowak, T.S. Delayed increases in regional brain quinolinic acid follow transient ischemia in the gerbil. J. Cereb. Blood Flow Metab. 1990, 10, 660–667. [Google Scholar] [CrossRef]
- Koo, Y.S.; Kim, H.; Park, J.H.; Kim, M.J.; Shin, Y.I.; Choi, B.T.; Lee, S.Y.; Shin, H.K. Indoleamine 2,3-dioxygenase-dependent neurotoxic kynurenine metabolism contributes to poststroke depression induced in mice by ischemic stroke along with spatial restraint stress. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Bosnyák, E.; Kamson, D.O.; Behen, M.E.; Barger, G.R.; Mittal, S.; Juhász, C. Imaging cerebral tryptophan metabolism in brain tumor-associated depression. EJNMMI Res. 2015, 5. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Cuartero, M.I.; Ballesteros, I.; De La Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernández-Salguero, P.; Corbí, Á.L.; Lizasoain, I.; Moro, M.A. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef]
- Kondrikov, D.; Elmansi, A.; Bragg, R.T.; Mobley, T.; Barrett, T.; Eisa, N.; Kondrikova, G.; Schoeinlein, P.; Aguilar-Perez, A.; Shi, X.M.; et al. Kynurenine inhibits autophagy and promotes senescence in aged bone marrow mesenchymal stem cells through the aryl hydrocarbon receptor pathway. Exp. Gerontol. 2020, 130, 110805. [Google Scholar] [CrossRef]
- García-Lara, L.; Pérez-Severiano, F.; González-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of aryl hydrocarbon receptors increases endogenous kynurenic acid levels and protects mouse brain against excitotoxic insult and oxidative stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacol. 2016, 300, 13–24. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO contribute to the malignancy of gliomas via the Kyn-AhR-AQP4 signaling pathway. Signal Transduct. Target. Ther. 2020, 5, 10. [Google Scholar] [CrossRef]
- Manni, G.; Mondanelli, G.; Scalisi, G.; Pallotta, M.T.; Nardi, D.; Padiglioni, E.; Romani, R.; Talesa, V.N.; Puccetti, P.; Fallarino, F.; et al. Pharmacologic Induction of Endotoxin Tolerance in Dendritic Cells by L-Kynurenine. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic Phenotype of IFN-γ–Induced IDO + Dendritic Cells Is Maintained via an Autocrine IDO–Kynurenine/AhR–IDO Loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef]
- Mondanelli, G.; Coletti, A.; Greco, F.A.; Pallotta, M.T.; Orabona, C.; Iacono, A.; Belladonna, M.L.; Albini, E.; Panfili, E.; Fallarino, F.; et al. Positive allosteric modulation of indoleamine 2,3-dioxygenase 1 restrains neuroinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 3848–3857. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hatabayashi, K.; Arita, M.; Yajima, N.; Takenaka, C.; Suzuki, T.; Takahashi, M.; Oshima, Y.; Hara, K.; Kagawa, K.; et al. Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Reyes Ocampo, J.; Lugo Huitrón, R.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; Pérez De La Cruz, V. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef]
- Kaiser, H.; Yu, K.; Pandya, C.; Mendhe, B.; Isales, C.M.; McGee-Lawrence, M.E.; Johnson, M.; Fulzele, S.; Hamrick, M.W. Kynurenine, a Tryptophan Metabolite That Increases with Age, Induces Muscle Atrophy and Lipid Peroxidation. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Morita, T.; Saito, K.; Takemura, M.; Maekawa, N.; Fujigaki, S.; Fujii, H.; Wada, H.; Takeuchi, S.; Noma, A.; Seishima, M. 3-Hydroxyanthranilic acid, an L-tryptophan metabolite, induces apoptosis in monocyte-derived cells stimulated by interferon-γ. Ann. Clin. Biochem. 2001, 38, 242–251. [Google Scholar] [CrossRef]
- Backhaus, C.; Rahman, H.; Scheffler, S.; Laatsch, H.; Hardeland, R. NO scavenging by 3-hydroxyanthranilic acid and 3-hydroxykynurenine: N-nitrosation leads via oxadiazoles to o-quinone diazides. Nitric Oxide Biol. Chem. 2008, 19, 237–244. [Google Scholar] [CrossRef]
- Song, P.; Ramprasath, T.; Wang, H.; Zou, M.H. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell. Mol. Life Sci. 2017, 74, 2899–2916. [Google Scholar] [CrossRef]
- Gargaro, M.; Vacca, C.; Massari, S.; Scalisi, G.; Manni, G.; Mondanelli, G.; Mazza, E.M.C.; Bicciato, S.; Pallotta, M.T.; Orabona, C.; et al. Engagement of nuclear coactivator 7 by 3-hydroxyanthranilic acid enhances activation of aryl hydrocarbon receptor in immunoregulatory dendritic cells. Front. Immunol. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the Central Nervous System. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- Eastman, C.L.; Guilarte, T.R. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem. Res. 1990, 15, 1101–1107. [Google Scholar] [CrossRef]
- Leipnitz, G.; Schumacher, C.; Dalcin, K.B.; Scussiato, K.; Solano, A.; Funchal, C.; Dutra-Filho, C.S.; Wyse, A.T.S.; Wannmacher, C.M.D.; Latini, A.; et al. In vitro evidence for an antioxidant role of 3-hydroxykynurenine and 3-hydroxyanthranilic acid in the brain. Neurochem. Int. 2007, 50, 83–94. [Google Scholar] [CrossRef]
- Fazio, F.; Zappulla, C.; Notartomaso, S.; Busceti, C.; Bessede, A.; Scarselli, P.; Vacca, C.; Gargaro, M.; Volpi, C.; Allegrucci, M.; et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology 2014, 81, 237–243. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Copeland, C.S.; Neale, S.A.; Bruno, V.; Battaglia, G.; Salt, T.E.; Nicoletti, F. Cinnabarinic acid and xanthurenic acid: Two kynurenine metabolites that interact with metabotropic glutamate receptors. Neuropharmacology 2017, 112, 365–372. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.R.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Lapin, I.P. Antagonism of kynurenine-induced seizures by picolinic, kynurenic and xanthurenic acids. J. Neural Transm. 1983, 56, 177–185. [Google Scholar] [CrossRef]
- Ferreira, F.S.; Biasibetti-Brendler, H.; Pierozan, P.; Schmitz, F.; Bertó, C.G.; Prezzi, C.A.; Manfredini, V.; Wyse, A.T.S. Kynurenic Acid Restores Nrf2 Levels and Prevents Quinolinic Acid-Induced Toxicity in Rat Striatal Slices. Mol. Neurobiol. 2018, 55, 8538–8549. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, J.A.; Klos, D.J.; Hamilton, P.D. Antiviral, cytotoxic and apoptotic activities of picolinic acid on human immunodeficiency virus-1 and human herpes simplex virus-2 infected cells. Anticancer Res. 2001, 21, 3773–3776. [Google Scholar] [PubMed]
- Cai, S.; Sato, K.; Shimizu, T.; Yamabe, S.; Hiraki, M.; Sano, C.; Tomioka, H. Antimicrobial activity of picolinic acid against extracellular and intracellular Mycobacterium avium complex and its combined activity with clarithromycin, rifampicin and fluoroquinolones. J. Antimicrob. Chemother. 2006, 57, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cioczek-Czuczwar, A.; Czuczwar, P.; Turski, W.A.; Parada-Turska, J. Influence of picolinic acid on seizure susceptibility in mice. Pharmacol. Reports 2017, 69, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Galván-Arzate, S.; Lisý, V.; Ali, S.F.; Duhart, H.M.; Osorio-Rico, L.; Ríos, C.; Sut’astný, F. Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport 2001, 12, 871–874. [Google Scholar] [CrossRef]
- Neale, S.A.; Copeland, C.S.; Salt, T.E. Effect of VGLUT inhibitors on glutamatergic synaptic transmission in the rodent hippocampus and prefrontal cortex. Neurochem. Int. 2014, 73, 159–165. [Google Scholar] [CrossRef]
- Neale, S.A.; Copeland, C.S.; Uebele, V.N.; Thomson, F.J.; Salt, T.E. Modulation of hippocampal synaptic transmission by the kynurenine pathway member xanthurenic acid and other vglut inhibitors. Neuropsychopharmacology 2013, 38, 1060–1067. [Google Scholar] [CrossRef]
- Kubicova, L.; Hadacek, F.; Bachmann, G.; Weckwerth, W.; Chobot, V. Coordination complex formation and redox properties of kynurenic and xanthurenic acid can affect brain tissue homeodynamics. Antioxidants 2019, 8, 476. [Google Scholar] [CrossRef] [PubMed]
- Speciale, C.; Schwarcz, R. Uptake of Kynurenine into Rat Brain Slices. J. Neurochem. 1990, 54, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Eastman, C.L.; Guilarte, T.R.; Lever, J.R. Uptake of 3-hydroxykynurenine measured in rat brain slices and in a neuronal cell line. Brain Res. 1992, 584, 110–116. [Google Scholar] [CrossRef]
- Sardar, A.M.; Bell, J.E.; Reynolds, G.P. Increased Concentrations of the Neurotoxin 3-Hydroxykynurenine in the Frontal Cortex of HIV-1-Positive Patients. J. Neurochem. 1995, 64, 932–935. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Colín-González, A.L.; Maya-López, M.; Pedraza-Chaverrí, J.; Ali, S.F.; Chavarría, A.; Santamaría, A. The Janus faces of 3-hydroxykynurenine: Dual redox modulatory activity and lack of neurotoxicity in the rat striatum. Brain Res. 2014, 1589, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, Y.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine toxicity on the rat striatum in vivo. Jpn. J. Pharmacol. 1996, 71, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Schwarcz, R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur. J. Neurosci. 1999, 11, 3857–3863. [Google Scholar] [CrossRef]
- Darlington, L.G.; Forrest, C.M.; Mackay, G.M.; Smith, R.A.; Smith, A.J.; Stoy, N.; Stone, T.W. On the biological importance of the 3-hydroxyanthranilic acid: Anthranilic acid ratio. Int. J. Tryptophan Res. 2010, 3, 51–59. [Google Scholar] [CrossRef]
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez De La Cruz, V. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Enhance the Toxicity Induced By Copper in Rat Astrocyte Culture. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Gadupudi, G.S.; Chung, K.T. Comparative genotoxicity of 3-hydroxyanthranilic acid and anthranilic acid in the presence of a metal cofactor Cu (II) in vitro. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2011, 726, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.; Suh, H.S.; Tarassishin, L.; Cui, Q.L.; Durafourt, B.A.; Choi, N.; Bauman, A.; Cosenza-Nashat, M.; Antel, J.P.; Zhao, M.L.; et al. The tryptophan metabolite 3-hydroxyanthranilic acid plays anti-inflammatory and neuroprotective roles during inflammation: Role of hemeoxygenase-1. Am. J. Pathol. 2011, 179, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ocampo, J.; Ramírez-Ortega, D.; Vázquez Cervantes, G.I.; Pineda, B.; Montes de Oca Balderas, P.; González-Esquivel, D.; Sánchez-Chapul, L.; Lugo-Huitrón, R.; Silva-Adaya, D.; Ríos, C.; et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: Non-dependent-effect of early reactive oxygen species production. Neurotoxicology 2015, 50, 81–91. [Google Scholar] [CrossRef]
- Berg, M.; Polyzos, K.A.; Agardh, H.; Baumgartner, R.; Forteza, M.J.; Kareinen, I.; Gisterå, A.; Bottcher, G.; Hurt-Camejo, E.; Hansson, G.K.; et al. 3-Hydroxyanthralinic acid metabolism controls the hepatic SREBP/lipoprotein axis, inhibits inflammasome activation in macrophages, and decreases atherosclerosis in Ldlr−/− mice. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid Immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Swartz, K.J.; Kowall, N.W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991, 11, 1649–1659. [Google Scholar] [CrossRef]
- Heyes, M.P. Elevated cerebrospinal fluid quinolinic acid levels are associated with region-specific cerebral volume loss in HIV infection. Brain 2001, 124, 1033–1042. [Google Scholar] [CrossRef]
- Yan, E.B.; Frugier, T.; Lim, C.K.; Heng, B.; Sundaram, G.; Tan, M.; Rosenfeld, J.V.; Walker, D.W.; Guillemin, G.J.; Morganti-Kossmann, M.C. Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflammation 2015, 12, 1–17. [Google Scholar] [CrossRef]
- Moroni, F.; Cozzi, A.; Carpendo, R.; Cipriani, G.; Veneroni, O.; Izzo, E. Kynurenine 3-mono-oxygenase inhibitors reduce glutamate concentration in the extracellular spaces of the basal ganglia but not in those of the cortex or hippocampus. Neuropharmacology 2005, 48, 788–795. [Google Scholar] [CrossRef]
- Brundin, L.; Sellgren, C.M.; Lim, C.K.; Grit, J.; Pålsson, E.; Landén, M.; Samuelsson, M.; Lundgren, K.; Brundin, P.; Fuchs, D.; et al. An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl. Psychiatry 2016, 6. [Google Scholar] [CrossRef]
- Rahman, A.; Rao, M.S.; Khan, K.M. Intraventricular infusion of quinolinic acid impairs spatial learning and memory in young rats: A novel mechanism of lead-induced neurotoxicity. J. Neuroinflammation 2018, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Latif-hernandez, A.; Shah, D.; Ahmed, T.; Lo, A.C.; Callaerts-, Z. Quinolinic acid injection in mouse medial prefrontal cortex affects reversal learning abilities, cortical connectivity and hippocampal synaptic plasticity. Nat. Publ. Gr. 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.F.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain. Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef]
- Prado de Carvalho, L.; Bochet, P.; Rossier, J. The endogeneous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between nmdar2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar] [CrossRef]
- Pierozan, P.; Ferreira, F.; de Lima, B.O.; Pessoa-Pureur, R. Quinolinic acid induces disrupts cytoskeletal homeostasis in striatal neurons. Protective role of astrocyte-neuron interaction. J. Neurosci. Res. 2015, 93, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Croitoru-Lamoury, J.; Dormont, D.; Armati, P.J.; Brew, B.J. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. Glia 2003, 41, 371–381. [Google Scholar] [CrossRef]
- Speciale, C.; Hares, K.; Schwarcz, R.; Brookes, N. High-affinity uptake of L-kynurenine by a Na+-independent transporter of neutral amino acids in astrocytes. J. Neurosci. 1989, 9, 2066–2072. [Google Scholar] [CrossRef]
- Ramos-Chávez, L.A.; Lugo Huitrón, R.; González Esquivel, D.; Pineda, B.; Ríos, C.; Silva-Adaya, D.; Sánchez-Chapul, L.; Roldán-Roldán, G.; Pérez de la Cruz, V. Relevance of alternative routes of kynurenic acid production in the brain. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- Beggiato, S.; Tanganelli, S.; Fuxe, K.; Antonelli, T.; Schwarcz, R.; Ferraro, L. Endogenous kynurenic acid regulates extracellular GABA levels in the rat prefrontal cortex. Neuropharmacology 2014, 82, 11–18. [Google Scholar] [CrossRef]
- Zmarowski, A.; Wu, H.Q.; Brooks, J.M.; Potter, M.C.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur. J. Neurosci. 2009, 29, 529–538. [Google Scholar] [CrossRef]
- Flores-Barrera, E.; Thomases, D.R.; Cass, D.K.; Bhandari, A.; Schwarcz, R.; Bruno, J.P.; Tseng, K.Y. Preferential disruption of prefrontal GABAergic function by nanomolar concentrations of the α7nACh negative modulator kynurenic acid. J. Neurosci. 2017, 37, 7921–7929. [Google Scholar] [CrossRef]
- Carpenedo, R.; Pittaluga, A.; Cozzi, A.; Attucci, S.; Galli, A.; Raiteri, M.; Moroni, F. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur. J. Neurosci. 2001, 13, 2141–2147. [Google Scholar] [CrossRef]
- Konradsson-Geuken, Å.; Wu, H.Q.; Gash, C.R.; Alexander, K.S.; Campbell, A.; Sozeri, Y.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience 2010, 169, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Wonodi, I.; Roberts, R.C.; Rassoulpour, A.; McMahon, R.P.; Schwarcz, R. Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr. Bull. 2011, 37, 1147–1156. [Google Scholar] [CrossRef]
- Alexander, K.S.; Wu, H.Q.; Schwarcz, R.; Bruno, J.P. Acute elevations of brain kynurenic acid impair cognitive flexibility: Normalization by the alpha7 positive modulator galantamine. Psychopharmacology 2012, 220, 627–637. [Google Scholar] [CrossRef]
- Banerjee, J.; Alkondon, M.; Pereira, E.F.R.; Albuquerque, E.X. Regulation of GABAergic inputs to CA1 pyramidal neurons by nicotinic receptors and kynurenic acid. J. Pharmacol. Exp. Ther. 2012, 341, 500–509. [Google Scholar] [CrossRef]
- Bortz, D.M.; Wu, H.Q.; Schwarcz, R.; Bruno, J.P. Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology 2017, 121, 69–78. [Google Scholar] [CrossRef]
- Koshy Cherian, A.; Gritton, H.; Johnson, D.E.; Young, D.; Kozak, R.; Sarter, M. A systemically-available kynurenine aminotransferase II (KAT II) inhibitor restores nicotine-evoked glutamatergic activity in the cortex of rats. Neuropharmacology 2014, 82, 41–48. [Google Scholar] [CrossRef]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.Q.; Schwarcz, R. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef]
- Erhardt, S.; Schwieler, L.; Emanuelsson, C.; Geyer, M. Endogenous kynurenic acid disrupts prepulse inhibition. Biol. Psychiatry 2004, 56, 255–260. [Google Scholar] [CrossRef]
- Linderholm, K.R.; Skogh, E.; Olsson, S.K.; Dahl, M.L.; Holtze, M.; Engberg, G.; Samuelsson, M.; Erhardt, S. Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr. Bull. 2012, 38, 426–432. [Google Scholar] [CrossRef]
- Olsson, S.K.; Samuelsson, M.; Saetre, P.; Lindström, L.; Jönsson, E.G.; Nordin, C.; Engberg, G.; Erhardt, S.; Landén, M. Elevated levels of kynurenic acid in the cerebrospinal fluid of patients with bipolar disorder. J. Psychiatry Neurosci. 2010, 35, 195–199. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Jungholm, O.; Perlis, R.H.; Engberg, G.; Schwieler, L.; Landen, M.; Erhardt, S. Peripheral and central levels of kynurenic acid in bipolar disorder subjects and healthy controls. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Berlinguer-Palmini, R.; Masi, A.; Narducci, R.; Cavone, L.; Maratea, D.; Cozzi, A.; Sili, M.; Moroni, F.; Mannaioni, G. GPR35 activation reduces Ca2+ transients and contributes to the kynurenic acid-dependent reduction of synaptic activity at CA3-CA1 synapses. PLoS ONE 2013, 8, e82180. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Wu, H.Q.; Potter, M.C.; Elmer, G.I.; Pellicciari, R.; Schwarcz, R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology 2011, 36, 2357–2367. [Google Scholar] [CrossRef]
- Vohra, M.; Lemieux, G.A.; Lin, L.; Ashrafi, K. Kynurenic acid accumulation underlies learning and memory impairment associated with aging. Genes Dev. 2018, 32, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Peyton, L.; Oliveros, A.; Tufvesson-Alm, M.; Schwieler, L.; Starski, P.; Engberg, G.; Erhardt, S.; Choi, D.S. Lipopolysaccharide Increases Cortical Kynurenic Acid and Deficits in Reference Memory in Mice. Int. J. Tryptophan Res. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wennström, M.; Nielsen, H.M.; Orhan, F.; Londos, E.; Minthon, L.; Erhardt, S. Kynurenic acid levels in cerebrospinal fluid from patients with alzheimer’s disease or dementia with lewy bodies. Int. J. Tryptophan Res. 2014, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, M.; Jiménez, J.; Narváez, A.; Antequera, D.; Llamas-Velasco, S.; Martín, A.H.S.; Arjona, J.A.M.; de Munain, A.L.; Bisa, A.L.; Marco, M.P.; et al. Kynurenic acid levels are increased in the CSF of Alzheimer’s disease patients. Biomolecules 2020, 10, 571. [Google Scholar] [CrossRef]
- Chatterjee, P.; Zetterberg, H.; Goozee, K.; Lim, C.K.; Jacobs, K.R.; Ashton, N.J.; Hye, A.; Pedrini, S.; Sohrabi, H.R.; Shah, T.; et al. Plasma neurofilament light chain and amyloid-β are associated with the kynurenine pathway metabolites in preclinical Alzheimer’s disease. J. Neuroinflammation 2019, 16, 1–12. [Google Scholar] [CrossRef]
- Giil, L.M.; Midttun, Ø.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef]
- Pierozan, P.; Biasibetti-brendler, H.; Schmitz, F.; Ferreira, F.; Pessoa-pureur, R.; Wyse, A.T.S. Kynurenic Acid Prevents Cytoskeletal Disorganization Induced by Quinolinic Acid in Mixed Cultures of Rat Striatum. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Gobaille, S.; Kemmel, V.; Brumaru, D.; Dugave, C.; Aunis, D.; Maitre, M. Xanthurenic acid distribution, transport, accumulation and release in the rat brain. J. Neurochem. 2008, 105, 982–993. [Google Scholar] [CrossRef]
- Uwai, Y.; Honjo, E. Transport of xanthurenic acid by rat/human organic anion transporters OAT1 and OAT3. Biosci. Biotechnol. Biochem. 2013, 77, 1517–1521. [Google Scholar] [CrossRef]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Actions of Xanthurenic Acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology 2013, 66, 133–142. [Google Scholar] [CrossRef]
- Hikichi, H.; Hiyoshi, T.; Marumo, T.; Tomishima, Y.; Kaku, A.; Iida, I.; Urabe, H.; Tamita, T.; Yasuhara, A.; Karasawa, J.I.; et al. Antipsychotic profiles of TASP0443294, a novel and orally active positive allosteric modulator of metabotropic glutamate 2 receptor. J. Pharmacol. Sci. 2015, 127, 352–361. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.W. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef]
- Fazio, F.; Carrizzo, A.; Lionetto, L.; Damato, A.; Capocci, L.; Ambrosio, M.; Battaglia, G.; Bruno, V.; Madonna, M.; Simmaco, M.; et al. Vasorelaxing action of the kynurenine metabolite, xanthurenic acid: The missing link in endotoxin-induced hypotension? Front. Pharmacol. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Nagamura, Y.; Uesugi, K.; Naito, J.; Ishiguro, I. Cinnabarinic acid was formed in damaged mitochondria and its effect on mitochondrial respiration. Adv. Exp. Med. Biol. 1999, 467, 419–423. [Google Scholar] [CrossRef]
- Joshi, A.D.; Hossain, E.; Elferink, C.J. Epigenetic regulation by agonist-specific aryl hydrocarbon receptor recruitment of metastasis-associated protein 2 selectively induces stanniocalcin 2 expression. Mol. Pharmacol. 2017, 92, 366–374. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Grant, R. Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female Wistar rats. FEBS J. 2011, 278, 4425–4434. [Google Scholar] [CrossRef]
- Grant, R.S.; Coggan, S.E.; Smythe, G.A. The physiological action of picolinic acid in the human brain. Int. J. Tryptophan Res. 2009, 2, 71–79. [Google Scholar] [CrossRef]
- Bosco, M.C.; Rapisarda, A.; Massazza, S.; Melillo, G.; Young, H.; Varesio, L. The Tryptophan Catabolite Picolinic Acid Selectively Induces the Chemokines Macrophage Inflammatory Protein-1α and -1β in Macrophages. J. Immunol. 2000, 164, 3283–3291. [Google Scholar] [CrossRef] [PubMed]
- Prodinger, J.; Loacker, L.J.; Schmidt, R.L.J.; Ratzinger, F.; Greiner, G.; Witzeneder, N.; Hoermann, G.; Jutz, S.; Pickl, W.F.; Steinberger, P.; et al. The tryptophan metabolite picolinic acid suppresses proliferation and metabolic activity of CD4+ T cells and inhibits c-Myc activation. J. Leukoc. Biol. 2016, 99, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Beskid, M.; Jachimowicz, J.; Taraszewska, A.; Kukulska, D. Histological and ultrastructural changes in the rat brain following systemic administration of picolinic acid. Exp. Toxicol. Pathol. 1995, 47, 25–30. [Google Scholar] [CrossRef]
- Vrooman, L.; Jhamandas, K.; Boegman, R.J.; Beninger, R.J. Picolinic acid modulates kainic acid-evoked glutamate release from the striatum in vitro. Brain Res. 1993, 627, 193–198. [Google Scholar] [CrossRef]
- Lapin, I.P. Stimulant and convulsive effects of kynurenines injected into brain ventricles in mice. J. Neural Transm. 1978, 42, 37–43. [Google Scholar] [CrossRef]
- Ryan, K.M.; Allers, K.A.; McLoughlin, D.M.; Harkin, A. Tryptophan metabolite concentrations in depressed patients before and after electroconvulsive therapy. Brain. Behav. Immun. 2020, 83, 153–162. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; Leclair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Morales, I.; Farías, G.A.; Cortes, N.; Maccioni, R.B. Neuroinflammation and Neurodegeneration. In Update on Dementia; InTech: London, UK, 2016; Volume i, p. 13. [Google Scholar]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 signaling: A double-edged sword in neural diseases. Front. Cell. Neurosci. 2018, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Moraga-Amaro, R.; Jerez-Baraona, J.M.; Simon, F.; Stehberg, J. Role of astrocytes in memory and psychiatric disorders. J. Physiol. Paris 2014, 108, 240–251. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar Amyloid-β Peptides Activate Microglia via TLR2: Implications for Alzheimer’s Disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 Is a Primary Receptor for Alzheimer’s Amyloid β Peptide To Trigger Neuroinflammatory Activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Souza, L.C.; Jesse, C.R.; Antunes, M.S.; Ruff, J.R.; de Oliveira Espinosa, D.; Gomes, N.S.; Donato, F.; Giacomeli, R.; Boeira, S.P. Indoleamine-2,3-dioxygenase mediates neurobehavioral alterations induced by an intracerebroventricular injection of amyloid-β1-42 peptide in mice. Brain. Behav. Immun. 2016, 56, 363–377. [Google Scholar] [CrossRef]
- Mazarei, G.; Budac, D.P.; Lu, G.; Lee, H.; Möller, T.; Leavitt, B.R. The absence of indoleamine 2,3-dioxygenase expression protects against NMDA receptor-mediated excitotoxicity in mouse brain. Exp. Neurol. 2013, 249, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, T.; Ullrich, C.; Sperner-Unterweger, B.; Humpel, C. Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2,3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 2011, 184, 128–138. [Google Scholar] [CrossRef]
- O’Farrell, K.; Fagan, E.; Connor, T.J.; Harkin, A. Inhibition of the kynurenine pathway protects against reactive microglial-associated reductions in the complexity of primary cortical neurons. Eur. J. Pharmacol. 2017, 810, 163–173. [Google Scholar] [CrossRef]
- Feng, W.; Wang, Y.; Qi, Z.; Xuan, L.; Han, R.; Zhu, Y. Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-α. Apoptosis 2017, 22, 696–709. [Google Scholar] [CrossRef]
- Mangas, A.; Yajeya, J.; González, N.; Ruiz, I.; Pernía, M.; Geffard, M.; Coveñas, R. Gemst: A taylor-made combination that reverts neuroanatomical changes in stroke. Eur. J. Histochem. 2017, 61, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Cai, L.; Yu, Y.; Gao, L.; Xiao, L.; He, Q.; Ren, Z.; Liu, Y. Activation of brain indoleamine 2,3-dioxygenase contributes to epilepsy-associated depressive-like behavior in rats with chronic temporal lobe epilepsy. J. Neuroinflammation 2014, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dobos, N.; de Vries, E.F.J.; Kema, I.P.; Patas, K.; Prins, M.; Nijholt, I.M.; Dierckx, R.A.; Korf, J.; den Boer, J.A.; Luiten, P.G.M.; et al. The Role of Indoleamine 2,3-Dioxygenase in a Mouse Model of Neuroinflammation-Induced Depression. J. Alzheimer’s Dis. 2012, 28, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Tao, B.-B.; Yang, Y.-Y.; Du, L.-S.; Yang, S.-S.; He, X.-J.; Zhu, Y.-W.; Yan, J.-K.; Yang, Q. The IDO Inhibitor Coptisine Ameliorates Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 43, 291–302. [Google Scholar] [CrossRef]
- Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Samadi, P.; Bédard, P.J.; Izzo, E.; Schwarcz, R.; Di Paolo, T. Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav. Brain Res. 2008, 186, 161–167. [Google Scholar] [CrossRef]
- Richter, A.; Hamann, M. The kynurenine 3-hydroxylase inhibitor Ro 61-8048 improves dystonia in a genetic model of paroxysmal dyskinesia. Eur. J. Pharmacol. 2003, 478, 47–52. [Google Scholar] [CrossRef]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef]
- Guidetti, P.; Wu, H.Q.; Schwarcz, R. In situ produced 7-chlorokynurenate provides protection against quinolinate- and malonate-induced neurotoxicity in the rat striatum. Exp. Neurol. 2000, 163, 123–130. [Google Scholar] [CrossRef]
- Zádori, D.; Klivényi, P.; Szalárdy, L.; Fülöp, F.; Toldi, J.; Vécsei, L. Mitochondrial disturbances, excitotoxicity, neuroinflammation and kynurenines: Novel therapeutic strategies for neurodegenerative disorders. J. Neurol. Sci. 2012, 322, 187–191. [Google Scholar] [CrossRef]
- Gellért, L.; Fuzik, J.; Göblös, A.; Sárközi, K.; Marosi, M.; Kis, Z.; Farkas, T.; Szatmári, I.; Fülöp, F.; Vécsei, L.; et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur. J. Pharmacol. 2011, 667, 182–187. [Google Scholar] [CrossRef]
- Demeter, I.; Nagy, K.; Gellért, L.; Vécsei, L.; Fülöp, F.; Toldi, J. A novel kynurenic acid analog (SZR104) inhibits pentylenetetrazole-induced epileptiform seizures. An electrophysiological study: Special issue related to Kynurenine. J. Neural Transm. 2012, 119, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, G.; Sherchan, P.; Li, Q.; Tang, J.; Zhang, J.H. Modification of kynurenine pathway via inhibition of kynurenine hydroxylase attenuates surgical brain injury complications in a male rat model. J. Neurosci. Res. 2020, 98, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, V.; Mrzljak, L.; Dijkman, U.; Freije, R.; Heins, M.; Rassoulpour, A.; Tombaugh, G.; Gelman, S.; Bradaia, A.; Steidl, E.; et al. The novel KMO inhibitor CHDI-340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease. Exp. Neurol. 2016, 282, 99–118. [Google Scholar] [CrossRef]
- Wu, H.Q.; Guidetti, P.; Goodman, J.H.; Varasi, M.; Ceresoli-Borroni, G.; Speciale, C.; Scharfman, H.E.; Schwarcz, R. Kynurenergic manipulations influence excitatory synaptic function and excitotoxic vulnerability in the rat hippocampus in vivo. Neuroscience 2000, 97, 243–251. [Google Scholar] [CrossRef]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef]
- Schwieler, L.; Samuelsson, M.; Frye, M.A.; Bhat, M.; Schuppe-Koistinen, I.; Jungholm, O.; Johansson, A.G.; Landén, M.; Sellgren, C.M.; Erhardt, S. Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J. Neuroinflammation 2016, 13, 4–13. [Google Scholar] [CrossRef]
- Guloksuz, S.; Arts, B.; Walter, S.; Drukker, M.; Rodriguez, L.; Myint, A.M.; Schwarz, M.J.; Ponds, R.; van Os, J.; Kenis, G.; et al. The impact of electroconvulsive therapy on the tryptophan-kynurenine metabolic pathway. Brain. Behav. Immun. 2015, 48, 48–52. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, W.; Liu, W.; Wang, C.; Zhan, Y.; Li, H.; Chen, L.; Li, M.; Ning, Y. Antidepressant effect of repeated ketamine administration on kynurenine pathway metabolites in patients with unipolar and bipolar depression. Brain. Behav. Immun. 2018, 74, 205–212. [Google Scholar] [CrossRef]
- Allen, A.P.; Naughton, M.; Dowling, J.; Walsh, A.; O’Shea, R.; Shorten, G.; Scott, L.; McLoughlin, D.M.; Cryan, J.F.; Clarke, G.; et al. Kynurenine pathway metabolism and the neurobiology of treatment-resistant depression: Comparison of multiple ketamine infusions and electroconvulsive therapy. J. Psychiatr. Res. 2018, 100, 24–32. [Google Scholar] [CrossRef]
- Wu, H.Q.; Lee, S.C.; Scharfman, H.E.; Schwarcz, R. L-4-chlorokynurenine attenuates kainate-induced seizures and lesions in the rat. Exp. Neurol. 2002, 177, 222–232. [Google Scholar] [CrossRef]
- Zanos, P.; Piantadosi, S.C.; Wu, H.Q.; Pribut, H.J.; Dell, M.J.; Can, A.; Snodgrass, H.R.; Zarate, C.A.; Schwarcz, R.; Gould, T.D. The prodrug 4-chlorokynurenine causes ketamine-like antidepressant effects, but not side effects, by NMDA/glycineB-site inhibitionS. J. Pharmacol. Exp. Ther. 2015, 355, 76–85. [Google Scholar] [CrossRef]
- Wallace, M.; White, A.; Grako, K.A.; Lane, R.; Cato, A. (Jo); Snodgrass, H.R. Randomized, double-blind, placebo-controlled, dose-escalation study: Investigation of the safety, pharmacokinetics, and antihyperalgesic activity of L-4-chlorokynurenine in healthy volunteers. Scand. J. Pain 2017, 17, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Halaris, A.; Myint, A.M.; Savant, V.; Meresh, E.; Lim, E.; Guillemin, G.; Hoppensteadt, D.; Fareed, J.; Sinacore, J. Does escitalopram reduce neurotoxicity in major depression? J. Psychiatr. Res. 2015, 66–67, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.F.R.; Murata, S.; Schwarz, M.; Schütze, G.; Moll, N.; Martin, B.; Burger, B.; Weidinger, E.; Mueller, N.; Halaris, A. Celecoxib augmentation of escitalopram in treatment-resistant bipolar depression and the effects on Quinolinic Acid. Neurol. Psychiatry Brain Res. 2019, 32, 22–29. [Google Scholar] [CrossRef]
- Kocki, T.; Urbańska, E.M.; Kocki, J.; Kloc, R.; Kocka, K.; Olajossy, M.; Owe-Larsson, B. Prolonged therapy with antidepressants increases hippocampal level of kynurenic acid and expression of Kat1 and Kat2 genes. Pharmacol. Reports 2018, 70, 737–745. [Google Scholar] [CrossRef]
- Réus, G.Z.; Becker, I.R.T.; Scaini, G.; Petronilho, F.; Oses, J.P.; Kaddurah-Daouk, R.; Ceretta, L.B.; Zugno, A.I.; Dal-Pizzol, F.; Quevedo, J.; et al. The inhibition of the kynurenine pathway prevents behavioral disturbances and oxidative stress in the brain of adult rats subjected to an animal model of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 81, 55–63. [Google Scholar] [CrossRef]
- Gibney, S.M.; Fagan, E.M.; Waldron, A.M.; O’Byrne, J.; Connor, T.J.; Harkin, A. Inhibition of stress-induced hepatic tryptophan 2,3-dioxygenase exhibits antidepressant activity in an animal model of depressive behaviour. Int. J. Neuropsychopharmacol. 2014, 17, 917–928. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef]
- Fuchs, D.; Reibnegger, G.; Werner, E.R.; Wachter, H.; Forsman, A.; Larsson, M.; Hagberg, L.; Norkrans, G. Immune Activation and Decreased Tryptophan in Patients with HIV-1 Infection. J. Interferon Res. 1990, 10, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Mailankot, M.; Stone, J.G.; Garrett, M.R.; Staniszewska, M.; Castellani, R.J.; Siedlak, S.L.; Zhu, X.; Lee, H.G.; Perry, G.; et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010, 15, 161–168. [Google Scholar] [CrossRef]
- Breda, C.; Sathyasaikumar, K.V.; Idrissi, S.S.; Notarangelo, F.M.; Estranero, J.G.; Moore, G.G.L.; Green, E.W.; Kyriacou, C.P.; Schwarcz, R.; Giorgini, F. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc. Natl. Acad. Sci. USA 2016, 113, 5435–5440. [Google Scholar] [CrossRef]
- Cozzi, A.; Carpenedo, R.; Moroni, F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: Studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2YL]-benzenesulfonamide (Ro 61-8048) in models of focal or global brain ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 771–777. [Google Scholar] [CrossRef]
- Zádori, D.; Nyiri, G.; Szonyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Ravaud, A.; Dantzer, R. Early depressive symptoms in cancer patients receiving interleukin 2 and/or interferon alfa-2b therapy. J. Clin. Oncol. 2000, 18, 2143–2151. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front. Psychiatry 2018, 9. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Salazar, A.; Gonzalez-Rivera, B.L.; Redus, L.; Parrott, J.M.; O’Connor, J.C. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm. Behav. 2012, 62, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Drevets, W.; Turecki, G.; Li, Q.S. The relationship between plasma serotonin and kynurenine pathway metabolite levels and the treatment response to escitalopram and desvenlafaxine. Brain. Behav. Immun. 2020. [Google Scholar] [CrossRef]
- Eskelund, A.; Li, Y.; Budac, D.P.; Müller, H.K.; Gulinello, M.; Sanchez, C.; Wegener, G. Drugs with antidepressant properties affect tryptophan metabolites differently in rodent models with depression-like behavior. J. Neurochem. 2017, 142, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Peng, Y.L.; Liu, L.; Wu, T.Y.; Zhang, Y.; Lian, Y.J.; Yang, Y.Y.; Kelley, K.W.; Jiang, C.L.; Wang, Y.X. TNFα mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur. Cytokine Netw. 2015, 26, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, D.; Tendilla-Beltrán, H.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C.; Caso, J.R. Chronic Mild Stress Alters Kynurenine Pathways Changing the Glutamate Neurotransmission in Frontal Cortex of Rats. Mol. Neurobiol. 2019, 56, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.M.; et al. Correction to Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 2013, 10, 1–9. [Google Scholar] [CrossRef][Green Version]
- Young, K.D.; Drevets, W.C.; Dantzer, R.; Teague, T.K.; Bodurka, J.; Savitz, J. Kynurenine pathway metabolites are associated with hippocampal activity during autobiographical memory recall in patients with depression. Brain. Behav. Immun. 2016, 56, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.T.; Ballard, E.D.; Bloch, M.H.; Mathew, S.J.; Murrough, J.W.; Feder, A.; Sos, P.; Wang, G.; Zarate, C.A.; Sanacora, G. The effect of a single dose of intravenous ketamine on suicidal ideation: A systematic review and individual participant data meta-analysis. Am. J. Psychiatry 2018, 175, 150–158. [Google Scholar] [CrossRef]
- Haroon, E.; Welle, J.R.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.; Patel, T.; Felger, J.C.; Miller, A.H. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; Mccabe, M.F. Contributes To the Comorbidity of Pain and Depression. J. Clin. Invest. 2012, 122, 2940–2954. [Google Scholar] [CrossRef]
- Hemmati, S.; Sadeghi, M.A.; Mohammad Jafari, R.; Yousefi-Manesh, H.; Dehpour, A.R. The antidepressant effects of GM-CSF are mediated by the reduction of TLR4/NF-KB-induced IDO expression. J. Neuroinflammation 2019, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Avila, C.; Khana, R.; Springer, E.; James, C.; Armstrong, D.; Marshall, J.; Collins, S.M.; Pinto-Sanchez, M.I.; Bercik, P. A67 Rapid Reduction In Anxiety Scores In Ibd Patients After Infliximab Infusion Is Associated With Changes In Kynurenine/Tryptophan Metabolism. J. Can. Assoc. Gastroenterol. 2018, 1, 106. [Google Scholar] [CrossRef][Green Version]
- Wonodi, I.; Stine, O.C.; Sathyasaikumar, K.V.; Roberts, R.C.; Mitchell, B.D.; Hong, L.E.; Kajii, Y.; Thaker, G.K.; Schwarcz, R. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch. Gen. Psychiatry 2011, 68, 665–674. [Google Scholar] [CrossRef]
- Wonodi, I.; McMahon, R.P.; Krishna, N.; Mitchell, B.D.; Liu, J.; Glassman, M.; Elliot Hong, L.; Gold, J.M. Influence of kynurenine 3-monooxygenase (KMO) gene polymorphism on cognitive function in schizophrenia. Schizophr. Res. 2014, 160, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lavebratt, C.; Olsson, S.; Backlund, L.; Frisén, L.; Sellgren, C.; Priebe, L.; Nikamo, P.; Träskman-Bendz, L.; Cichon, S.; Vawter, M.P.; et al. The KMO allele encoding Arg 452 is associated with psychotic features in bipolar disorder type 1, and with increased CSF KYNA level and reduced KMO expression. Mol. Psychiatry 2014, 19, 334–341. [Google Scholar] [CrossRef]
- Aoyama, N.; Takahashi, N.; Saito, S.; Maeno, N.; Ishihara, R.; Ji, X.; Miura, H.; Ikeda, M.; Suzuki, T.; Kitajima, T.; et al. Association study between kynurenine 3-monooxygenase gene and schizophrenia in the Japanese population. Genes Brain Behav. 2006, 5, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Lezheiko, T.V.; Golimbet, V.E.; Andryushchenko, A.V.; Melik-Pashayan, A.E.; Mironova, E.V. Studies of the Association between the Kynurenine-3-Monooxygenase Gene and Depression. Neurosci. Behav. Physiol. 2018, 48, 416–419. [Google Scholar] [CrossRef]
- Douet, V.; Tanizaki, N.; Franke, A.; Li, X.; Chang, L. Polymorphism of Kynurenine Pathway-Related Genes, Kynurenic Acid, and Psychopathological Symptoms in HIV. J. Neuroimmune Pharmacol. 2016, 11, 549–561. [Google Scholar] [CrossRef]
- Claes, S.; Myint, A.M.; Domschke, K.; Del-Favero, J.; Entrich, K.; Engelborghs, S.; De Deyn, P.; Mueller, N.; Baune, B.; Rothermundt, M. The kynurenine pathway in major depression: Haplotype analysis of three related functional candidate genes. Psychiatry Res. 2011, 188, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cutler, J.A.; Rush, A.J.; McMahon, F.J.; Laje, G. Common genetic variation in the indoleamine-2,3-dioxygenase genes and antidepressant treatment outcome in major depressive disorder. J. Psychopharmacol. 2012, 26, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Duan, K.M.; Tan, X.F.; Yin, J.Y.; Mao, X.Y.; Zheng, W.; Wang, C.Y.; Yang, M.; Peng, C.; Zhou, H.H.; et al. Genetic variants of the kynurenine-3-monooxygenase and postpartum depressive symptoms after cesarean section in Chinese women. J. Affect. Disord. 2017, 215, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.M.; Wang, S.Y.; Yin, J.Y.; Li, X.; Ma, J.H.; Huang, Z.D.; Zhou, Y.Y.; Yu, H.Y.; Yang, M.; Zhou, H.H.; et al. The IDO genetic polymorphisms and postpartum depressive symptoms: An association study in Chinese parturients who underwent cesarean section. Arch. Womens. Ment. Health 2019, 22, 339–348. [Google Scholar] [CrossRef]
- Quan, C.; Wang, S.; Duan, K.; Ma, J.; Yu, H.; Yang, M.; Hu, N.; Long, G.; Zeng, G.; Huang, Z. The role of kynurenine pathway and kynurenic aminotransferase alleles in postpartum depression following cesarean section in Chinese women. Brain Behav. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Martí-Massó, J.F.; Bergareche, A.; Makarov, V.; Ruiz-Martinez, J.; Gorostidi, A.; De Munain, A.L.; Poza, J.J.; Striano, P.; Buxbaum, J.D.; Paisán-Ruiz, C. The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. J. Mol. Med. 2013, 91, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Vilas, D.; Fernández-Santiago, R.; Sanchez, E.; Azcona, L.J.; Santos-Montes, M.; Casquero, P.; Argandoña, L.; Tolosa, E.; Paisán-Ruiz, C. A Novel p.Glu298Lys Mutation in the ACMSD Gene in Sporadic Parkinson’s Disease. J. Parkinsons. Dis. 2017, 7, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; Sveinbjörnsdóttir, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nabi, R.; Serajee, F.J.; Chugani, D.C.; Zhong, H.; Huq, A.H.M.M. Association of tryptophan 2,3 dioxygenase gene polymorphism with autism. Am. J. Med. Genet. 2004, 125B, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.K.; Simon, J.S.; Gustafson, E.L.; Noviello, S.; Cubells, J.F.; Epstein, M.P.; Devlin, D.J.; Qiu, P.; Albrecht, J.K.; Brass, C.A.; et al. Association of a polymorphism in the indoleamine-2,3-dioxygenase gene and interferon-α-induced depression in patients with chronic hepatitis C. Mol. Psychiatry 2012, 17, 781–789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holtze, M.; Saetre, P.; Engberg, G.; Schwieler, L.; Werge, T.; Andreassen, O.A.; Hall, H.; Terenius, L.; Agartz, I.; Jönsson, E.G.; et al. Kynurenine 3-monooxygenase polymorphisms: Relevance for kynurenic acid synthesis in patients with schizophrenia and healthy controls. J. Psychiatry Neurosci. 2012, 37, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Golimbet, V.E.; Lezheiko, T.V.; Alfimova, M.V.; Abramova, L.I.; Kondrat’ev, N.V. Association of kynurenine-3-monooxygenase gene with schizophrenia. Russ. J. Genet. 2014, 50, 634–637. [Google Scholar] [CrossRef]
- Zhang, S.; Sakuma, M.; Deora, G.S.; Levy, C.W.; Klausing, A.; Breda, C.; Read, K.D.; Edlin, C.D.; Ross, B.P.; Wright Muelas, M.; et al. A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites. Commun. Biol. 2019, 2. [Google Scholar] [CrossRef]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Allen, A.P.; O’Neill, A.; Quigley, E.M.M.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Acute tryptophan depletion reduces kynurenine levels: Implications for treatment of impaired visuospatial memory performance in irritable bowel syndrome. Psychopharmacology 2015, 232, 1357–1371. [Google Scholar] [CrossRef]
- Kazemi, A.; Noorbala, A.A.; Azam, K.; Eskandari, M.H.; Djafarian, K. Effect of probiotic and prebiotic vs placebo on psychological outcomes in patients with major depressive disorder: A randomized clinical trial. Clin. Nutr. 2019, 38, 522–528. [Google Scholar] [CrossRef]
- Moloney, G.M.; O’Leary, O.F.; Salvo-Romero, E.; Desbonnet, L.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. Microbial regulation of hippocampal miRNA expression: Implications for transcription of kynurenine pathway enzymes. Behav. Brain Res. 2017, 334, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Valente-Silva, P.; Ruas, J.L. Tryptophan-Kynurenine Metabolites in Exercise and Mental Health BT—Hormones, Metabolism and the Benefits of Exercise; Spiegelman, B., Ed.; Springer International Publishing: Cham, Germany, 2017; pp. 83–91. ISBN 978-3-319-72790-5. [Google Scholar]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Günther, J.; Däbritz, J.; Wirthgen, E. Limitations and Off-Target Effects of Tryptophan-Related IDO Inhibitors in Cancer Treatment. Front. Immunol. 2019, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma—An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef]



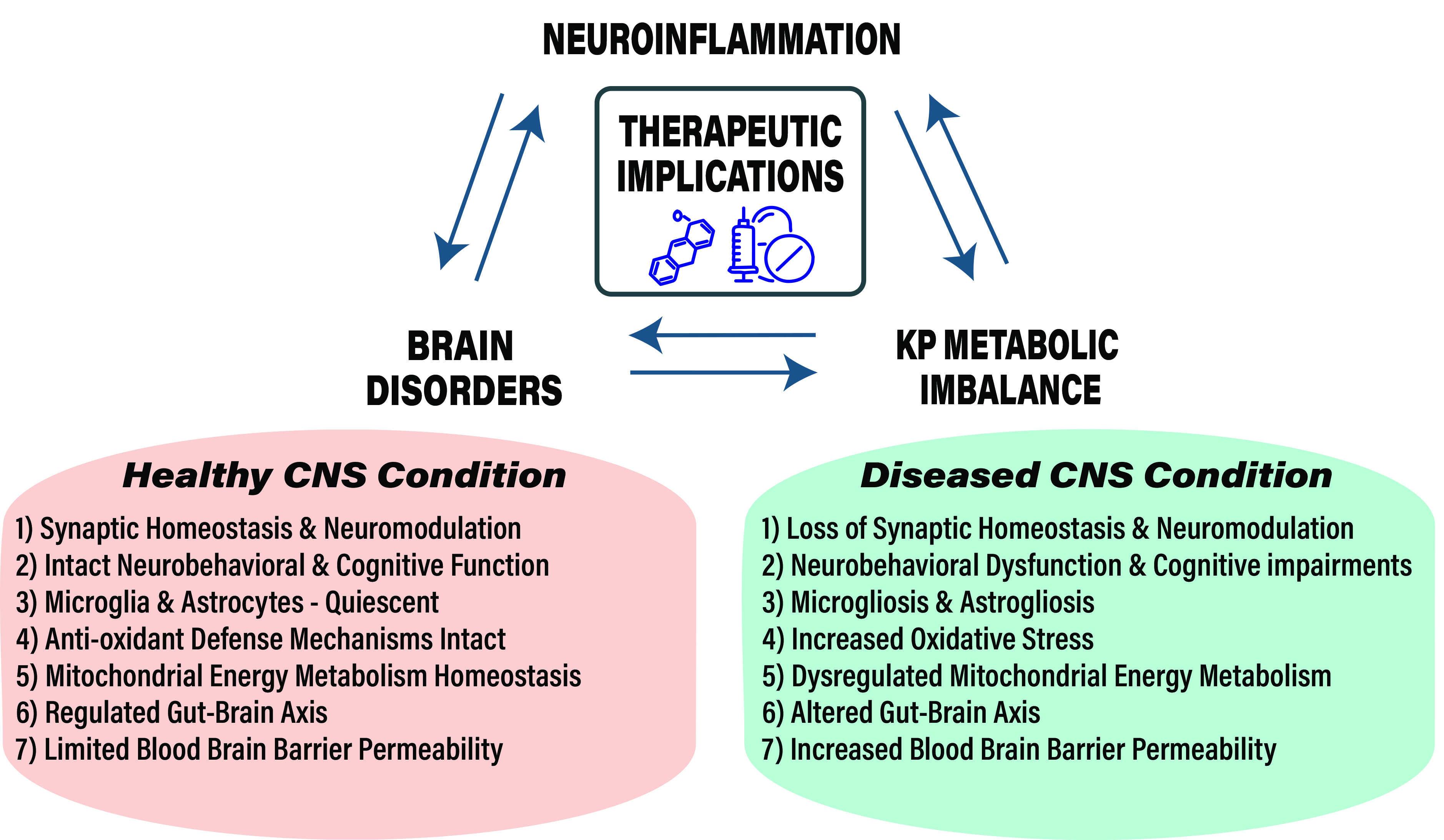{kind=link}
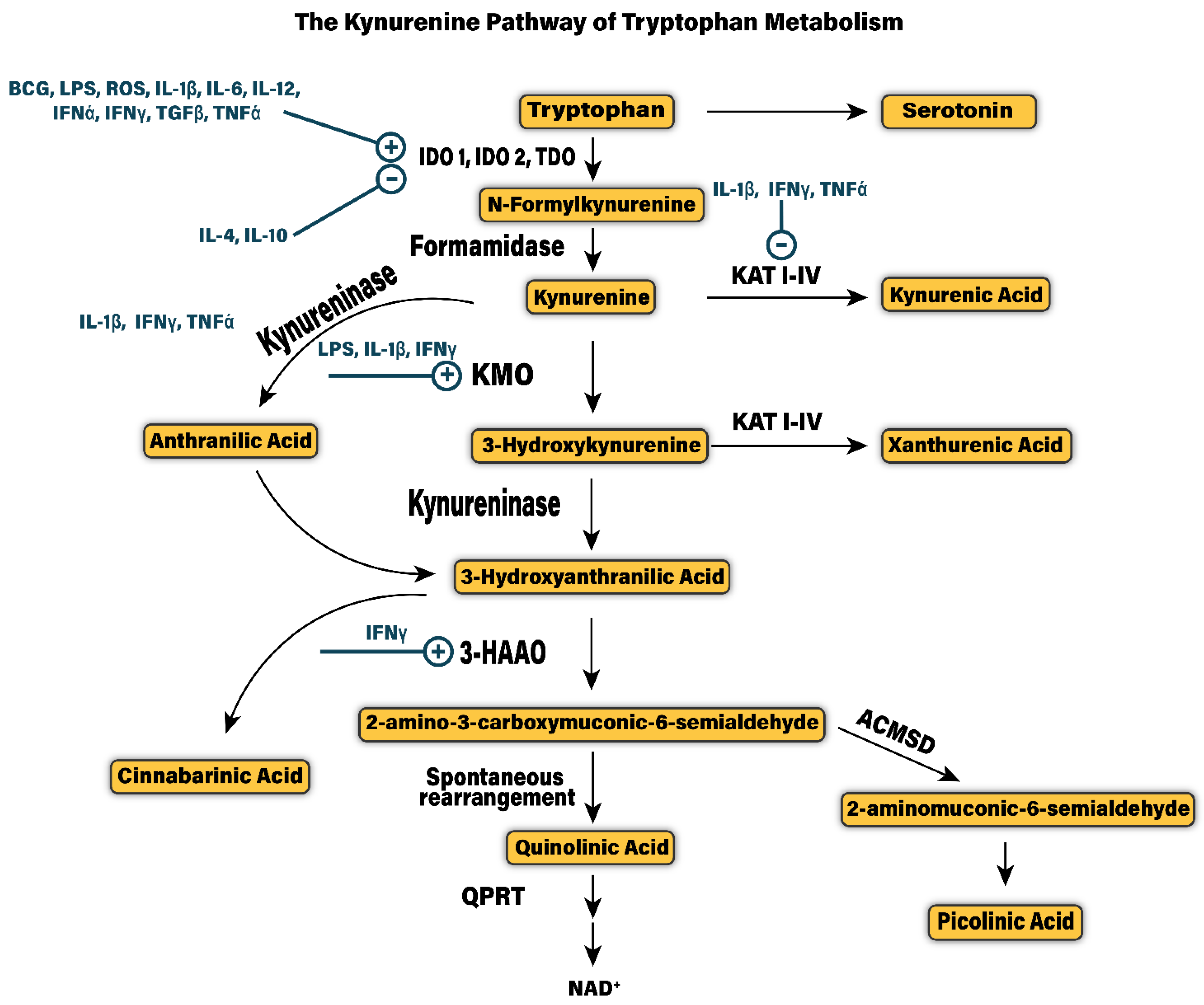{kind=link}
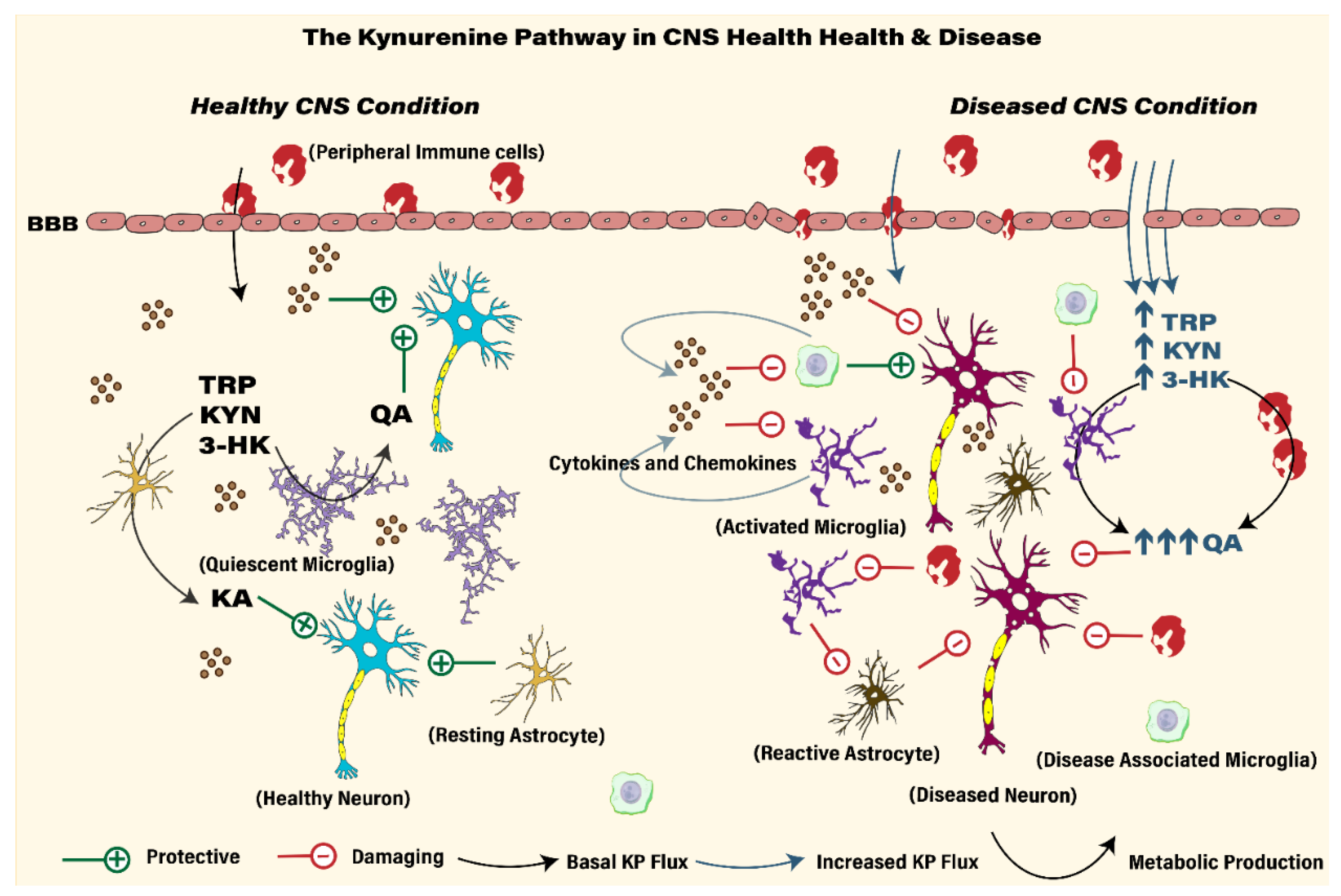{kind=link}
| Metabolite | Metabolite-Receptor Target | Neurotransmitter Activity | Biological Functions | Ref |
|---|---|---|---|---|
| 3-Hydroxyanthranilic Acid | Unknown | Unknown | Anti-Inflammatory, Oxidative stress regulation | [114,115,116,117] |
| 3-Hydroxykynurenine | Unknown | Unknown | Oxidative Stress Regulation | [118,119,120] |
| Cinnabarinic Acid | mGLUR4, AhR | Glutamate | Immunomodulation | [65,121,122] |
| Kynurenine | AhR | Unknown | Transcription factor, Immunomodulation, Anti-Cancer, Oxidative stress regulation | [103,111,112,113] |
| Kynurenic Acid | α7nAChR, AhR, NMDAR, GPR35 | Glutamate, GABA and Nicotinic | Anti-Oxidant, Immunomodulation, Anti-convulsant | [123,124,125,126,127] |
| Picolinic Acid | Unknown | Glutamate | Anti-convulsant, Anti-viral, Anti-microbial, Immunomodulation | [125,128,129,130] |
| Quinolinic Acid | NMDAR | Glutamate | Pro-convulsant, Pro-oxidant | [46,56,131] |
| Xanthurenic Acid | mGLUR 2/3 * | Glutamate | Anti-convulsant, Anti-oxidant | [63,132,133,134] |
| Drug | Target | Disease Model | Pre/Clinical | Effect | Study |
|---|---|---|---|---|---|
| Gemst | Anti-inflammatory Poly-l-lysine compound | Stroke | Rat model of ischemia | Treatment down-regulates IDO pathway, reduction of microglia activation and gliosis in late phase of stroke | [224] |
| 1-MT | IDO inhibitor | epilepsy-induced depression | Pilocarpine induced Chronic temporal lobe epilepsy in rats | Prevented depressive-like behavior but did not influence spontaneous seizures | [225] |
| 1-MT | IDO inhibitor | Inflammation-induced depression | Mice treated with LPS (ICV) | Treatment prevented depressive-like behavior | [226] |
| Coptisine | IDO inhibitor | Alzheimer’s Disease | Alzheimer’s transgenic AβPP/PS mice | Prevented neuronal cell loss and cognitive impairment, reduced amyloid plague development; decreased activation of microglia and astrocytes | [227] |
| N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride-SZR-72 | KA analogue | Huntington’s Disease | HD N171-82Q transgenic mouse model | Treatment increased survival, restored locomotor activity, increased body weight and prevented striatum neuronal loss | [228] |
| 2-(2-N,N-dimethylaminoethylamine-1carbonyl)-1H-quinolin-4-one hydrochloride | KA analogue | Stroke | Rat model of global forebrain ischemia | Treatment prevents neuronal cell loss in CA1 region; preserves induction of LTP | [229] |
| SZR104 | KA analogue | Epilepsy | Pentylenetetrazole induced epileptiform seizures in rats | Treatment decreased population spike activity and epileptiform seizures | [230] |
| Ro-61-8048 | KMO inhibitor | Parkinson’s | MPTP cynomolgus monkey | Chronic treatment with Ro-61-8048 reduced levodopa-induced dyskinesia’s | [228] |
| Ro-61-8048 | KMO inhibitor | Paroxysmal dystonia | dtsz mutant Syrian golden hamsters | Anti-dystonic; reduced severity of dystonic attacks | [231] |
| Ro-61-8048 | KMO inhibitor | Brain injury/Neurotrauma | Surgical brain injury in rats | Treatment reduced brain edema and improved long-term neurological function after SBI | [232] |
| Ro 61-8048 | KMO inhibitor | Spared nerve injury model | Neuropathic pain and depression like behavior in SNI mice | Anti-depressant effect following treatment but no effect on improving mechanical allodynia | [233] |
| Ro-61-8048, mNBA | KMO inhibitor | Stroke | Rat and gerbil models of ischemia | Treatment prevented neuronal loss and decreased infarct volume | [234] |
| JM6 | KMO inhibitor | Alzheimer’s and Huntington’s | AD (APPtg) and HD (R6/2) transgenic mouse models | Prevents AD and HD behavioral deficits and synaptic loss; increased life span of HD mice | [235] |
| CHDI-340246 | KMO inhibitor | Huntington’s Disease | Multiple HD transgenic mouse models | Treatment ameliorates electrophysiological alterations in striatum, no improvement in behavioral phenotype | [236] |
| PNU 156561A | KMO inhibitor | Huntington’s Disease | Chemical model of HD in rat; QA induced neurodegeneration and seizures | Anti-convulsant and neuroprotective | [237] |
| Leucine | LAT1 competitor | Inflammation-induced depression | LPS induced depressive like behavior | Anti-depressant effect due to blockade of kynurenine entry to the brain. | [238] |
| Electroconvulsive therapy | NA | Depression | Humans with MDD | Treatment improved depressive symptoms; reduced kyn and QA levels in plasma, significantly reduced the QA/KA ratio | [239] |
| Electroconvulsive therapy | NA | Depression | Humans with MDD | Treatment improved depressive symptoms, increased serum KA levels, increased the KYN/TRP, KA/KYN, and KA/3-HK ratios | [240] |
| Ketamine | NMDAR antagonist | Inflammation-induced depression; Treatment Resistant Depression | Mice treated with LPS; humans with TRD | Reduces QA and elevates KA in mice treated with LPS, reduced depressive behaviors | [34] |
| Ketamine | NMDAR antagonist | Treatment Resistant Depression | Humans with TRD | Treatment reduced depressive symptoms; elevated KYNA and KA/KYN ratio in responders | [241] |
| Ketamine | NMDAR antagonist | Treatment Resistant Depression | Humans with TRD | Treatment reduced depressive symptoms; statistical non-significant trend of elevating KA and KA/KYN | [242] |
| AV-101 (4-Chloro-kynurenine) | selective GlyB-NMDA receptor antagonist | Epilepsy | Kainate induced seizures and lesion | Anti-convulsant and neuroprotective effects, prevents seizures and lesion in rats | [243] |
| AV-101 (4-Chloro-kynurenine) | selective GlyB-NMDA receptor antagonist | Depression | Healthy C57BL/6J mice | Anti-depressant like effects on behavioral paradigms used in mice | [244] |
| AV-101 (4-Chloro-kynurenine) | selective GlyB-NMDA receptor antagonist | Pain | Capsaicin induced experimental pain in humans | Treatment trends to decrease in algesic response, feeling of wellbeing in small cohort of patients | [245] |
| Escitalopram | SSRI | Depression | Humans with MDD | Treatment improved depressive symptoms, reduced QA and 3-HK, increased KA/QA ratio | [246] |
| Escitalopram + celecoxib | SSRI, COX-2 inhibitor | Bipolar disorder | Treatment resistant Bipolar depression | Combination of drugs improved treatment response, statistically non-significant trend of reduced peripheral QA | [247] |
| amitriptyline, imipramine, fluoxetine, citalopram | SSRI, TCAs | Depression | Humans with MDD | All antidepressants increase KA level in hippocampus and increased KAT1 and KAT2 expression in hippocampus | [248] |
| D-1MT, allopurinol, Ro 61-8048 | IDO inhibitor, TDO inhibitor, KMO inhibitor | Schizophrenia | Ketamine-induced increased locomotor activity in rats | All inhibitors reverse the increased locomotor activity in response to ketamine | [249] |
| Allopurinol | TDO inhibitor | Depression | Rat model of chronic restraint stress | Reduced immobility time in forced swim test, prevented increase in kynurenine levels induced by stress | [250] |
| Gene Allele | SNP | Disease Model | Nature of Association with SNP | Study |
|---|---|---|---|---|
| TDO2 | rs3755910 | Autism | Polymorphism in TDO2 promoter region; CC genotype associated with autism | [286] |
| KMO | rs1053230 | Bipolar disorder | Associated with elevated CSF KA in bipolar patients and common in bipolar patients with psychotic features during manic episode | [274] |
| KMO | rs1053230, rs2275163 | Depression | No association between rs2275163 and depression. Association trend for rs 1053230 G/G genotype and depression | [276] |
| IDO2 | rs2929115, rs2929116 | Depression | Nature of association unclear, SNP may alter SSRI citalopram response | [279] |
| KATII, KMO | KATII-rs1480544, KMO-rs1053230 | Depression in HIV patients | C-allele in KATII-rs1480544 appears to be protective, KATII-TT-carriers at risk | [277] |
| IDO1 | rs9657182 | Inflammation-induced depression | Associated with depressive symptoms development post immune therapy; CC genotype more at risk that TT genotype | [287] |
| KAT III, KMO | KAT III-rs12729558, KMO-rs1053230 | Major Depressive Disorder and Bipolar Disorder | Associated with bipolar disorder and trend for association with MDD | [278] |
| ACMSD | p.Trp26Stop codon | Familial cortical myoclonic tremor and epilepsy | ACMSD p.Trp26Stop mutation associated with family history of FCMTE | [283] |
| ACMSD | rs775129424 | Parkinson’s Disease | Risk associated mutation found in patient with PD; affects response to treatment | [284] |
| ACMSD | rs6710823 | Parkinson’s Disease | Meta-analysis of GWAS reveals ACMSD SNP predicts PD risk | [285] |
| KMO | rs1053230 | Post-partum depression | Associated with elevated levels of 3-HK and higher 3-HK/KYN ratio in serum of women in post-partum depression. Significantly associated with post-partum depression | [280] |
| IDO1 | rs10108662 | Post-partum depression | Associated with post-partum depression | [281] |
| KATI, KATII, KATIII | NA | Post-partum depression | No association between eight KAT I–III SNPs and PPD | [282] |
| KMO | rs1053230 | Schizophrenia | SNP associated with Increased KA in the CSF of schizophrenia patient | [288] |
| KMO | rs1053230, rs2275163 | Schizophrenia | Increased risk were the GG genotype (rs1053230) and the T*GG diplotype (rs2275163 and rs1053230) | [289] |
| KMO | rs2275163 | Schizophrenia | Associated with oculomotor measures of predictive pursuit and visuospatial working memory deficits in schizophrenic patients | [272] |
| KMO | rs2275163, rs1053230 | Schizophrenia | rs2275163C>T allele is associated with poor performance on battery of cognitive tests in schizophrenia with decrease cognitive scores associated but rs1053230 is not | [273] |
| KMO | rs2275163 | Schizophrenia | Associated with schizophrenia in one cohort, but effect was not replicated in second larger cohort | [275] |
| ACMSD | rs2121337 | Suicide | SNP associated with higher QA in suicidal patients and reduced PA production | [154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’Connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. https://doi.org/10.3390/cells10061548
Mithaiwala MN, Santana-Coelho D, Porter GA, O’Connor JC. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells. 2021; 10(6):1548. https://doi.org/10.3390/cells10061548
Chicago/Turabian StyleMithaiwala, Mustafa N., Danielle Santana-Coelho, Grace A. Porter, and Jason C. O’Connor. 2021. "Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications" Cells 10, no. 6: 1548. https://doi.org/10.3390/cells10061548
APA StyleMithaiwala, M. N., Santana-Coelho, D., Porter, G. A., & O’Connor, J. C. (2021). Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells, 10(6), 1548. https://doi.org/10.3390/cells10061548