
Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics


Abstract
1. Introduction
2. Fibrosis
3. Extracellular Vesicles
3.1. EV Definition
3.2. EV Biogenesis
3.3. EVs in the Biology of Fibrosis
4. Pulmonary Fibrosis
4.1. Causes and Pathological Features of Pulmonary Fibrosis
4.2. Mechanistic Aspects of EVs in Pulmonary Fibrosis
4.2.1. Production and Action of EVs from Lung Tissues or Pulmonary Fibroblasts
4.2.2. Production and Action of EVs from Lung Epithelial Cells or Macrophages
4.2.3. Production and Action of EVs from Endothelial Cells
4.3. Therapeutic Actions of EVs in Pulmonary Fibrosis
4.3.1. EVs from Adult Stem Cells
4.3.2. EVs from Lung Fibroblasts or Macrophages
4.3.3. EVs from Serum
4.4. EVs as Biomarkers for Pulmonary Fibrosis
5. Renal Fibrosis
5.1. Causes and Pathological Features of Renal Fibrosis
5.2. Mechanistic Aspects of EVs in Renal Fibrosis
5.2.1. Production and Action of EVs from Epithelial Cells
5.2.2. Production and Action of EVs from Podocytes, Endothelial Cells, Mesangial Cells, or Macrophages
5.3. Therapeutic Actions of EVs in Renal Fibrosis
5.3.1. EVs from Adult Stem Cells
5.3.2. EVs from Differentiated Cells or Urine
5.4. EVs as Biomarkers in Renal Fibrosis
6. Cardiac Fibrosis
6.1. Causes and Pathological Features of Cardiac Fibrosis
6.2. Mechanistic Aspects of EVs in Cardiac Fibrosis
6.2.1. Production and Action of EVs from Cardiomyocytes
6.2.2. Production and Action of EVs by Fibroblasts
6.2.3. Production and Action of EVs from T Cells
6.2.4. Production and Action of EVs from Macrophages
6.2.5. Production and Action of EVs from the Circulation
6.3. Therapeutic Actions of EVs in Cardiac Fibrosis
6.3.1. EVs from Adult Stem Cells
6.3.2. EVs from Induced Pluripotent Stem Cells (iPSC)
6.3.3. EVs from Embryonic stem Cells (ESC)
6.3.4. EVs from Cardiomyocytes or Skeletal Muscle
6.3.5. EVs from Vascular Endothelial or Smooth Muscle Cells
6.3.6. EVs from the Circulation
6.4. EVs as Biomarkers in Cardiac Fibrosis
7. Hepatic Fibrosis
7.1. Causes and Pathological Features of Hepatic Fibrosis
7.2. Mechanistic Aspects of EVs in Hepatic Fibrosis
7.2.1. Production and Action of EVs from Hepatocytes
7.2.2. Production and Action of EVs from Cholangiocytes
7.2.3. Production and Action of EVs from HSC or Fibroblasts
7.2.4. Production and Action of EVs from Endothelial Cells or Macrophages
7.2.5. Action of EVs from the Circulation
7.3. Therapeutic Actions of EVs in Hepatic Fibrosis
7.3.1. EVs from Adult Stem Cells
7.3.2. EVs from iPSCs
7.3.3. EVs from ESC
7.3.4. EVs from Hepatocytes
7.3.5. EVs from HSC
7.3.6. EVs from T Cells, Macrophages or Natural Killer Cells
7.3.7. EVs from Serum
7.4. EVs as Biomarkers in Hepatic Fibrosis
8. Pancreatic Fibrosis
8.1. Causes and Pathological Features of Pancreatic Fibrosis
8.2. Mechanistic Aspects of EVs in Pancreatic Fibrosis
9. Skin Fibrosis
9.1. Causes and Pathological Features of Skin Fibrosis
9.2. Mechanistic Aspects of EVs in Skin Fibrosis
9.3. Therapeutic Actions of EVs in Skin Fibrosis
EVs from Adult Stem Cells
10. Perspective and Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002, 7, d793–d807. [Google Scholar] [CrossRef]
- Ismaeel, A.; Kim, J.S.; Kirk, J.S.; Smith, R.S.; Bohannon, W.T.; Koutakis, P. Role of Transforming Growth Factor-beta in Skeletal Muscle Fibrosis: A Review. Int. J. Mol. Sci. 2019, 20, 2446. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Signaling in fibrosis: Targeting the TGF beta, endothelin-1 and CCN2 axis in scleroderma. Front. Biosci. 2009, 1, 115–122. [Google Scholar]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Horie, M.; Nagase, T. TGF-beta Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-beta signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Burgy, O.; Konigshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/beta-catenin signaling: A promising new target for fibrosis diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar] [CrossRef]
- Miao, C.G.; Yang, Y.Y.; He, X.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.W.; Jin, Y.; Li, J. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie 2013, 95, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.J.; Shi, K.H.; Li, J. Wnt signaling pathway in cardiac fibrosis: New insights and directions. Metabolism 2016, 65, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Liu, Y. New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis. Nephrology 2018, 23 (Suppl. 4), 38–43. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Friedman, S.L. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr. Opin. Pharmacol. 2019, 49, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef]
- Juan, T.; Furthauer, M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Chairoungdua, A.; Smith, D.L.; Pochard, P.; Hull, M.; Caplan, M.J. Exosome release of beta-catenin: A novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 2010, 190, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef]
- Charrier, A.; Chen, R.; Chen, L.; Kemper, S.; Hattori, T.; Takigawa, M.; Brigstock, D.R. Exosomes mediate intercellular transfer of pro-fibrogenic connective tissue growth factor (CCN2) between hepatic stellate cells, the principal fibrotic cells in the liver. Surgery 2014, 156, 548–555. [Google Scholar] [CrossRef]
- Chen, L.; Charrier, A.; Zhou, Y.; Chen, R.; Yu, B.; Agarwal, K.; Tsukamoto, H.; Lee, L.J.; Paulaitis, M.E.; Brigstock, D.R. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 2014, 59, 1118–1129. [Google Scholar] [CrossRef]
- Lang, J.K.; Young, R.F.; Ashraf, H.; Canty, J.M., Jr. Inhibiting Extracellular Vesicle Release from Human Cardiosphere Derived Cells with Lentiviral Knockdown of nSMase2 Differentially Effects Proliferation and Apoptosis in Cardiomyocytes, Fibroblasts and Endothelial Cells In Vitro. PLoS ONE 2016, 11, e0165926. [Google Scholar] [CrossRef]
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Glick, B.S. The mechanisms of vesicle budding and fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef]
- Tkach, M.; Kowal, J.; Thery, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160479. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Rilla, K.; Mustonen, A.M.; Arasu, U.T.; Harkonen, K.; Matilainen, J.; Nieminen, P. Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix Biol. 2019, 75–76, 201–219. [Google Scholar] [CrossRef]
- Lenzini, S.; Bargi, R.; Chung, G.; Shin, J.W. Matrix mechanics and water permeation regulate extracellular vesicle transport. Nat. Nanotechnol. 2020, 15, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romero, N.; Esteban-Rubio, S.; Rackov, G.; Carrion-Navarro, J.; Belda-Iniesta, C.; Ayuso-Sacido, A. Extracellular vesicles compartment in liquid biopsies: Clinical application. Mol. Asp. Med. 2018, 60, 27–37. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 5, 78. [Google Scholar] [CrossRef]
- Pollard, K.M. Silica, Silicosis, and Autoimmunity. Front. Immunol. 2016, 7, 97. [Google Scholar] [CrossRef]
- Fujimura, N. Pathology and pathophysiology of pneumoconiosis. Curr. Opin. Pulm. Med. 2000, 6, 140–144. [Google Scholar] [CrossRef]
- Bhandari, J.; Thada, P.K.; Sedhai, Y.R. Asbestosis; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lareau, S.C.; Fahy, B.; Meek, P.; Wang, A. Chronic Obstructive Pulmonary Disease (COPD). Am. J. Respir. Crit. Care Med. 2019, 199, P1–P2. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Hwang, J.S.; Rehan, V.K. Recent Advances in Bronchopulmonary Dysplasia: Pathophysiology, Prevention, and Treatment. Lung 2018, 196, 129–138. [Google Scholar] [CrossRef]
- Savani, R.C. Modulators of inflammation in Bronchopulmonary Dysplasia. Semin. Perinatol. 2018, 42, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Moore, B.B. Animal Models of Pulmonary Fibrosis. Methods Mol. Biol. 2018, 1809, 363–378. [Google Scholar] [CrossRef]
- Martin-Medina, A.; Lehmann, M.; Burgy, O.; Hermann, S.; Baarsma, H.A.; Wagner, D.E.; De Santis, M.M.; Ciolek, F.; Hofer, T.P.; Frankenberger, M.; et al. Increased Extracellular Vesicles Mediate WNT-5A Signaling in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 1527–1538. [Google Scholar] [CrossRef]
- Xie, H.; Gao, Y.M.; Zhang, Y.C.; Jia, M.W.; Peng, F.; Meng, Q.H.; Wang, Y.C. Low let-7d exosomes from pulmonary vascular endothelial cells drive lung pericyte fibrosis through the TGFbetaRI/FoxM1/Smad/beta-catenin pathway. J. Cell. Mol. Med. 2020, 24, 13913–13926. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Habiel, D.M.; Ge, L.; Bora, S.A.; Brauer, R.; Evans, C.M.; Xie, T.; Alonso-Valenteen, F.; Medina-Kauwe, L.K.; et al. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 2019, 5, e129359. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol. Asp. Med. 2018, 60, 92–103. [Google Scholar] [CrossRef]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef]
- Kang, J.H.; Jung, M.Y.; Choudhury, M.; Leof, E.B. Transforming growth factor beta induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J. 2020, 34, 2213–2226. [Google Scholar] [CrossRef]
- Yao, M.Y.; Zhang, W.H.; Ma, W.T.; Liu, Q.H.; Xing, L.H.; Zhao, G.F. microRNA-328 in exosomes derived from M2 macrophages exerts a promotive effect on the progression of pulmonary fibrosis via FAM13A in a rat model. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Ye, C.; Li, H.; Bao, M.; Zhuo, R.; Jiang, G.; Wang, W. Alveolar macrophage-derived exosomes modulate severity and outcome of acute lung injury. Aging 2020, 12, 6120–6128. [Google Scholar] [CrossRef]
- Munson, P.; Lam, Y.W.; Dragon, J.; MacPherson, M.; Shukla, A. Exosomes from asbestos-exposed cells modulate gene expression in mesothelial cells. FASEB J. 2018, 32, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Munson, P.; Lam, Y.W.; MacPherson, M.; Beuschel, S.; Shukla, A. Mouse serum exosomal proteomic signature in response to asbestos exposure. J. Cell. Biochem. 2018, 119, 6266–6273. [Google Scholar] [CrossRef]
- Wang, D.; Hao, C.; Zhang, L.; Zhang, J.; Liu, S.; Li, Y.; Qu, Y.; Zhao, Y.; Huang, R.; Wei, J.; et al. Exosomal miR-125a-5p derived from silica-exposed macrophages induces fibroblast transdifferentiation. Ecotoxicol. Environ. Saf. 2020, 192, 110253. [Google Scholar] [CrossRef] [PubMed]
- Kuse, N.; Kamio, K.; Azuma, A.; Matsuda, K.; Inomata, M.; Usuki, J.; Morinaga, A.; Tanaka, T.; Kashiwada, T.; Atsumi, K.; et al. Exosome-Derived microRNA-22 Ameliorates Pulmonary Fibrosis by Regulating Fibroblast-to-Myofibroblast Differentiation in Vitro and in Vivo. J. Nippon. Med. Sch. 2020, 87, 118–128. [Google Scholar] [CrossRef]
- Wang, Y.C.; Xie, H.; Zhang, Y.C.; Meng, Q.H.; Xiong, M.M.; Jia, M.W.; Peng, F.; Tang, D.L. Exosomal miR-107 antagonizes profibrotic phenotypes of pericytes by targeting a pathway involving HIF-1alpha/Notch1/PDGFRbeta/YAP1/Twist1 axis in vitro. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H520–H534. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4, e128060. [Google Scholar] [CrossRef]
- Willis, G.R.; Fernandez-Gonzalez, A.; Anastas, J.; Vitali, S.H.; Liu, X.; Ericsson, M.; Kwong, A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation. Am. J. Respir. Crit. Care Med. 2018, 197, 104–116. [Google Scholar] [CrossRef]
- Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Yeung, V.; Liu, X.; Ericsson, M.; Andrews, N.A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell-derived small extracellular vesicles restore lung architecture and improve exercise capacity in a model of neonatal hyperoxia-induced lung injury. J. Extracell. Vesicles 2020, 9, 1790874. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, L.; Zhang, S.; Wang, J.; Jiang, W.; Xin, Q.; Luan, Y. The protective effects of MSC-EXO against pulmonary hypertension through regulating Wnt5a/BMP signalling pathway. J. Cell. Mol. Med. 2020, 24, 13938–13948. [Google Scholar] [CrossRef]
- Wan, X.; Chen, S.; Fang, Y.; Zuo, W.; Cui, J.; Xie, S. Mesenchymal stem cell-derived extracellular vesicles suppress the fibroblast proliferation by downregulating FZD6 expression in fibroblasts via micrRNA-29b-3p in idiopathic pulmonary fibrosis. J. Cell. Physiol. 2020, 235, 8613–8625. [Google Scholar] [CrossRef]
- Xiao, K.; He, W.; Guan, W.; Hou, F.; Yan, P.; Xu, J.; Zhou, T.; Liu, Y.; Xie, L. Mesenchymal stem cells reverse EMT process through blocking the activation of NF-kappaB and Hedgehog pathways in LPS-induced acute lung injury. Cell Death Dis. 2020, 11, 863. [Google Scholar] [CrossRef]
- Xu, C.; Zhao, J.; Li, Q.; Hou, L.; Wang, Y.; Li, S.; Jiang, F.; Zhu, Z.; Tian, L. Exosomes derived from three-dimensional cultured human umbilical cord mesenchymal stem cells ameliorate pulmonary fibrosis in a mouse silicosis model. Stem Cell Res. Ther. 2020, 11, 503. [Google Scholar] [CrossRef]
- Lei, X.; He, N.; Zhu, L.; Zhou, M.; Zhang, K.; Wang, C.; Huang, H.; Chen, S.; Li, Y.; Liu, Q.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Radiation-Induced Lung Injury via miRNA-214-3p. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, J.; Dong, C.; Zhao, M.; Hu, Y.; Jin, F. Extracellular Vesicles Derived from Adipose Mesenchymal Stem Cells Alleviate PM2.5-Induced Lung Injury and Pulmonary Fibrosis. Med. Sci. Monit. 2020, 26, e922782. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Patel, K.P.; Mao, W.; Zhu, D.; Lim, R.; Samuel, C.S. Serelaxin enhances the therapeutic effects of human amnion epithelial cell-derived exosomes in experimental models of lung disease. Br. J. Pharmacol. 2019, 176, 2195–2208. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Lau, S.N.; Leaw, B.; Nguyen, H.P.T.; Salamonsen, L.A.; Saad, M.I.; Chan, S.T.; Zhu, D.; Krause, M.; Kim, C.; et al. Amnion Epithelial Cell-Derived Exosomes Restrict Lung Injury and Enhance Endogenous Lung Repair. Stem Cells Transl. Med. 2018, 7, 180–196. [Google Scholar] [CrossRef]
- Dinh, P.C.; Paudel, D.; Brochu, H.; Popowski, K.D.; Gracieux, M.C.; Cores, J.; Huang, K.; Hensley, M.T.; Harrell, E.; Vandergriff, A.C.; et al. Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis. Nat. Commun. 2020, 11, 1064. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhu, M.; Feng, W.; Lin, Y.; Yin, J.; Jin, J.; Wang, Y. Exosomal miRNA Let-7 from Menstrual Blood-Derived Endometrial Stem Cells Alleviates Pulmonary Fibrosis through Regulating Mitochondrial DNA Damage. Oxidative Med. Cell. Longev. 2019, 2019, 4506303. [Google Scholar] [CrossRef]
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax 2020, 75, 870–881. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Xie, W.; Li, G.; Yao, L.; Zhang, R.; Xu, B. miR-425 reduction causes aberrant proliferation and collagen synthesis through modulating TGF-beta/Smad signaling in acute respiratory distress syndrome. Int. J. Clin. Exp. Pathol. 2019, 12, 2604–2612. [Google Scholar]
- Inomata, M.; Kamio, K.; Azuma, A.; Matsuda, K.; Usuki, J.; Morinaga, A.; Tanaka, T.; Kashiwada, T.; Atsumi, K.; Hayashi, H.; et al. Rictor-targeting exosomal microRNA-16 ameliorates lung fibrosis by inhibiting the mTORC2-SPARC axis. Exp. Cell Res. 2021, 398, 112416. [Google Scholar] [CrossRef]
- Makiguchi, T.; Yamada, M.; Yoshioka, Y.; Sugiura, H.; Koarai, A.; Chiba, S.; Fujino, N.; Tojo, Y.; Ota, C.; Kubo, H.; et al. Serum extracellular vesicular miR-21-5p is a predictor of the prognosis in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 110. [Google Scholar] [CrossRef]
- Njock, M.S.; Guiot, J.; Henket, M.A.; Nivelles, O.; Thiry, M.; Dequiedt, F.; Corhay, J.L.; Louis, R.E.; Struman, I. Sputum exosomes: Promising biomarkers for idiopathic pulmonary fibrosis. Thorax 2019, 74, 309–312. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M.; Martinez-Salgado, C.; Rodriguez-Pena, A.B.; Lopez-Hernandez, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Molitch, M.E. Diabetic Kidney Disease: Much Progress, But Still More to Do. Diabetes Spectr. 2015, 28, 154–156. [Google Scholar] [CrossRef]
- Rao, V.R.A.L.B.V.; Tan, S.H.; Candasamy, M.; Bhattamisra, S.K. Diabetic nephropathy: An update on pathogenesis and drug development. Diabetes Metab. Syndr. 2019, 13, 754–762. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef]
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 2009, 53, 961–973. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Li, S.; Zuo, B.; Zhang, X.; Wang, F.; Sun, D. Exosomes derived from GDNF-modified human adipose mesenchymal stem cells ameliorate peritubular capillary loss in tubulointerstitial fibrosis by activating the SIRT1/eNOS signaling pathway. Theranostics 2020, 10, 9425–9442. [Google Scholar] [CrossRef]
- Liu, X.; Miao, J.; Wang, C.; Zhou, S.; Chen, S.; Ren, Q.; Hong, X.; Wang, Y.; Hou, F.F.; Zhou, L.; et al. Tubule-derived exosomes play a central role in fibroblast activation and kidney fibrosis. Kidney Int. 2020, 97, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xiong, M.; Fang, L.; Jiang, L.; Wen, P.; Dai, C.; Zhang, C.Y.; Yang, J. miR-21-containing microvesicles from injured tubular epithelial cells promote tubular phenotype transition by targeting PTEN protein. Am. J. Pathol. 2013, 183, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Ma, Z.; Livingston, M.J.; Zhang, W.; Yuan, Y.; Guo, C.; Liu, Y.; Fu, P.; Dong, Z. Decreased secretion and profibrotic activity of tubular exosomes in diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 2020, 319, F664–F673. [Google Scholar] [CrossRef]
- Liu, J.R.; Cai, G.Y.; Ning, Y.C.; Wang, J.C.; Lv, Y.; Guo, Y.N.; Fu, B.; Hong, Q.; Sun, X.F.; Chen, X.M. Caloric restriction alleviates aging-related fibrosis of kidney through downregulation of miR-21 in extracellular vesicles. Aging 2020, 12, 18052–18072. [Google Scholar] [CrossRef]
- Borges, F.T.; Melo, S.A.; Ozdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Gattone, V.H., 2nd; LeBleu, V.S.; Kalluri, R. TGF-beta1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. Am. Soc. Nephrol. 2013, 24, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Furini, G.; Schroeder, N.; Huang, L.; Boocock, D.; Scarpellini, A.; Coveney, C.; Tonoli, E.; Ramaswamy, R.; Ball, G.; Verderio, C.; et al. Proteomic Profiling Reveals the Transglutaminase-2 Externalization Pathway in Kidneys after Unilateral Ureteric Obstruction. J. Am. Soc. Nephrol. 2018, 29, 880–905. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Li, X.; Peng, C.; Gao, R.; Ma, L.; Hu, J.; Luo, T.; Qing, H.; Wang, Y.; Ge, Q.; et al. miR-196b-5p-enriched extracellular vesicles from tubular epithelial cells mediated aldosterone-induced renal fibrosis in mice with diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001101. [Google Scholar] [CrossRef] [PubMed]
- Prunotto, M.; Farina, A.; Lane, L.; Pernin, A.; Schifferli, J.; Hochstrasser, D.F.; Lescuyer, P.; Moll, S. Proteomic analysis of podocyte exosome-enriched fraction from normal human urine. J. Proteom. 2013, 82, 193–229. [Google Scholar] [CrossRef] [PubMed]
- Munkonda, M.N.; Akbari, S.; Landry, C.; Sun, S.; Xiao, F.; Turner, M.; Holterman, C.E.; Nasrallah, R.; Hebert, R.L.; Kennedy, C.R.J.; et al. Podocyte-derived microparticles promote proximal tubule fibrotic signaling via p38 MAPK and CD36. J. Extracell. Vesicles 2018, 7, 1432206. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gao, Y.; Xu, L.; Dang, W.; Yan, H.; Zou, D.; Zhu, Z.; Luo, L.; Tian, N.; Wang, X.; et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci. Rep. 2017, 7, 9371. [Google Scholar] [CrossRef]
- Wu, X.M.; Gao, Y.B.; Xu, L.P.; Zou, D.W.; Zhu, Z.Y.; Wang, X.L.; Yao, W.J.; Luo, L.T.; Tong, Y.; Tian, N.X.; et al. Tongxinluo Inhibits Renal Fibrosis in Diabetic Nephropathy: Involvement of the Suppression of Intercellular Transfer of TGF-β1-Containing Exosomes from GECs to GMCs. Am. J. Chin. Med. 2017, 45, 1075–1092. [Google Scholar] [CrossRef]
- Ling, L.; Tan, Z.; Zhang, C.; Gui, S.; Cui, Y.; Hu, Y.; Chen, L. CircRNAs in exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells. Am. J. Transl. Res. 2019, 11, 4667–4682. [Google Scholar] [PubMed]
- Wang, Y.Y.; Tang, L.Q.; Wei, W. Berberine attenuates podocytes injury caused by exosomes derived from high glucose-induced mesangial cells through TGFbeta1-PI3K/AKT pathway. Eur. J. Pharmacol. 2018, 824, 185–192. [Google Scholar] [CrossRef]
- Da Silva Novaes, A.; Borges, F.T.; Maquigussa, E.; Varela, V.A.; Dias, M.V.S.; Boim, M.A. Influence of high glucose on mesangial cell-derived exosome composition, secretion and cell communication. Sci. Rep. 2019, 9, 6270. [Google Scholar] [CrossRef]
- Zhu, Q.J.; Zhu, M.; Xu, X.X.; Meng, X.M.; Wu, Y.G. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-beta1/Smad3 pathway in vivo and in vitro. FASEB J. 2019, 33, 9279–9290. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Sun, X.; Qi, X.; Xia, L.; Wu, Y. Exosomes from high glucose-treated macrophages activate macrophages andinduce inflammatory responses via NF-kappaB signaling pathway in vitro and in vivo. Int. Immunopharmacol. 2020, 84, 106551. [Google Scholar] [CrossRef] [PubMed]
- Nagaishi, K.; Mizue, Y.; Chikenji, T.; Otani, M.; Nakano, M.; Konari, N.; Fujimiya, M. Mesenchymal stem cell therapy ameliorates diabetic nephropathy via the paracrine effect of renal trophic factors including exosomes. Sci. Rep. 2016, 6, 34842. [Google Scholar] [CrossRef]
- Ebrahim, N.; Ahmed, I.A.; Hussien, N.I.; Dessouky, A.A.; Farid, A.S.; Elshazly, A.M.; Mostafa, O.; Gazzar, W.B.E.; Sorour, S.M.; Seleem, Y.; et al. Mesenchymal Stem Cell-Derived Exosomes Ameliorated Diabetic Nephropathy by Autophagy Induction through the mTOR Signaling Pathway. Cells 2018, 7, 226. [Google Scholar] [CrossRef]
- Grange, C.; Tritta, S.; Tapparo, M.; Cedrino, M.; Tetta, C.; Camussi, G.; Brizzi, M.F. Stem cell-derived extracellular vesicles inhibit and revert fibrosis progression in a mouse model of diabetic nephropathy. Sci. Rep. 2019, 9, 4468. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wang, Q.; Zhang, Y.; Jiang, D. Extracellular vesicles produced by bone marrow mesenchymal stem cells attenuate renal fibrosis, in part by inhibiting the RhoA/ROCK pathway, in a UUO rat model. Stem Cell Res. Ther. 2020, 11, 253. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.F.; Fu, G.P.; Guan, C.; Zhang, X.; Yang, D.G.; Shi, Y.C. Protective effect of miRNA-containing extracellular vesicles derived from mesenchymal stromal cells of old rats on renal function in chronic kidney disease. Stem Cell Res. Ther. 2020, 11, 274. [Google Scholar] [CrossRef]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301. [Google Scholar] [CrossRef]
- He, J.; Jiang, Y.L.; Wang, Y.; Tian, X.J.; Sun, S.R. Micro-vesicles from mesenchymal stem cells over-expressing miR-34a inhibit transforming growth factor-beta1-induced epithelial-mesenchymal transition in renal tubular epithelial cells in vitro. Chin. Med. J. (Engl.) 2020, 133, 800–807. [Google Scholar] [CrossRef]
- Eirin, A.; Zhu, X.Y.; Puranik, A.S.; Tang, H.; McGurren, K.A.; van Wijnen, A.J.; Lerman, A.; Lerman, L.O. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017, 92, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Eirin, A.; Zhu, X.; Zhao, Y.; Krier, J.D.; Tang, H.; Jordan, K.L.; Woollard, J.R.; Taner, T.; Lerman, A.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Induce Regulatory T Cells to Ameliorate Chronic Kidney Injury. Hypertension 2020, 75, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Farahani, R.A.; Zhu, X.Y.; Tang, H.; Jordan, K.L.; Lerman, A.; Lerman, L.O.; Eirin, A. Metabolic Syndrome Alters the Cargo of Mitochondria-Related microRNAs in Swine Mesenchymal Stem Cell-Derived Extracellular Vesicles, Impairing Their Capacity to Repair the Stenotic Kidney. Stem Cells Int. 2020, 2020, 8845635. [Google Scholar] [CrossRef] [PubMed]
- Lindoso, R.S.; Lopes, J.A.; Binato, R.; Abdelhay, E.; Takiya, C.M.; Miranda, K.R.; Lara, L.S.; Viola, A.; Bussolati, B.; Vieyra, A.; et al. Adipose Mesenchymal Cells-Derived EVs Alleviate DOCA-Salt-Induced Hypertension by Promoting Cardio-Renal Protection. Mol. Ther. Methods Clin. Dev. 2020, 16, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hu, D.; Zhou, Y.; Yu, Y.; Shen, L.; Long, C.; Butnaru, D.; Timashev, P.; He, D.; Lin, T.; et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against renal interstitial fibrosis through ROS-mediated P38MAPK/ERK signaling pathway. Am. J. Transl. Res. 2020, 12, 4998–5014. [Google Scholar]
- Ji, C.; Zhang, J.; Zhu, Y.; Shi, H.; Yin, S.; Sun, F.; Wang, Q.; Zhang, L.; Yan, Y.; Zhang, X.; et al. Exosomes derived from hucMSC attenuate renal fibrosis through CK1delta/beta-TRCP-mediated YAP degradation. Cell Death Dis. 2020, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Hou, Y.P.; Zou, X.Y.; Xing, X.Y.; Ju, G.Q.; Zhong, L.; Sun, J. Oct-4 Enhanced the Therapeutic Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles in Acute Kidney Injury. Kidney Blood Press. Res. 2020, 45, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Gu, D.; Xing, X.; Cheng, Z.; Gong, D.; Zhang, G.; Zhu, Y. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am. J. Transl. Res. 2016, 8, 4289–4299. [Google Scholar]
- Xiang, E.; Han, B.; Zhang, Q.; Rao, W.; Wang, Z.; Chang, C.; Zhang, Y.; Tu, C.; Li, C.; Wu, D. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res. Ther. 2020, 11, 336. [Google Scholar] [CrossRef]
- Li, H.; Rong, P.; Ma, X.; Nie, W.; Chen, Y.; Zhang, J.; Dong, Q.; Yang, M.; Wang, W. Mouse Umbilical Cord Mesenchymal Stem Cell Paracrine Alleviates Renal Fibrosis in Diabetic Nephropathy by Reducing Myofibroblast Transdifferentiation and Cell Proliferation and Upregulating MMPs in Mesangial Cells. J. Diabetes Res. 2020, 2020, 3847171. [Google Scholar] [CrossRef]
- Kholia, S.; Herrera Sanchez, M.B.; Cedrino, M.; Papadimitriou, E.; Tapparo, M.; Deregibus, M.C.; Brizzi, M.F.; Tetta, C.; Camussi, G. Human Liver Stem Cell-Derived Extracellular Vesicles Prevent Aristolochic Acid-Induced Kidney Fibrosis. Front. Immunol. 2018, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Kholia, S.; Herrera Sanchez, M.B.; Cedrino, M.; Papadimitriou, E.; Tapparo, M.; Deregibus, M.C.; Bruno, S.; Antico, F.; Brizzi, M.F.; Quesenberry, P.J.; et al. Mesenchymal Stem Cell Derived Extracellular Vesicles Ameliorate Kidney Injury in Aristolochic Acid Nephropathy. Front. Cell Dev. Biol. 2020, 8, 188. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Han, C.; Xian, S.; Zhuang, F.; Ding, F.; Zhang, W.; Liu, Y. Placental Mesenchymal Stromal Cells (PMSCs) and PMSC-Derived Extracellular Vesicles (PMSC-EVs) Attenuated Renal Fibrosis in Rats with Unilateral Ureteral Obstruction (UUO) by Regulating CD4(+) T Cell Polarization. Stem Cells Int. 2020, 2020, 2685820. [Google Scholar] [CrossRef]
- Cambier, L.; Giani, J.F.; Liu, W.; Ijichi, T.; Echavez, A.K.; Valle, J.; Marban, E. Angiotensin II-Induced End-Organ Damage in Mice Is Attenuated by Human Exosomes and by an Exosomal Y RNA Fragment. Hypertension 2018, 72, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.H.; Liu, Y.; Gao, H.; Dominguez, J.M., 2nd; Xie, D.; Kelly, K.J. Renal Tubular Cell-Derived Extracellular Vesicles Accelerate the Recovery of Established Renal Ischemia Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 3533–3544. [Google Scholar] [CrossRef]
- Dominguez, J.M., 2nd; Dominguez, J.H.; Xie, D.; Kelly, K.J. Human extracellular microvesicles from renal tubules reverse kidney ischemia-reperfusion injury in rats. PLoS ONE 2018, 13, e0202550. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, C.H.; Song, S.H.; Um, J.E.; Kim, H.S.; Yun, J.S.; Han, D.; Cho, E.S.; Nam, B.Y.; Yook, J.I.; et al. Micellized Protein Transduction Domain-Bone Morphogenetic Protein-7 Efficiently Blocks Renal Fibrosis Via Inhibition of Transforming Growth Factor-Beta-Mediated Epithelial-Mesenchymal Transition. Front. Pharmacol. 2020, 11, 591275. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Chun-Yan, L.; Zi-Yi, Z.; Tian-Lin, Y.; Yi-Li, W.; Bao, L.; Jiao, L.; Wei-Jun, D. Liquid biopsy biomarkers of renal interstitial fibrosis based on urinary exosome. Exp. Mol. Pathol. 2018, 105, 223–228. [Google Scholar] [CrossRef]
- Lv, C.Y.; Ding, W.J.; Wang, Y.L.; Zhao, Z.Y.; Li, J.H.; Chen, Y.; Lv, J. A PEG-based method for the isolation of urinary exosomes and its application in renal fibrosis diagnostics using cargo miR-29c and miR-21 analysis. Int. Urol. Nephrol. 2018, 50, 973–982. [Google Scholar] [CrossRef]
- Lv, L.L.; Cao, Y.H.; Ni, H.F.; Xu, M.; Liu, D.; Liu, H.; Chen, P.S.; Liu, B.C. MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am. J. Physiol. Ren. Physiol. 2013, 305, F1220–F1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; He, W.; Zhao, Y.; Zhang, A.; Liu, Y.; Hassounah, F.; Ma, F.; Klein, J.D.; Wang, X.H.; et al. Exogenous miR-29a Attenuates Muscle Atrophy and Kidney Fibrosis in Unilateral Ureteral Obstruction Mice. Hum. Gene Ther. 2020, 31, 367–375. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, H.; Wang, B.; Yuan, Y.; Klein, J.D.; Wang, X.H. Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. FASEB J. 2019, 33, 13590–13601. [Google Scholar] [CrossRef] [PubMed]
- Sole, C.; Cortes-Hernandez, J.; Felip, M.L.; Vidal, M.; Ordi-Ros, J. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol. Dial. Transplant. 2015, 30, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, J.; Martinez-Arroyo, O.; Ortega, A.; Galera, M.; Solis-Salguero, M.A.; Chaves, F.J.; Redon, J.; Forner, M.J.; Cortes, R. Urinary exosomal miR-146a as a marker of albuminuria, activity changes and disease flares in lupus nephritis. J. Nephrol. 2020. [Google Scholar] [CrossRef]
- Garcia-Vives, E.; Sole, C.; Moline, T.; Vidal, M.; Agraz, I.; Ordi-Ros, J.; Cortes-Hernandez, J. The Urinary Exosomal miRNA Expression Profile is Predictive of Clinical Response in Lupus Nephritis. Int. J. Mol. Sci. 2020, 21, 1372. [Google Scholar] [CrossRef]
- Khurana, R.; Ranches, G.; Schafferer, S.; Lukasser, M.; Rudnicki, M.; Mayer, G.; Huttenhofer, A. Identification of urinary exosomal noncoding RNAs as novel biomarkers in chronic kidney disease. RNA 2017, 23, 142–152. [Google Scholar] [CrossRef]
- Lv, L.L.; Cao, Y.H.; Pan, M.M.; Liu, H.; Tang, R.N.; Ma, K.L.; Chen, P.S.; Liu, B.C. CD2AP mRNA in urinary exosome as biomarker of kidney disease. Clin. Chim. Acta 2014, 428, 26–31. [Google Scholar] [CrossRef]
- Carreras-Planella, L.; Cucchiari, D.; Canas, L.; Juega, J.; Franquesa, M.; Bonet, J.; Revuelta, I.; Diekmann, F.; Taco, O.; Lauzurica, R.; et al. Urinary vitronectin identifies patients with high levels of fibrosis in kidney grafts. J. Nephrol. 2020, 34, 861–874. [Google Scholar] [CrossRef]
- Saejong, S.; Townamchai, N.; Somparn, P.; Tangtanatakul, P.; Ondee, T.; Hirankarn, N.; Leelahavanichkul, A. MicroRNA-21 in plasma exosome, but not from whole plasma, as a biomarker for the severe interstitial fibrosis and tubular atrophy (IF/TA) in post-renal transplantation. Asian Pac. J. Allergy Immunol. 2020. [Google Scholar] [CrossRef]
- Xie, J.X.; Fan, X.; Drummond, C.A.; Majumder, R.; Xie, Y.; Chen, T.; Liu, L.; Haller, S.T.; Brewster, P.S.; Dworkin, L.D.; et al. MicroRNA profiling in kidney disease: Plasma versus plasma-derived exosomes. Gene 2017, 627, 1–8. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Knight, D.A.; Boyle, A.J. The Processes and Mechanisms of Cardiac and Pulmonary Fibrosis. Front. Physiol. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Sharma, P.; Agrawal, S.; Agrawal, D.K. Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research. Mol. Cell. Biochem. 2017, 424, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, X.; Xue, F.; Li, Y.; Liu, W.; Zhang, S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am. J. Transl. Res. 2018, 10, 4350–4366. [Google Scholar]
- Kenneweg, F.; Bang, C.; Xiao, K.; Boulanger, C.M.; Loyer, X.; Mazlan, S.; Schroen, B.; Hermans-Beijnsberger, S.; Foinquinos, A.; Hirt, M.N.; et al. Long Noncoding RNA-Enriched Vesicles Secreted by Hypoxic Cardiomyocytes Drive Cardiac Fibrosis. Mol. Ther. Nucleic Acids 2019, 18, 363–374. [Google Scholar] [CrossRef]
- Danielson, K.M.; Shah, R.; Yeri, A.; Liu, X.; Camacho Garcia, F.; Silverman, M.; Tanriverdi, K.; Das, A.; Xiao, C.; Jerosch-Herold, M.; et al. Plasma Circulating Extracellular RNAs in Left Ventricular Remodeling Post-Myocardial Infarction. EBioMedicine 2018, 32, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.W.; Wang, S.B. Microvesicles containing microRNA-21 induce myocardial fibrosis via AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4634–4641. [Google Scholar] [CrossRef]
- Kuo, H.F.; Hsieh, C.C.; Wang, S.C.; Chang, C.Y.; Hung, C.H.; Kuo, P.L.; Liu, Y.R.; Li, C.Y.; Liu, P.L. Simvastatin Attenuates Cardiac Fibrosis via Regulation of Cardiomyocyte-Derived Exosome Secretion. J. Clin. Med. 2019, 8, 794. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.; Ikeda, G.; Tada, Y.; Wahlquist, C.; Mercola, M.; Yang, P.C. Sacubitril/Valsartan Improves Cardiac Function and Decreases Myocardial Fibrosis Via Downregulation of Exosomal miR-181a in a Rodent Chronic Myocardial Infarction Model. J. Am. Heart Assoc. 2020, 9, e015640. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef] [PubMed]
- Dzialo, E.; Rudnik, M.; Koning, R.I.; Czepiel, M.; Tkacz, K.; Baj-Krzyworzeka, M.; Distler, O.; Siedlar, M.; Kania, G.; Blyszczuk, P. WNT3a and WNT5a Transported by Exosomes Activate WNT Signaling Pathways in Human Cardiac Fibroblasts. Int. J. Mol. Sci. 2019, 20, 1436. [Google Scholar] [CrossRef]
- Frieler, R.A.; Mortensen, R.M. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 2015, 131, 1019–1030. [Google Scholar] [CrossRef]
- Cai, L.; Chao, G.; Li, W.; Zhu, J.; Li, F.; Qi, B.; Wei, Y.; Chen, S.; Zhou, G.; Lu, X.; et al. Activated CD4(+) T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging 2020, 12, 7380–7396. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Patil, M.; Garikipati, V.N.S.; Verma, S.K.; Saheera, S.; Narasimhan, G.; Zhu, W.; Kishore, R.; Zhang, J.; Krishnamurthy, P. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 2020, 34, 2238–2251. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Z.M.; Ji, J.L.; Gan, W.; Zhang, A.; Shi, H.J.; Wang, H.; Lv, L.; Li, Z.; Tang, T.; et al. Macrophage-Derived Exosomal Mir-155 Regulating Cardiomyocyte Pyroptosis and Hypertrophy in Uremic Cardiomyopathy. JACC Basic Transl. Sci. 2020, 5, 148–166. [Google Scholar] [CrossRef]
- Yang, J.; Yu, X.F.; Li, Y.Y.; Xue, F.T.; Zhang, S. Decreased HSP70 expression on serum exosomes contributes to cardiac fibrosis during senescence. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3993–4001. [Google Scholar] [CrossRef]
- Shao, L.; Zhang, Y.; Pan, X.; Liu, B.; Liang, C.; Zhang, Y.; Wang, Y.; Yan, B.; Xie, W.; Sun, Y.; et al. Knockout of beta-2 microglobulin enhances cardiac repair by modulating exosome imprinting and inhibiting stem cell-induced immune rejection. Cell. Mol. Life Sci. 2020, 77, 937–952. [Google Scholar] [CrossRef]
- Wang, J.; Lee, C.J.; Deci, M.B.; Jasiewicz, N.; Verma, A.; Canty, J.M.; Nguyen, J. MiR-101a loaded extracellular nanovesicles as bioactive carriers for cardiac repair. Nanomedicine 2020, 27, 102201. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shen, H.; Shao, L.; Teng, X.; Chen, Y.; Liu, X.; Yang, Z.; Shen, Z. HIF-1alpha overexpression in mesenchymal stem cell-derived exosomes mediates cardioprotection in myocardial infarction by enhanced angiogenesis. Stem Cell Res. Ther. 2020, 11, 373. [Google Scholar] [CrossRef]
- Sun, L.; Zhu, W.; Zhao, P.; Zhang, J.; Lu, Y.; Zhu, Y.; Zhao, W.; Liu, Y.; Chen, Q.; Zhang, F. Down-Regulated Exosomal MicroRNA-221—3p Derived from Senescent Mesenchymal Stem Cells Impairs Heart Repair. Front. Cell Dev. Biol. 2020, 8, 263. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Zhao, Z.; Meng, Q.; Yu, Y.; Sun, J.; Yang, Z.; Chen, Y.; Li, J.; Ma, T.; et al. Engineered Exosomes with Ischemic Myocardium-Targeting Peptide for Targeted Therapy in Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008737. [Google Scholar] [CrossRef]
- Firoozi, S.; Pahlavan, S.; Ghanian, M.H.; Rabbani, S.; Barekat, M.; Nazari, A.; Pakzad, M.; Shekari, F.; Hassani, S.N.; Moslem, F.; et al. Mesenchymal stem cell-derived extracellular vesicles alone or in conjunction with a SDKP-conjugated self-assembling peptide improve a rat model of myocardial infarction. Biochem. Biophys. Res. Commun. 2020, 524, 903–909. [Google Scholar] [CrossRef]
- Chen, F.; Li, X.; Zhao, J.; Geng, J.; Xie, J.; Xu, B. Bone marrow mesenchymal stem cell-derived exosomes attenuate cardiac hypertrophy and fibrosis in pressure overload induced remodeling. In Vitro Cell. Dev. Biol. Anim. 2020, 56, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, W.; Wani, M.; Yu, X.; Ashraf, M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS ONE 2014, 9, e88685. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, K.; Zhang, N.; Zhao, Y.; Ma, Q.; Shen, J.; Lin, Y.; Xiang, P.; Tang, Y.; Hu, X.; et al. Myocardial reparative functions of exosomes from mesenchymal stem cells are enhanced by hypoxia treatment of the cells via transferring microRNA-210 in an nSMase2-dependent way. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1659–1670. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, J.; Yan, W.; Li, Y.; Shen, Z.; Asahara, T. Pretreatment of Cardiac Stem Cells with Exosomes Derived from Mesenchymal Stem Cells Enhances Myocardial Repair. J. Am. Heart Assoc. 2016, 5, e002856. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, A.; Wang, H.; Klein, J.D.; Tan, L.; Wang, Z.M.; Du, J.; Naqvi, N.; Liu, B.C.; Wang, X.H. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019, 9, 1864–1877. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Cao, W.; Ma, J.; Sun, L.; Qian, H.; Zhu, W.; Xu, W. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Relieve Acute Myocardial Ischemic Injury. Stem Cells Int. 2015, 2015, 761643. [Google Scholar] [CrossRef]
- Han, C.; Zhou, J.; Liang, C.; Liu, B.; Pan, X.; Zhang, Y.; Wang, Y.; Yan, B.; Xie, W.; Liu, F.; et al. Human umbilical cord mesenchymal stem cell derived exosomes encapsulated in functional peptide hydrogels promote cardiac repair. Biomater. Sci. 2019, 7, 2920–2933. [Google Scholar] [CrossRef]
- Zhang, N.; Zhu, J.; Ma, Q.; Zhao, Y.; Wang, Y.; Hu, X.; Chen, J.; Zhu, W.; Han, Z.; Yu, H. Exosomes derived from human umbilical cord MSCs rejuvenate aged MSCs and enhance their functions for myocardial repair. Stem Cell Res. Ther. 2020, 11, 273. [Google Scholar] [CrossRef]
- Venkat, P.; Cui, C.; Chen, Z.; Chopp, M.; Zacharek, A.; Landschoot-Ward, J.; Culmone, L.; Yang, X.P.; Xu, J.; Chen, J. CD133+Exosome Treatment Improves Cardiac Function after Stroke in Type 2 Diabetic Mice. Transl. Stroke Res. 2020, 12, 112–124. [Google Scholar] [CrossRef]
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564. [Google Scholar] [CrossRef]
- Luo, Q.; Guo, D.; Liu, G.; Chen, G.; Hang, M.; Jin, M. Exosomes from MiR-126-Overexpressing Adscs Are Therapeutic in Relieving Acute Myocardial Ischaemic Injury. Cell Physiol. Biochem. 2017, 44, 2105–2116. [Google Scholar] [CrossRef]
- Pan, J.; Alimujiang, M.; Chen, Q.; Shi, H.; Luo, X. Exosomes derived from miR-146a-modified adipose-derived stem cells attenuate acute myocardial infarction-induced myocardial damage via downregulation of early growth response factor 1. J. Cell. Biochem. 2019, 120, 4433–4443. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, X.Y.; Zhao, Y.; Eirin, A.; Liu, L.; Ferguson, C.M.; Tang, H.; Lerman, A.; Lerman, L.O. Selective intrarenal delivery of mesenchymal stem cell-derived extracellular vesicles attenuates myocardial injury in experimental metabolic renovascular disease. Basic Res. Cardiol. 2020, 115, 16. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, W.; Meng, W.; Jegga, A.G.; Wang, Y.; Cai, W.; Kim, H.W.; Pasha, Z.; Wen, Z.; Rao, F.; et al. Heat shock improves Sca-1+ stem cell survival and directs ischemic cardiomyocytes toward a prosurvival phenotype via exosomal transfer: A critical role for HSF1/miR-34a/HSP70 pathway. Stem Cells 2014, 32, 462–472. [Google Scholar] [CrossRef]
- Xuan, W.; Khan, M.; Ashraf, M. Extracellular Vesicles from Notch Activated Cardiac Mesenchymal Stem Cells Promote Myocyte Proliferation and Neovasculogenesis. Front. Cell Dev. Biol. 2020, 8, 11. [Google Scholar] [CrossRef]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 2020, 116, 383–392. [Google Scholar] [CrossRef]
- Kompa, A.R.; Greening, D.W.; Kong, A.M.; McMillan, P.J.; Fang, H.; Saxena, R.; Wong, R.C.B.; Lees, J.G.; Sivakumaran, P.; Newcomb, A.E.; et al. Sustained subcutaneous delivery of secretome of human cardiac stem cells promotes cardiac repair following myocardial infarction. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Tseliou, E.; Fouad, J.; Reich, H.; Slipczuk, L.; de Couto, G.; Aminzadeh, M.; Middleton, R.; Valle, J.; Weixin, L.; Marban, E. Fibroblasts Rendered Antifibrotic, Antiapoptotic, and Angiogenic by Priming with Cardiosphere-Derived Extracellular Membrane Vesicles. J. Am. Coll. Cardiol. 2015, 66, 599–611. [Google Scholar] [CrossRef]
- Gallet, R.; Dawkins, J.; Valle, J.; Simsolo, E.; de Couto, G.; Middleton, R.; Tseliou, E.; Luthringer, D.; Kreke, M.; Smith, R.R.; et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur. Heart J. 2017, 38, 201–211. [Google Scholar] [CrossRef]
- Hirai, K.; Ousaka, D.; Fukushima, Y.; Kondo, M.; Eitoku, T.; Shigemitsu, Y.; Hara, M.; Baba, K.; Iwasaki, T.; Kasahara, S.; et al. Cardiosphere-derived exosomal microRNAs for myocardial repair in pediatric dilated cardiomyopathy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Ibrahim, A.G.E.; Li, C.; Rogers, R.; Fournier, M.; Li, L.; Vaturi, S.D.; Antes, T.; Sanchez, L.; Akhmerov, A.; Moseley, J.J.; et al. Augmenting canonical Wnt signalling in therapeutically inert cells converts them into therapeutically potent exosome factories. Nat. Biomed. Eng. 2019, 3, 695–705. [Google Scholar] [CrossRef]
- Vandergriff, A.C.; de Andrade, J.B.; Tang, J.; Hensley, M.T.; Piedrahita, J.A.; Caranasos, T.G.; Cheng, K. Intravenous Cardiac Stem Cell-Derived Exosomes Ameliorate Cardiac Dysfunction in Doxorubicin Induced Dilated Cardiomyopathy. Stem Cells Int. 2015, 2015, 960926. [Google Scholar] [CrossRef]
- Vandergriff, A.; Huang, K.; Shen, D.; Hu, S.; Hensley, M.T.; Caranasos, T.G.; Qian, L.; Cheng, K. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics 2018, 8, 1869–1878. [Google Scholar] [CrossRef]
- Ni, J.; Liu, Y.; Kang, L.; Wang, L.; Han, Z.; Wang, K.; Xu, B.; Gu, R. Human trophoblast-derived exosomes attenuate doxorubicin-induced cardiac injury by regulating miR-200b and downstream Zeb1. J. Nanobiotechnol. 2020, 18, 171. [Google Scholar] [CrossRef]
- Ke, X.; Yang, D.; Liang, J.; Wang, X.; Wu, S.; Wang, X.; Hu, C. Human Endothelial Progenitor Cell-Derived Exosomes Increase Proliferation and Angiogenesis in Cardiac Fibroblasts by Promoting the Mesenchymal-Endothelial Transition and Reducing High Mobility Group Box 1 Protein B1 Expression. DNA Cell Biol. 2017, 36, 1018–1028. [Google Scholar] [CrossRef]
- Lin, F.; Zeng, Z.; Song, Y.; Li, L.; Wu, Z.; Zhang, X.; Li, Z.; Ke, X.; Hu, X. YBX-1 mediated sorting of miR-133 into hypoxia/reoxygenation-induced EPC-derived exosomes to increase fibroblast angiogenesis and MEndoT. Stem Cell Res. Ther. 2019, 10, 263. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, H.; Mai, J.; Chen, Z.; Huang, T.; Wang, S.; Chen, Y.; Wang, J. Distinct Anti-Fibrotic Effects of Exosomes Derived from Endothelial Colony-Forming Cells Cultured Under Normoxia and Hypoxia. Med. Sci. Monit. 2018, 24, 6187–6199. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Xue, F.; Liu, W.; Zhang, S. Exosomes derived from cardiac telocytes exert positive effects on endothelial cells. Am. J. Transl. Res. 2017, 9, 5375–5387. [Google Scholar]
- Su, X.; Jin, Y.; Shen, Y.; Ju, C.; Cai, J.; Liu, Y.; Kim, I.M.; Wang, Y.; Yu, H.; Weintraub, N.L.; et al. Exosome-Derived Dystrophin from Allograft Myogenic Progenitors Improves Cardiac Function in Duchenne Muscular Dystrophic Mice. J. Cardiovasc. Transl. Res. 2018, 11, 412–419. [Google Scholar] [CrossRef]
- Santoso, M.R.; Ikeda, G.; Tada, Y.; Jung, J.H.; Vaskova, E.; Sierra, R.G.; Gati, C.; Goldstone, A.B.; von Bornstaedt, D.; Shukla, P.; et al. Exosomes from Induced Pluripotent Stem Cell-Derived Cardiomyocytes Promote Autophagy for Myocardial Repair. J. Am. Heart Assoc. 2020, 9, e014345. [Google Scholar] [CrossRef]
- Xuan, W.; Wang, L.; Xu, M.; Weintraub, N.L.; Ashraf, M. miRNAs in Extracellular Vesicles from iPS-Derived Cardiac Progenitor Cells Effectively Reduce Fibrosis and Promote Angiogenesis in Infarcted Heart. Stem Cells Int. 2019, 2019, 3726392. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Kervadec, A.; Bellamy, V.; El Harane, N.; Arakelian, L.; Vanneaux, V.; Cacciapuoti, I.; Nemetalla, H.; Perier, M.C.; Toeg, H.D.; Richart, A.; et al. Cardiovascular progenitor-derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J. Heart Lung Transplant. 2016, 35, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Charles, C.J.; Li, R.R.; Yeung, T.; Mazlan, S.M.I.; Lai, R.C.; de Kleijn, D.P.V.; Lim, S.K.; Richards, A.M. Systemic Mesenchymal Stem Cell-Derived Exosomes Reduce Myocardial Infarct Size: Characterization with MRI in a Porcine Model. Front. Cardiovasc. Med. 2020, 7, 601990. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Kalani, A.; Medina, I.; Familtseva, A.; Tyagi, S.C. Cardiosome mediated regulation of MMP9 in diabetic heart: Role of mir29b and mir455 in exercise. J. Cell. Mol. Med. 2015, 19, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 2018, 8, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Izumi, Y.; Nakamura, Y.; Yamazaki, T.; Shiota, M.; Sano, S.; Tanaka, M.; Osada-Oka, M.; Shimada, K.; Miura, K.; et al. Repeated remote ischemic conditioning attenuates left ventricular remodeling via exosome-mediated intercellular communication on chronic heart failure after myocardial infarction. Int. J. Cardiol. 2015, 178, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Gollmann-Tepekoylu, C.; Polzl, L.; Graber, M.; Hirsch, J.; Nagele, F.; Lobenwein, D.; Hess, M.W.; Blumer, M.J.; Kirchmair, E.; Zipperle, J.; et al. miR-19a-3p containing exosomes improve function of ischemic myocardium upon shock wave therapy. Cardiovasc. Res. 2020, 116, 1226–1236. [Google Scholar] [CrossRef]
- Paudel, K.R.; Kim, D.W. Microparticles-Mediated Vascular Inflammation and its Amelioration by Antioxidant Activity of Baicalin. Antioxidants 2020, 9, 890. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Yokoya, M.; Kodama, T.; Hori, K.; Matsumoto, K.; Okada, M.; Yamawaki, H. Plasma exosomes regulate systemic blood pressure in rats. Biochem. Biophys. Res. Commun. 2018, 503, 776–783. [Google Scholar] [CrossRef]
- Kang, J.Y.; Park, H.; Kim, H.; Mun, D.; Park, H.; Yun, N.; Joung, B. Human peripheral bloodderived exosomes for microRNA delivery. Int. J. Mol. Med. 2019, 43, 2319–2328. [Google Scholar] [CrossRef]
- Mun, D.; Kim, H.; Kang, J.Y.; Park, H.; Park, H.; Lee, S.H.; Yun, N.; Joung, B. Expression of miRNAs in circulating exosomes derived from patients with persistent atrial fibrillation. FASEB J. 2019, 33, 5979–5989. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.; Shu, J.; Tang, C.E.; Jiang, Y.; Luo, F. Identification of microRNAs enriched in exosomes in human pericardial fluid of patients with atrial fibrillation based on bioinformatic analysis. J. Thorac. Dis. 2020, 12, 5617–5627. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Xu, B.; Liu, Y.L.; Liu, Z. Reduced exosome miR-425 and miR-744 in the plasma represents the progression of fibrosis and heart failure. Kaohsiung J. Med. Sci. 2018, 34, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.Y.; Xu, J.; et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Kabbany, M.N.; Conjeevaram Selvakumar, P.K.; Watt, K.; Lopez, R.; Akras, Z.; Zein, N.; Carey, W.; Alkhouri, N. Prevalence of Nonalcoholic Steatohepatitis-Associated Cirrhosis in the United States: An Analysis of National Health and Nutrition Examination Survey Data. Am. J. Gastroenterol. 2017, 112, 581–587. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Valencia-Rodriguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodriguez, G.; Zamora-Valdes, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed. Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Furuta, K.; Lucien, F.; Gutierrez Sanchez, L.H.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, S.Y.; Ko, E.; Lee, J.H.; Yi, H.S.; Yoo, Y.J.; Je, J.; Suh, S.J.; Jung, Y.K.; Kim, J.H.; et al. Exosomes derived from palmitic acid-treated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci. Rep. 2017, 7, 3710. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Panera, N.; Eguchi, A.; Johnson, C.D.; Papouchado, B.G.; de Araujo Horcel, L.; Pinatel, E.M.; Alisi, A.; Nobili, V.; Feldstein, A.E. Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 646–663.e4. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Morello, E.; Bocca, C.; Foglia, B.; Benetti, E.; Novo, E.; Chiazza, F.; Rogazzo, M.; Fantozzi, R.; Povero, D.; et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0172575. [Google Scholar] [CrossRef]
- Povero, D.; Eguchi, A.; Li, H.; Johnson, C.D.; Papouchado, B.G.; Wree, A.; Messer, K.; Feldstein, A.E. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS ONE 2014, 9, e113651. [Google Scholar] [CrossRef]
- McCommis, K.S.; Hodges, W.T.; Brunt, E.M.; Nalbantoglu, I.; McDonald, W.G.; Holley, C.; Fujiwara, H.; Schaffer, J.E.; Colca, J.R.; Finck, B.N. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017, 65, 1543–1556. [Google Scholar] [CrossRef]
- Eguchi, A.; Yan, R.; Pan, S.Q.; Wu, R.; Kim, J.; Chen, Y.; Ansong, C.; Smith, R.D.; Tempaku, M.; Ohno-Machado, L.; et al. Comprehensive characterization of hepatocyte-derived extracellular vesicles identifies direct miRNA-based regulation of hepatic stellate cells and DAMP-based hepatic macrophage IL-1beta and IL-17 upregulation in alcoholic hepatitis mice. J. Mol. Med. 2020, 98, 1021–1034. [Google Scholar] [CrossRef]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef]
- Zhang, X.W.; Zhou, J.C.; Peng, D.; Hua, F.; Li, K.; Yu, J.J.; Lv, X.X.; Cui, B.; Liu, S.S.; Yu, J.M.; et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy 2020, 16, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Chen, C.; Xue, J.; Xiao, T.; Mostofa, G.; Wang, D.; Chen, X.; Xu, H.; Sun, Q.; Li, J.; et al. Exosomal MALAT1 derived from hepatic cells is involved in the activation of hepatic stellate cells via miRNA-26b in fibrosis induced by arsenite. Toxicol. Lett. 2019, 316, 73–84. [Google Scholar] [CrossRef]
- Dhanraj, P.; Venter, C.; Bester, M.J.; Oberholzer, H.M. Induction of hepatic portal fibrosis, mitochondria damage, and extracellular vesicle formation in Sprague-Dawley rats exposed to copper, manganese, and mercury, alone and in combination. Ultrastruct. Pathol. 2020, 44, 182–192. [Google Scholar] [CrossRef]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-Mediated Intercellular Communication between Hepatitis C Virus-Infected Hepatocytes and Hepatic Stellate Cells. J. Virol. 2017, 91, e02225-16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, C.H.; Lee, S.W. Exosomal Transmission of MicroRNA from HCV Replicating Cells Stimulates Transdifferentiation in Hepatic Stellate Cells. Mol. Ther. Nucleic Acids 2019, 14, 483–497. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Huang, Z.; Gurley, E.C.; Wang, X.; Wang, J.; He, H.; Yang, H.; Lai, G.; Zhang, L.; et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology 2018, 68, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Wang, Y.; Zhu, W.; Zhao, D.; Wang, X.; Yang, H.; Gurley, E.C.; Chen, W.; Hylemon, P.B.; et al. Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells 2020, 9, 190. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef]
- Al Suraih, M.S.; Trussoni, C.E.; Splinter, P.L.; LaRusso, N.F.; O’Hara, S.P. Senescent cholangiocytes release extracellular vesicles that alter target cell phenotype via the epidermal growth factor receptor. Liver Int. 2020, 40, 2455–2468. [Google Scholar] [CrossRef]
- Kostallari, E.; Hirsova, P.; Prasnicka, A.; Verma, V.K.; Yaqoob, U.; Wongjarupong, N.; Roberts, L.R.; Shah, V.H. Hepatic stellate cell-derived platelet-derived growth factor receptor-alpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology 2018, 68, 333–348. [Google Scholar] [CrossRef]
- Gao, J.; Wei, B.; de Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J. Hepatol. 2020, 73, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, Y.; Li, S.; Liu, Y.; Zhou, S.; Cao, S.; Yin, J.; Li, G. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J. Cell. Mol. Med. 2017, 21, 3679–3692. [Google Scholar] [CrossRef]
- Wan, L.; Xia, T.; Du, Y.; Liu, J.; Xie, Y.; Zhang, Y.; Guan, F.; Wu, J.; Wang, X.; Shi, C. Exosomes from activated hepatic stellate cells contain GLUT1 and PKM2: A role for exosomes in metabolic switch of liver nonparenchymal cells. FASEB J. 2019, 33, 8530–8542. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Brigstock, D.R. Integrins and heparan sulfate proteoglycans on hepatic stellate cells (HSC) are novel receptors for HSC-derived exosomes. FEBS Lett. 2016, 590, 4263–4274. [Google Scholar] [CrossRef]
- Fang, P.P.; Pan, C.W.; Lin, W.; Li, J.; Huang, S.S.; Zhou, G.Y.; Du, W.J.; Li, Q. ASK1 Enhances Angiotensin II-Induced Liver Fibrosis In Vitro by Mediating Endoplasmic Reticulum Stress-Dependent Exosomes. Mediat. Inflamm. 2020, 2020, 8183713. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, R.; Kemper, S.; Brigstock, D.R. Dynamic Changes in Function and Proteomic Composition of Extracellular Vesicles from Hepatic Stellate Cells during Cellular Activation. Cells 2020, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Cadoret, A.; Rautou, P.E.; El Mourabit, H.; Ratziu, V.; Corpechot, C.; Rey, C.; Bosselut, N.; Barbu, V.; Wendum, D.; et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 2015, 61, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ding, Q.; Yaqoob, U.; de Assuncao, T.M.; Verma, V.K.; Hirsova, P.; Cao, S.; Mukhopadhyay, D.; Huebert, R.C.; Shah, V.H. Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-dependent Migration. J. Biol. Chem. 2015, 290, 30684–30696. [Google Scholar] [CrossRef]
- Chen, L.; Yao, X.; Yao, H.; Ji, Q.; Ding, G.; Liu, X. Exosomal miR-103-3p from LPS-activated THP-1 macrophage contributes to the activation of hepatic stellate cells. FASEB J. 2020, 34, 5178–5192. [Google Scholar] [CrossRef]
- Khodayari, N.; Oshins, R.; Holliday, L.S.; Clark, V.; Xiao, Q.; Marek, G.; Mehrad, B.; Brantly, M. Alpha-1 antitrypsin deficient individuals have circulating extracellular vesicles with profibrogenic cargo. Cell Commun. Signal. 2020, 18, 140. [Google Scholar] [CrossRef]
- Kim, D.K.; Cho, Y.E.; Komarow, H.D.; Bandara, G.; Song, B.J.; Olivera, A.; Metcalfe, D.D. Mastocytosis-derived extracellular vesicles exhibit a mast cell signature, transfer KIT to stellate cells, and promote their activation. Proc. Natl. Acad. Sci. USA 2018, 115, E10692–E10701. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Liu, J.; Yao, X.; Jiang, T.; Wang, Y.; Xie, F. Human bone marrow mesenchymal stem cells-derived exosomes alleviate liver fibrosis through the Wnt/beta-catenin pathway. Stem Cell Res. Ther. 2019, 10, 98. [Google Scholar] [CrossRef]
- Nishi, M.; Matsumoto, T.; Fujisawa, K.; Suehiro, Y.; Takami, T.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Mesenchymal Stem Cells Induce a Fibrolytic Phenotype by Regulating mmu-miR-6769b-5p Expression in Macrophages. Stem Cells Dev. 2020, 29, 1457–1466. [Google Scholar] [CrossRef]
- Lou, G.; Yang, Y.; Liu, F.; Ye, B.; Chen, Z.; Zheng, M.; Liu, Y. MiR-122 modification enhances the therapeutic efficacy of adipose tissue-derived mesenchymal stem cells against liver fibrosis. J. Cell. Mol. Med. 2017, 21, 2963–2973. [Google Scholar] [CrossRef]
- Du, Z.; Wu, T.; Liu, L.; Luo, B.; Wei, C. Extracellular vesicles-derived miR-150-5p secreted by adipose-derived mesenchymal stem cells inhibits CXCL1 expression to attenuate hepatic fibrosis. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, Q.; Cai, X.; Li, F.; Ma, Z.; Xu, M.; Lu, L. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell. Mol. Med. 2017, 21, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, X.; Li, W.; Wang, L. Exosomes derived from mmu_circ_0000623-modified ADSCs prevent liver fibrosis via activating autophagy. Hum. Exp. Toxicol. 2020, 39, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Tan, Y.; Cai, M.; Zhao, T.; Mao, F.; Zhang, X.; Xu, W.; Yan, Z.; Qian, H.; Yan, Y. Human Umbilical Cord MSC-Derived Exosomes Suppress the Development of CCl4-Induced Liver Injury through Antioxidant Effect. Stem Cells Int. 2018, 2018, 6079642. [Google Scholar] [CrossRef]
- Li, T.; Yan, Y.; Wang, B.; Qian, H.; Zhang, X.; Shen, L.; Wang, M.; Zhou, Y.; Zhu, W.; Li, W.; et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013, 22, 845–854. [Google Scholar] [CrossRef]
- Kim, J.; Lee, C.; Shin, Y.; Wang, S.; Han, J.; Kim, M.; Kim, J.M.; Shin, S.C.; Lee, B.J.; Kim, T.J.; et al. sEVs from tonsil-derived mesenchymal stromal cells alleviate activation of hepatic stellate cells and liver fibrosis through miR-486-5p. Mol. Ther. 2020, 29, 1471–1486. [Google Scholar] [CrossRef]
- Fiore, E.; Dominguez, L.M.; Bayo, J.; Malvicini, M.; Atorrasagasti, C.; Rodriguez, M.; Cantero, M.J.; Garcia, M.; Yannarelli, G.; Mazzolini, G. Human umbilical cord perivascular cells-derived extracellular vesicles mediate the transfer of IGF-I to the liver and ameliorate hepatic fibrogenesis in mice. Gene Ther. 2020, 27, 62–73. [Google Scholar] [CrossRef]
- Alhomrani, M.; Correia, J.; Zavou, M.; Leaw, B.; Kuk, N.; Xu, R.; Saad, M.I.; Hodge, A.; Greening, D.W.; Lim, R.; et al. The Human Amnion Epithelial Cell Secretome Decreases Hepatic Fibrosis in Mice with Chronic Liver Fibrosis. Front. Pharmacol. 2017, 8, 748. [Google Scholar] [CrossRef]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Yuyama, K.; Nakamura, H.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Extracellular Vesicles from Amnion-Derived Mesenchymal Stem Cells Ameliorate Hepatic Inflammation and Fibrosis in Rats. Stem Cells Int. 2018, 2018, 3212643. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Pasquino, C.; Herrera Sanchez, M.B.; Tapparo, M.; Figliolini, F.; Grange, C.; Chiabotto, G.; Cedrino, M.; Deregibus, M.C.; Tetta, C.; et al. HLSC-Derived Extracellular Vesicles Attenuate Liver Fibrosis and Inflammation in a Murine Model of Non-alcoholic Steatohepatitis. Mol. Ther. 2020, 28, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Shiha, G.; Nabil, A.; Lotfy, A.; Soliman, R.; Hassan, A.A.; Ali, I.S.; Gad, D.F.; Zahran, F. Antifibrotic Effect of Combination of Nilotinib and Stem Cell-Conditioned Media on CCl4-Induced Liver Fibrosis. Stem Cells Int. 2020, 2020, 6574010. [Google Scholar] [CrossRef]
- McDaniel, K.; Wu, N.; Zhou, T.; Huang, L.; Sato, K.; Venter, J.; Ceci, L.; Chen, D.; Ramos-Lorenzo, S.; Invernizzi, P.; et al. Amelioration of Ductular Reaction by Stem Cell Derived Extracellular Vesicles in MDR2 Knockout Mice via Lethal-7 microRNA. Hepatology 2019, 69, 2562–2578. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Pinatel, E.M.; Leszczynska, A.; Goyal, N.P.; Nishio, T.; Kim, J.; Kneiber, D.; de Araujo Horcel, L.; Eguchi, A.; Ordonez, P.M.; et al. Human induced pluripotent stem cell-derived extracellular vesicles reduce hepatic stellate cell activation and liver fibrosis. JCI Insight 2019, 5, e125652. [Google Scholar] [CrossRef] [PubMed]
- Mardpour, S.; Hassani, S.N.; Mardpour, S.; Sayahpour, F.; Vosough, M.; Ai, J.; Aghdami, N.; Hamidieh, A.A.; Baharvand, H. Extracellular vesicles derived from human embryonic stem cell-MSCs ameliorate cirrhosis in thioacetamide-induced chronic liver injury. J. Cell Physiol. 2018, 233, 9330–9344. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Brigstock, D.R. Pathways of production and delivery of hepatocyte exosomes. J. Cell Commun. Signal. 2018, 12, 343–357. [Google Scholar] [CrossRef]
- Li, X.; Chen, R.; Kemper, S.; Brigstock, D.R. Extracellular Vesicles from Hepatocytes Are Therapeutic for Toxin-Mediated Fibrosis and Gene Expression in the Liver. Front. Cell Dev. Biol. 2019, 7, 368. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Li, J.; Chen, Y.; Xu, Y.; Wei, Y.; Lu, J.; Dong, R. Hepatic stellate cell reprogramming via exosome-mediated CRISPR/dCas9-VP64 delivery. Drug Deliv. 2021, 28, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Velazquez, V.M.; Brigstock, D.R. Fibrogenic Signaling Is Suppressed in Hepatic Stellate Cells through Targeting of Connective Tissue Growth Factor (CCN2) by Cellular or Exosomal MicroRNA-199a-5p. Am. J. Pathol. 2016, 186, 2921–2933. [Google Scholar] [CrossRef] [PubMed]
- Kornek, M.; Popov, Y.; Libermann, T.A.; Afdhal, N.H.; Schuppan, D. Human T cell microparticles circulate in blood of hepatitis patients and induce fibrolytic activation of hepatic stellate cells. Hepatology 2011, 53, 230–242. [Google Scholar] [CrossRef]
- Hou, X.; Yin, S.; Ren, R.; Liu, S.; Yong, L.; Liu, Y.; Li, Y.; Zheng, M.H.; Kunos, G.; Gao, B.; et al. Myeloid cell-specific IL-6 signaling promotes miR-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology 2020. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Quan, J. Exosomes derived from natural killer cells inhibit hepatic stellate cell activation and liver fibrosis. Hum. Cell 2020, 33, 582–589. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Quan, J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol. Med. 2020, 26, 81. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Cong, M.; You, H.; Brigstock, D.R. Therapeutic effects of serum extracellular vesicles in liver fibrosis. J. Extracell. Vesicles 2018, 7, 1461505. [Google Scholar] [CrossRef]
- Welsh, J.A.; Scorletti, E.; Clough, G.F.; Englyst, N.A.; Byrne, C.D. Leukocyte extracellular vesicle concentration is inversely associated with liver fibrosis severity in NAFLD. J. Leukoc. Biol. 2018, 104, 631–639. [Google Scholar] [CrossRef]
- Murakami, Y.; Toyoda, H.; Tanahashi, T.; Tanaka, J.; Kumada, T.; Yoshioka, Y.; Kosaka, N.; Ochiya, T.; Taguchi, Y.H. Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS ONE 2012, 7, e48366. [Google Scholar] [CrossRef]
- Lambrecht, J.; Jan Poortmans, P.; Verhulst, S.; Reynaert, H.; Mannaerts, I.; van Grunsven, L.A. Circulating ECV-Associated miRNAs as Potential Clinical Biomarkers in Early Stage HBV and HCV Induced Liver Fibrosis. Front. Pharmacol. 2017, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Aizawa, N.; Enomoto, H.; Nishiguchi, S.; Toyoda, H.; Kumada, T.; Iio, E.; Ito, K.; Ogawa, S.; Isogawa, M.; et al. Circulating let-7 Levels in Serum Correlate with the Severity of Hepatic Fibrosis in Chronic Hepatitis C. Open Forum Infect. Dis. 2018, 5, ofy268. [Google Scholar] [CrossRef]
- Broermann, A.; Schmid, R.; Gabrielyan, O.; Sakowski, M.; Eisele, C.; Keller, S.; Wolff, M.; Baum, P.; Stierstorfer, B.; Huber, J.; et al. Exosomal miRNAs as Potential Biomarkers to Monitor Phosphodiesterase 5 Inhibitor Induced Anti-Fibrotic Effects on CCl4 Treated Rats. Int. J. Mol. Sci. 2020, 22, 382. [Google Scholar] [CrossRef]
- Cai, P.; Mu, Y.; Olveda, R.M.; Ross, A.G.; Olveda, D.U.; McManus, D.P. Serum Exosomal miRNAs for Grading Hepatic Fibrosis Due to Schistosomiasis. Int. J. Mol. Sci. 2020, 21, 3560. [Google Scholar] [CrossRef]
- Lunardi, S.; Muschel, R.J.; Brunner, T.B. The stromal compartments in pancreatic cancer: Are there any therapeutic targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.; Afghani, E.; Pandol, S. Chronic Pancreatitis: Current Status and Challenges for Prevention and Treatment. Dig. Dis. Sci. 2017, 62, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A Rising Star in Pancreatic Diseases: Pancreatic Stellate Cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Korc, M. Pancreatic cancer-associated stroma production. Am. J. Surg. 2007, 194, S84–S86. [Google Scholar] [CrossRef]
- Farran, B.; Nagaraju, G.P. The dynamic interactions between the stroma, pancreatic stellate cells and pancreatic tumor development: Novel therapeutic targets. Cytokine Growth Factor Rev. 2019, 48, 11–23. [Google Scholar] [CrossRef]
- Ali, S.; Suresh, R.; Banerjee, S.; Bao, B.; Xu, Z.; Wilson, J.; Philip, P.A.; Apte, M.; Sarkar, F.H. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am. J. Cancer Res. 2015, 5, 1251–1264. [Google Scholar]
- Takikawa, T.; Masamune, A.; Yoshida, N.; Hamada, S.; Kogure, T.; Shimosegawa, T. Exosomes Derived from Pancreatic Stellate Cells: MicroRNA Signature and Effects on Pancreatic Cancer Cells. Pancreas 2017, 46, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, H.; Wang, Q.; Chen, K.; Marko, K.; Tian, X.; Yang, Y. Pancreatic stellate cells derived exosomal miR-5703 promotes pancreatic cancer by downregulating CMTM4 and activating PI3K/Akt pathway. Cancer Lett. 2020, 490, 20–30. [Google Scholar] [CrossRef]
- Ma, Q.; Wu, H.; Xiao, Y.; Liang, Z.; Liu, T. Upregulation of exosomal microRNA21 in pancreatic stellate cells promotes pancreatic cancer cell migration and enhances Ras/ERK pathway activity. Int. J. Oncol. 2020, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Zhou, Y.Z.; Zhang, B.; Huang, S.F.; Li, P.P.; He, X.M.; Cao, G.D.; Kang, M.X.; Dong, X.; Wu, Y.L. Pancreatic cancer-derived exosomes promoted pancreatic stellate cells recruitment by pancreatic cancer. J. Cancer 2019, 10, 4397–4407. [Google Scholar] [CrossRef]
- Masamune, A.; Yoshida, N.; Hamada, S.; Takikawa, T.; Nabeshima, T.; Shimosegawa, T. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2018, 495, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef]
- Lingzhi, Z.; Meirong, L.; Xiaobing, F. Biological approaches for hypertrophic scars. Int. Wound J. 2020, 17, 405–418. [Google Scholar] [CrossRef]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol. Surg. 2017, 43 (Suppl. 1), S3–S18. [Google Scholar] [CrossRef]
- Mari, W.; Alsabri, S.G.; Tabal, N.; Younes, S.; Sherif, A.; Simman, R. Novel Insights on Understanding of Keloid Scar: Article Review. J. Am. Coll. Clin. Wound Spec. 2015, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Zeiser, R.; Blazar, B.R. Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N. Engl. J. Med. 2017, 377, 2565–2579. [Google Scholar] [CrossRef]
- Iversen, L.V.; Ullman, S.; Ostergaard, O.; Nielsen, C.T.; Halberg, P.; Karlsmark, T.; Heegaard, N.H.; Jacobsen, S. Cross-sectional study of soluble selectins, fractions of circulating microparticles and their relationship to lung and skin involvement in systemic sclerosis. BMC Musculoskelet. Disord. 2015, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Jinnin, M.; Harada, M.; Kudo, H.; Nakayama, W.; Inoue, K.; Ogata, A.; Kajihara, I.; Fukushima, S.; Ihn, H. Altered expression of CD63 and exosomes in scleroderma dermal fibroblasts. J. Dermatol. Sci. 2016, 84, 30–39. [Google Scholar] [CrossRef]
- Li, L.; Zuo, X.; Xiao, Y.; Liu, D.; Luo, H.; Zhu, H. Neutrophil-derived exosome from systemic sclerosis inhibits the proliferation and migration of endothelial cells. Biochem. Biophys. Res. Commun. 2020, 526, 334–340. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Piera-Velazquez, S.; Jimenez, S.A. Exosomes isolated from serum of systemic sclerosis patients display alterations in their content of profibrotic and antifibrotic microRNA and induce a profibrotic phenotype in cultured normal dermal fibroblasts. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 21–30. [Google Scholar]
- Li, L.; Zuo, X.; Liu, D.; Luo, H.; Zhu, H. The profiles of miRNAs and lncRNAs in peripheral blood neutrophils exosomes of diffuse cutaneous systemic sclerosis. J. Dermatol. Sci. 2020, 98, 88–97. [Google Scholar] [CrossRef]
- Moulin, V.J.; Mayrand, D.; Messier, H.; Martinez, M.C.; Lopez-Valle, C.A.; Genest, H. Shedding of microparticles by myofibroblasts as mediator of cellular cross-talk during normal wound healing. J. Cell. Physiol. 2010, 225, 734–740. [Google Scholar] [CrossRef]
- Lai, P.; Chen, X.; Guo, L.; Wang, Y.; Liu, X.; Liu, Y.; Zhou, T.; Huang, T.; Geng, S.; Luo, C.; et al. A potent immunomodulatory role of exosomes derived from mesenchymal stromal cells in preventing cGVHD. J. Hematol. Oncol. 2018, 11, 135. [Google Scholar] [CrossRef]
- Fang, S.; Xu, C.; Zhang, Y.; Xue, C.; Yang, C.; Bi, H.; Qian, X.; Wu, M.; Ji, K.; Zhao, Y.; et al. Umbilical Cord-Derived Mesenchymal Stem Cell-Derived Exosomal MicroRNAs Suppress Myofibroblast Differentiation by Inhibiting the Transforming Growth Factor-beta/SMAD2 Pathway During Wound Healing. Stem Cells Transl. Med. 2016, 5, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lai, P.; Wang, Y.; Huang, T.; Chen, X.; Geng, S.; Huang, X.; Luo, C.; Wu, S.; Ling, W.; et al. Extracellular vesicles derived from mesenchymal stem cells prevent skin fibrosis in the cGVHD mouse model by suppressing the activation of macrophages and B cells immune response. Int. Immunopharmacol. 2020, 84, 106541. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, Y.; Han, S.; Zhang, W.; Zhou, Q.; Guan, H.; Liu, J.; Shi, J.; Su, L.; Hu, D. Exosomes derived from human amniotic epithelial cells accelerate wound healing and inhibit scar formation. J. Mol. Histol. 2017, 48, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Kholia, S.; Deregibus, M.C.; Camussi, G. The Role of Extracellular Vesicles as Paracrine Effectors in Stem Cell-Based Therapies. Adv. Exp. Med. Biol. 2019, 1201, 175–193. [Google Scholar] [CrossRef]
- Shao, L.; Zhang, Y.; Lan, B.; Wang, J.; Zhang, Z.; Zhang, L.; Xiao, P.; Meng, Q.; Geng, Y.J.; Yu, X.Y.; et al. MiRNA-Sequence Indicates That Mesenchymal Stem Cells and Exosomes Have Similar Mechanism to Enhance Cardiac Repair. Biomed. Res. Int. 2017, 2017, 4150705. [Google Scholar] [CrossRef]
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Tan, J.; Miao, Y.; Lv, Y.; Zhang, Q. Crosstalk between exosomes and autophagy: A review of molecular mechanisms and therapies. J. Cell. Mol. Med. 2021, 25, 2297–2308. [Google Scholar] [CrossRef]
- Ye, H.L.; Zhang, J.W.; Chen, X.Z.; Wu, P.B.; Chen, L.; Zhang, G. Ursodeoxycholic acid alleviates experimental liver fibrosis involving inhibition of autophagy. Life Sci. 2020, 242, 117175. [Google Scholar] [CrossRef]
- Toh, W.S.; Lai, R.C.; Zhang, B.; Lim, S.K. MSC exosome works through a protein-based mechanism of action. Biochem. Soc. Trans. 2018, 46, 843–853. [Google Scholar] [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed]
- Gho, Y.S.; Lee, C. Emergent properties of extracellular vesicles: A holistic approach to decode the complexity of intercellular communication networks. Mol. Biosyst. 2017, 13, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; De Giorgi, V.; Schechterly, C.; Wang, R.Y.; Farci, P.; Tanaka, Y.; Alter, H.J. Circulating let-7 levels in plasma and extracellular vesicles correlate with hepatic fibrosis progression in chronic hepatitis C. Hepatology 2016, 64, 732–745. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
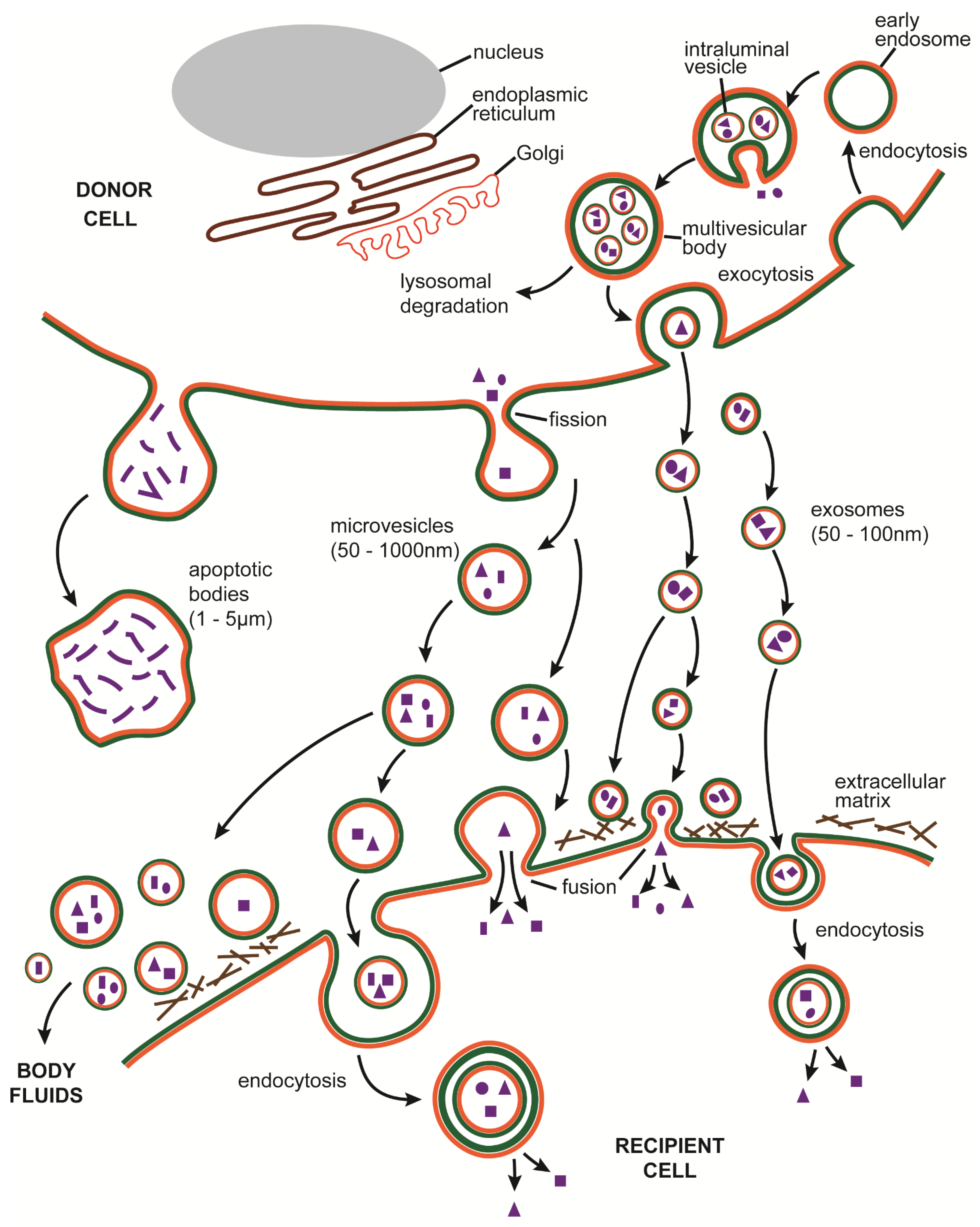{kind=link}
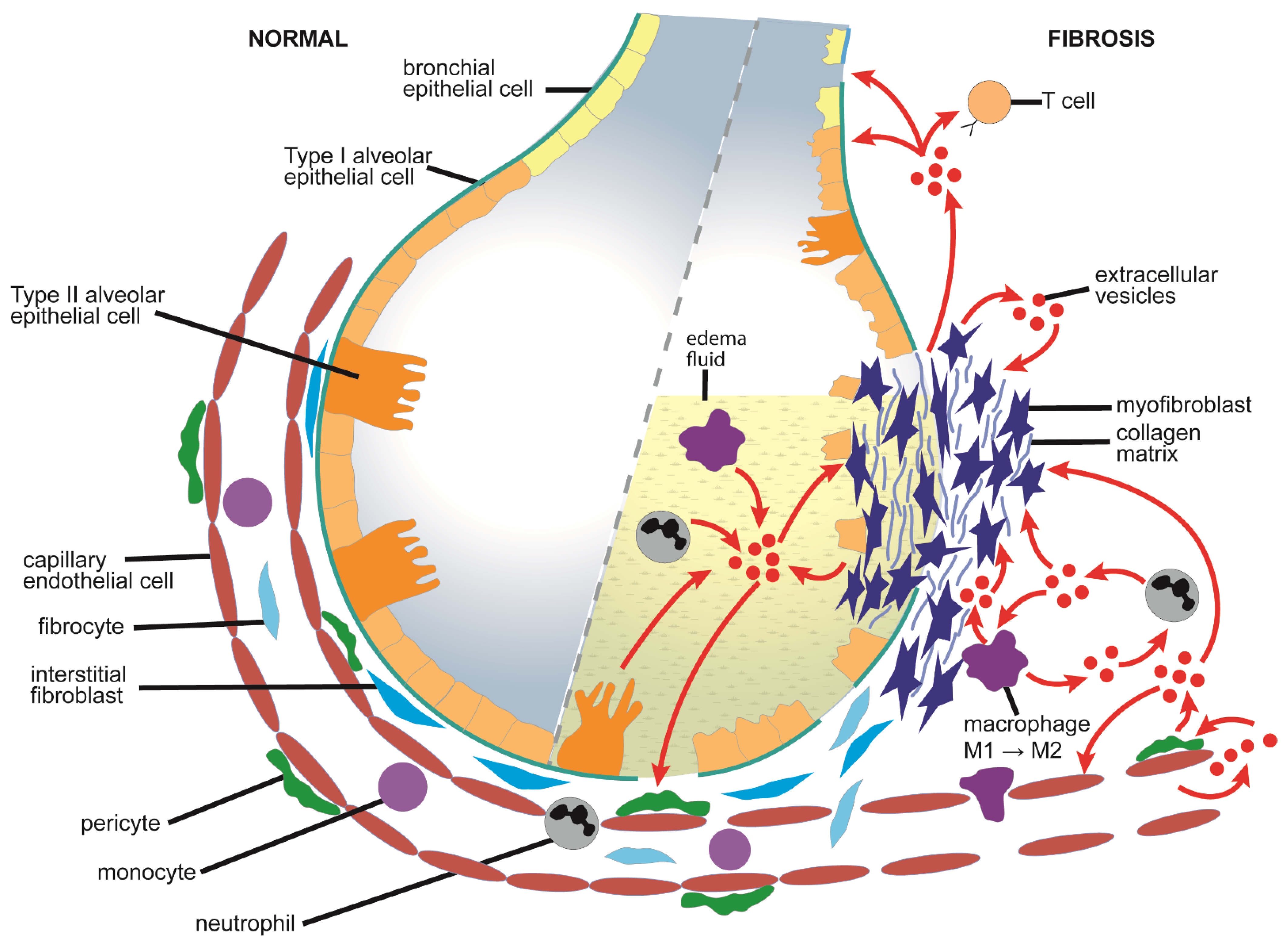{kind=link}
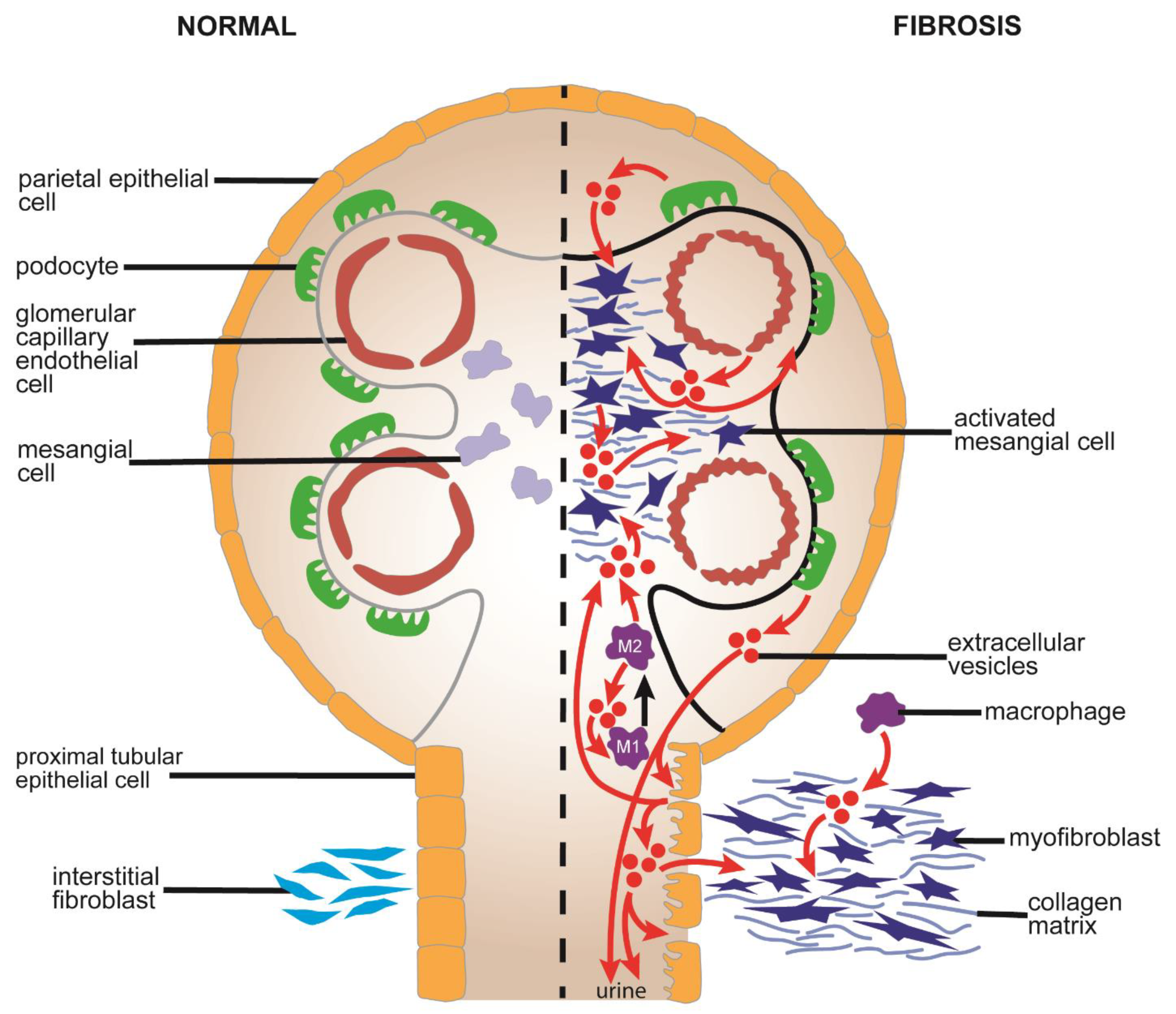{kind=link}
{kind=link}
{kind=link}
| EV-Targeted Signaling Component | Lung | Kidney | Heart | Liver | Pancreas | Skin | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fibrosis | Therapy | Fibrosis | Therapy | Fibrosis | Therapy | Fibrosis | Therapy | Fibrosis | Fibrosis | |
| Wnt/β-catenin | Martin-Medina et al., 2018 [66] | Wu et al., 2017 [118] | Ibrahim et al., 2019 [210] | Rong et al., 2019 [273] | Li et al., 2020 [329] | |||||
| Xie et al., 2020 [67] | Dzialo et al., 2019 [177] | |||||||||
| Parimon et al., 2019 [68] | Cai et al., 2020 [179] | |||||||||
| Zhang et al., 2020 [82] | ||||||||||
| SIRT-1/-3 | Kadota et al., 2020 [70] | Chen et al., 2020 [108] | ||||||||
| PDL1 | Kang et al., 2020 [71] | |||||||||
| Notch | Kadota et al., 2020 [70] | He et al., 2020 [131] | Xuan et al., 2020 [204] | Li et al., 2020 [329] | ||||||
| Wang et al., 2021 [78] | ||||||||||
| YAP/TAZ | Wang et al., 2021 [78] | Ji et al., 2020 [137] | Hou et al., 2020 [296] | |||||||
| PTEN | Zhou et al., 2013 [110] | Xia et al., 2018 [173] | Sun et al., 2020 [187] | |||||||
| Kang et al., 2019 [230] | ||||||||||
| AKT | Zhou et al., 2013 [110] | Xia et al., 2018 [173] | Shao et al., 2020 [184] | Wang et al., 2015 [269] | Li et al., 2020 [314] | |||||
| Sun et al., 2020 [187] | ||||||||||
| PPAR | Liu et al., 2020 [112] | Povero et al., 2015 [246] | Rong et al., 2019 [273] | |||||||
| HIF | Wang et al., 2021 [78] | Liu et al., 2020 [112] | Zou et al., 2016 [139] | Sun et al., 2020 [186] | Wan et al., 2019 [264] | |||||
| Dyrk2 | Yang et al., 2018 [170] | |||||||||
| Nrf2 | Tian et al., 2018 [176] | |||||||||
| Neat 1 | Kenneweg et al., 2019 [171] | |||||||||
| HuR | Govindappa et al., 2020 [180] | |||||||||
| Foxo3a | Wang et al., 2020 [182] | |||||||||
| HSP70,72 | Yang et al., 2019 [183] | Feng et al., 2014 [203] | Alhomrani et al., 2017 [283] | |||||||
| Ohara et al., 2018 [284] | ||||||||||
| NF-κB | Xiao et al., 2020 [84] | Cannito et al., 2017 [247] | ||||||||
| Khodayari et al., 2020 [271] | ||||||||||
| NLRP3 | Sun et al., 2019 [91] | Cannito et al., 2017 [247] | ||||||||
| ROCK | Shi et al., 2020 [128] | Xuan et al., 2019 [220] | Hirsova et al., 2016 [244] | |||||||
| Lee et al., 2017 [245] | ||||||||||
| Povero et al., 2015 [246] | ||||||||||
| TLR | Eguchi et al., 2020 [250] | |||||||||
| JAK/STAT | Devhare et al., 2017 [255] Khodayari et al., 2020 [271] | Qu et al., 2017 [277] | ||||||||
| KLF4 | Chen et al., 2020 [270] | |||||||||
| CXCR/CXCL | Khodayari et al., 2020 [271] | Du et al., 2020 [276] | Takikawa et al., 2017 [313] | |||||||
| KIT | Kim et al., 2018 [272] | |||||||||
| SNAIL | Zhang et al., 2020 [138] | |||||||||
| Bcl2 | Zhao et al. (2015) [195] | Qu et al., 2017 [277] | ||||||||
| MAPK/ERK | Liu et al., 2020 [136] | Yuan et al., 2018 [225] | Korneck et al., 2011 [295] | Ma et al., 2020 [315] | ||||||
| EV Source | Organ Targeted | Fibrosis Model | Principal EV Effects | EV Features Associated with Therapy | Reference |
|---|---|---|---|---|---|
| Bone marrow MSC | lung | bleomycin |
| Targeting of FZD6 by miR-29b-3p | [79] |
| [83] | ||||
| hyperoxia-induced BPD |
| [80] | |||
| LPS-induced acute lung injury |
| Inactivation of NFκB and Hh pathways by EV miR-182-5p and miR-23a-3p | [84] | ||
| kidney | Stz DN |
| [125] | ||
| [126] | ||||
| EV miRs predicted to target TGF-β, EGFR, PDGFR, ARF-6, mTOR, VEGF, p53-apoptosis, ZTM, TNF pathways | [127] | |||
| AA CKD |
| [143] | |||
| UUO |
| EVs were experimentally augmented with miR-Let7c | [130] | ||
| Inhibition of Rho/ROCK by EV MFG-E8 | [128] | |||
| heart | MI |
| EVs were from miR-22-transfected MSC since ischemic conditioning of MSC led to elevated miR-22 in EVs. | [191] | |
| EVs were from ischemic conditioned MSC and contained elevated miR-210. Cardioprotective effects were associated with miR-210 | [192] | |||
| EVs were experimentally augmented with Lamp2b-IMPT to target ischemic myocardium | [188] | |||
| EV therapy enhanced by enrichment with miR-101 which blocks autophagy | [185] | |||
| EV treatment of CSC before CSC therapy. In CSC, EVs caused increased expression of genes associated with angiogenesis, heart development, cell proliferation, migration and differentiation | [193] | |||
| EVs were experimentally augmented with HIF-1α | [186] | |||
| EV miR-221-3p inhibited PTEN leading to stimulation of AKT and suppression of apoptosis | [187] | |||
| Pressure-overload |
| EV-mediated senescence in cardiac myofibroblasts | [190] | ||
| liver | CCl4 |
| [273] | ||
| skin | cGVDH |
| [331] | ||
| Adipose tissue MSC | lung | Particulate inhalation |
| Suppression of TGF-βR1 by EV let-7d-5p | [87] |
| kidney | Metabolic syndrome + renal artery stenosis |
| EV IL-10 mRNA expressed 100x higher than in producer cells. EVs non-therapeutic upon knockdown of IL-10 in MSC. | [132] | |
| EVs from GDNF-transfected AD-MSCs; enhanced SIRT-1 and eNOS in endothelial cells | [108] | |||
| EV TGF-β associated with anti-inflammation; EV mitochondrial-regulating miRs associated with reduced oxidative stress | [133] | |||
| UUO |
| EV delivery of casein kinase 1δ and E3 ubiquitin ligase β-TRCP, targeting YAP and collagen | [136,137] | ||
| kidney & heart | DOCA-salt hypertension |
| [135] | ||
| heart | MI |
| [199] | ||
| EVs were experimentally augmented with miR-126 | [200] | |||
| Evs were experimentally augmented with miR-146a | [201] | |||
| EVs administered in free form or encapsulated within peptide hydrogel | [189] | |||
| As compared to producer MSC, EVs had increased miR-29 or -24 and decreased miR-15, -21, -34, -130, or -378 | [336] | |||
| liver | CCl4 |
| EVs were experimentally augmented with miR-122 | [275] | |
| EVs were experimentally augmented with miR-181-5p | [277] | |||
| EVs were experimentally augmented with miR-150-5p which targeted CXCL1 | [276] | |||
| EVs were experimentally augmented with mmu_circ_0000623 which targeted miR-125/ATG4D-mediated autophagy | [278] | |||
| Tonsil-derived MSC | liver | CCl4 |
| EVs enriched in miR-486.5p which suppressed Hh signaling in HSC by targeting the Smo Hh receptor | [281] |
| Liver stem cells | kidney | AA CKD |
| [142] | |
| liver | NASH |
| [285] | ||
| CCl4 |
| EV effects were substantially improved by co-administration of Nilotinib | [286] | ||
| MDR2-/- primary sclerosing cholangitis |
| EVs enriched in let-7 | [287] | ||
| Amnion epithelial cells | lung | bleomycin |
| EV enriched with fibrosis-related miRs (PI3-AKT, MAPK, Ras, Hippo, TGF-β, FAK) and proteins associated with apoptosis, developmental growth, MAPK, inflammation, EGF, PDGF, FGF. | [89] |
| Bleomycin or OVA/NA |
| Some EV effects augmented by serelaxin | [88] | ||
| liver | CCl4 fibrosis |
| EVs contain MFGE8, HSP72, SOD1 which suppress TGF-β signaling | [283] | |
| NASH |
| [284] | |||
| skin | Full thickness wounds |
| [334] | ||
| Lung spheroid cells | lung | Silicableomycin |
| EVs enriched for miR-30a, let-7, miR-99 | [90] |
| Cardiosphere-derived cells CDC | kidney & heart | Ang II |
| Therapeutic effects mimicked by EV-FF1, the most abundant small RNA constituent in CDC EVs | [145] |
| heart | MI |
| EVs enriched in miR-146a-5p, 302b-3p, 181b-5p, 155-5p | [207] | |
| [208] | ||||
| Pediatric dilated cardiomyopathy |
| Antifibrotic effects associated with suppression of TRAF6/Smad4/FOS by EV miR-146a-5p | [209] | ||
| Doxorubicin |
| Therapy improved by conjugation of cardiac homing peptide to EV | [209,211,212] | ||
| Cardiac MSC (Sca-1+) | heart | MI |
| EVs enriched in proteins associated with ECM, adhesion, collagens, integrins. EVs from Notch1-overexpressing C-MSC were tested therapeutically | [204] |
| Cardiac resident progenitor cells | heart | Doxorubicin/trastuzumab |
| EVs were purified from CRPCs derived from outgrowths of primary atrial tissue explants. EVs enriched in redox-regulating proteins and miR-146-5p | [205] |
| MI |
|
W8B2+ CRPCs were obtained from outgrowth of primary atrial tissue explants. EVs contained proteins associated with inflammation, immunoregulation, tissue remodelling, fibrosis. | [206] | ||
| Umbilical cord-derived MSC | lung | Hyperoxia-induced BPD |
| EVs isolated from Wharton’s jelly | [81] |
| Monocrotaline |
| Wnt5a/BMP regulated by Wharton’s jelly EVs | [82] | ||
| silicosis |
| [85] | |||
| kidney | I/R AKI |
| VEGF delivered directly by EVs and insensitive to RNase. All other effects abrogated by RNase treatment of EVs | [139] | |
| Delivery of EV oct-4 | [138] | |||
| heart | MI |
| EVs regulate Bcl-2 expression | [195] | |
| EVs encapsulated in functional peptide hydrogel | [196] | |||
| EVs more effective when purified from cells lacking human leuckocyte antigen light chain β2-microglobulin (B2M) to avoid T cell immunity EV enriched in miR-24 | [184] | |||
| Targeting of apoptotic peptidase activating factor by EV miR-136 | [197] | |||
| Type 2 diabetes stroke |
| [198] | |||
| Metabolic syndrome + renal artery stenosis |
| [202] | |||
| liver | CCl4 |
| [280] | ||
| [279] | ||||
| skin | Full thickness wound |
| EVs enriched in miR-21, 23a, 100, 125b, 145 | [332] | |
| cGVHD |
| [333] | |||
| Placental MSC | lung | radiation |
| EVs enriched in miR-214-3p which targeted ATM/p53/p21 | [86] |
| kidney | UUO |
| [144] | ||
| Menstrual blood-derived stem cells | lung | bleomycin |
| Targeting of LOX1 and NLRP3 apoptosis by EV let-7 | [91] |
| Embryonic stem cells | heart | MI |
| EVs enriched in miR290-295 cluster | [221] |
| EVs purified from human embryonic stem cell-derived CPCs | [222] | |||
| [223] | ||||
| liver | TAA |
| [289] | ||
| Telocytes | heart | MI |
| [217] | |
| Induced pluripotent stem cells (iPSC) | heart | MI |
| EVs from iPSC-derived cardiomyocytes | [219] |
| EVs from CPCs induced from iPSCs with ISX-9. EVs enriched in miR-373/-520 family members | [220] | |||
| liver | CCL4, BDL |
| EVs from iPSC that were reprogrammed from primary fibroblasts EVs enriched in anti-fibrotic miRs (miR-92a-5p, -10b-5p, -302-3p, -92b-3p) | [288] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brigstock, D.R. Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics. Cells 2021, 10, 1596. https://doi.org/10.3390/cells10071596
Brigstock DR. Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics. Cells. 2021; 10(7):1596. https://doi.org/10.3390/cells10071596
Chicago/Turabian StyleBrigstock, David R. 2021. "Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics" Cells 10, no. 7: 1596. https://doi.org/10.3390/cells10071596
APA StyleBrigstock, D. R. (2021). Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics. Cells, 10(7), 1596. https://doi.org/10.3390/cells10071596