
Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability

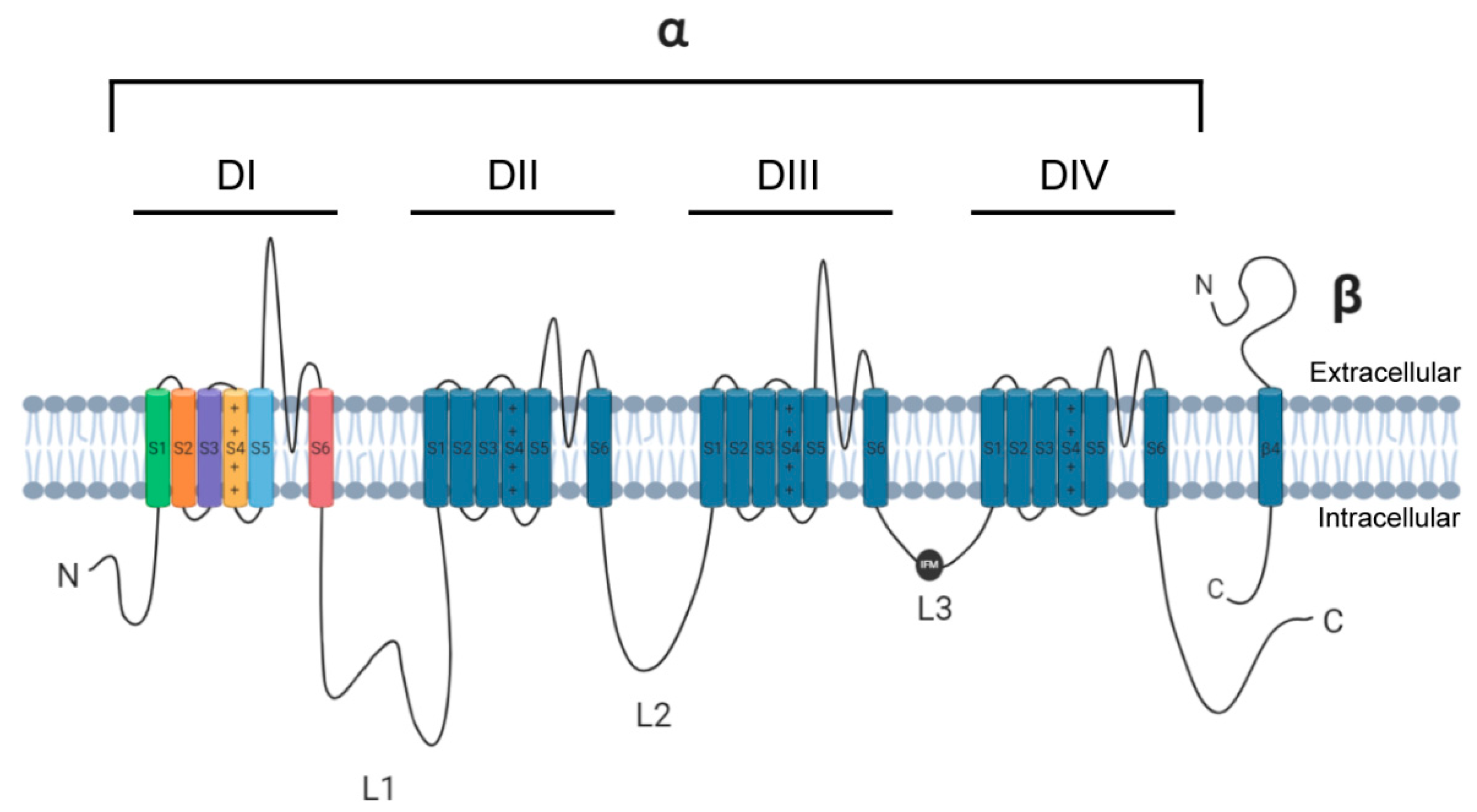{kind=link}
{kind=link}
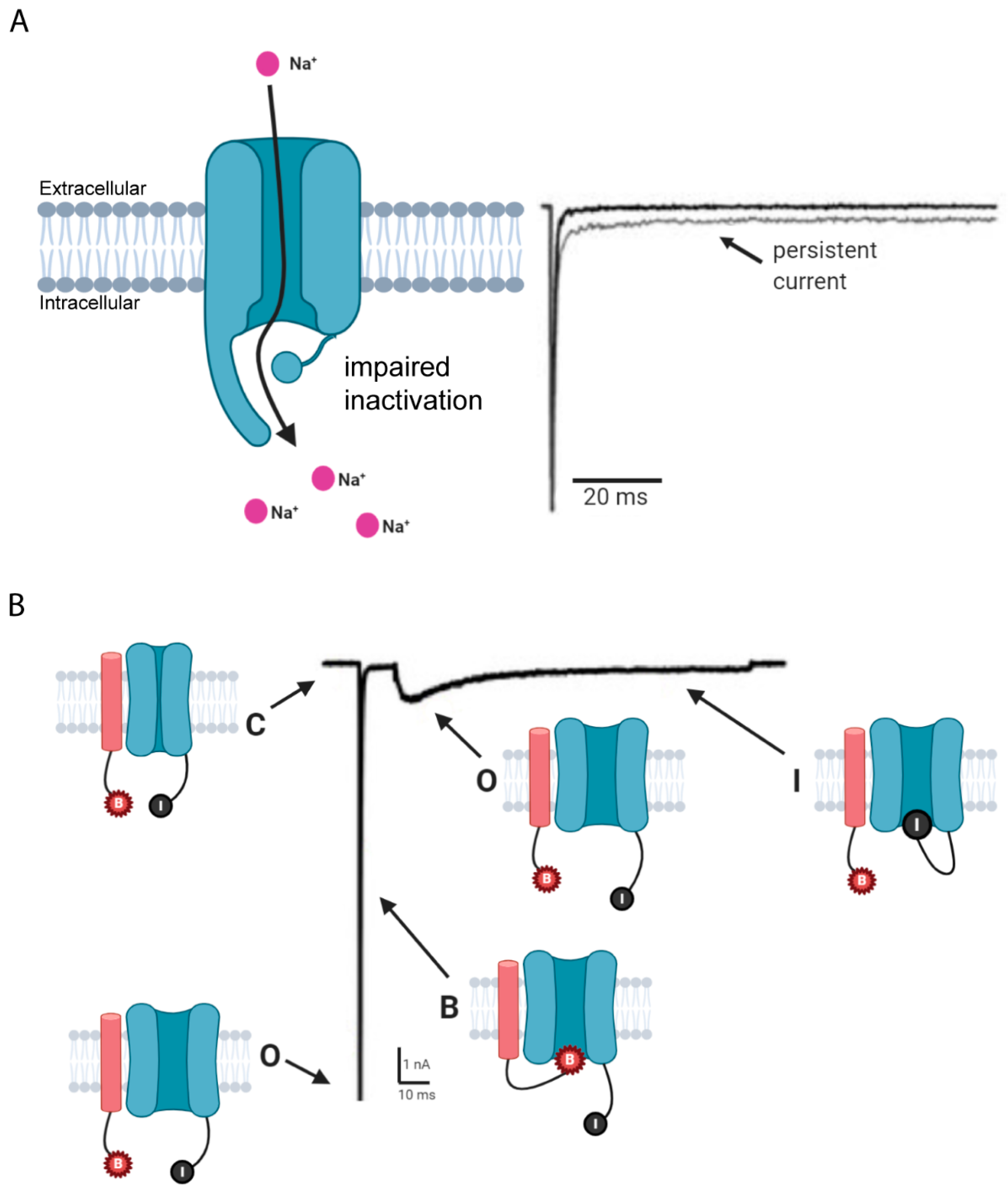{kind=link}
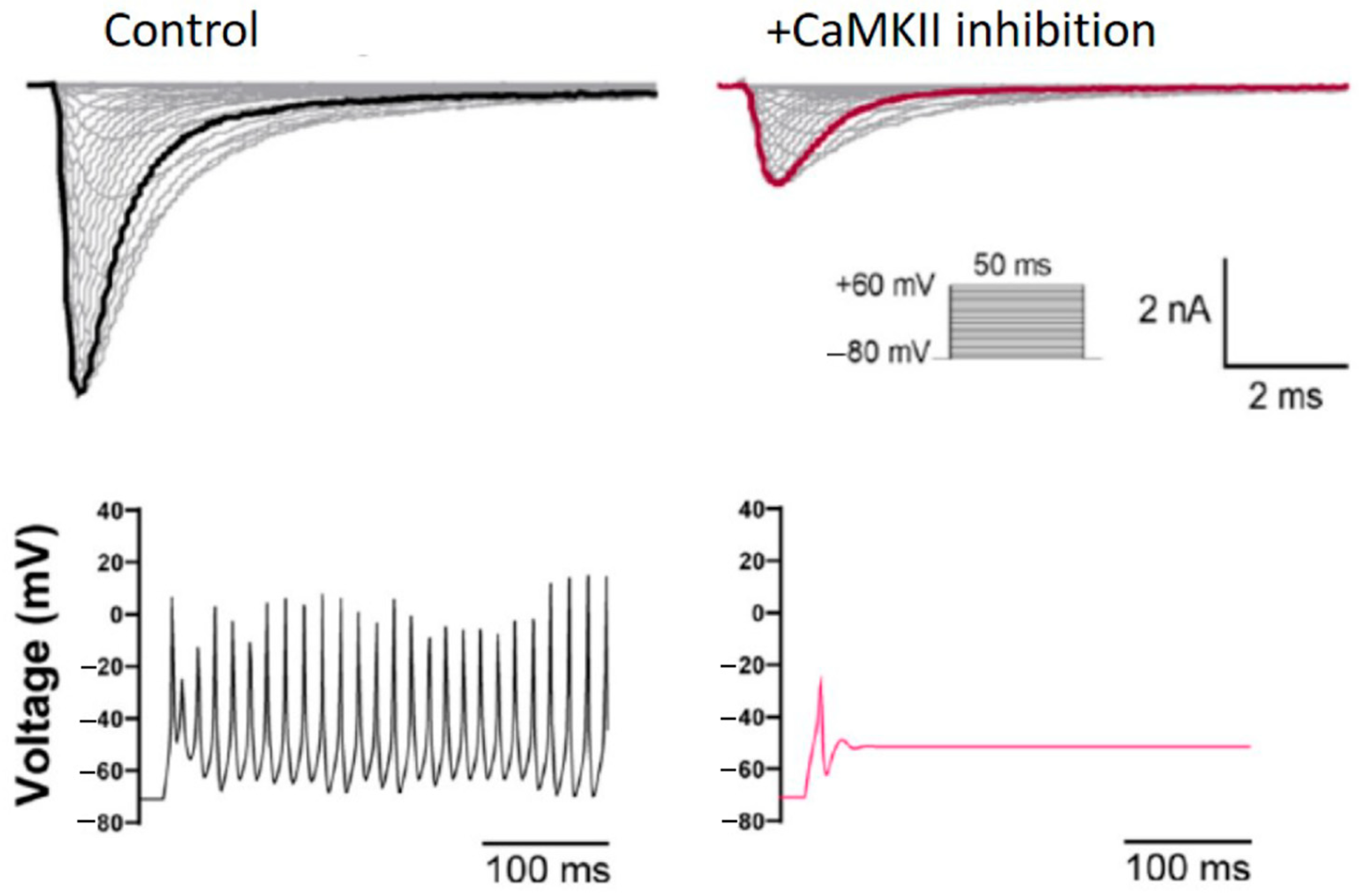{kind=link}
{kind=link}
Abstract
1. Introduction
2. Nav1.6 Overview
2.1. Discovery of Nav1.6
2.2. Nav1.6 Expression and Distribution
2.3. Nav1.6 Subcellular Localization in Neurons
2.4. Unique Biophysical Properties
2.5. Pathophysiology
3. Nav1.6 Regulation by Protein-Protein Interactions
3.1. Sodium Channel β Subunits
3.2. Fibroblast Growth Factor Homologous Factors
3.3. Ca2+ and Calmodulin
4. Post-Translational Regulation of Nav1.6
4.1. Glycosylation
4.2. Uniquitination
4.3. Palmitoylation
4.4. Phosphorylation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef]
- Noda, M.; Ikeda, T.; Suzuki, H.; Takeshima, H.; Takahashi, T.; Kuno, M.; Numa, S. Expression of functional sodium channels from cloned cDNA. Nature 1986, 322, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Shimizu, S.; Tanabe, T.; Takai, T.; Kayano, T.; Ikeda, T.; Takahashi, H.; Nakayama, H.; Kanaoka, Y.; Minamino, N.; et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 1984, 312, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Stühmer, W.; Conti, F.; Suzuki, H.; Wang, X.D.; Noda, M.; Yahagi, N.; Kubo, H.; Numa, S. Structural parts involved in activation and inactivation of the sodium channel. Nature 1989, 339, 597–603. [Google Scholar] [CrossRef]
- Noda, M.; Suzuki, H.; Numa, S.; Stühmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989, 259, 213–216. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Bezanilla, F.; Rojas, E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J. Gen. Physiol. 1973, 62, 375–391. [Google Scholar] [CrossRef]
- Vassilev, P.M.; Scheuer, T.; Catterall, W.A. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 1988, 241, 1658–1661. [Google Scholar] [CrossRef]
- Vassilev, P.; Scheuer, T.; Catterall, W.A. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc. Nat. Acad. Sci. USA 1989, 86, 8147–8151. [Google Scholar] [CrossRef]
- West, J.W.; Patton, D.E.; Scheuer, T.; Wang, Y.; Goldin, A.L.; Catterall, W.A. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Nat. Acad. Sci. USA 1992, 89, 10910–10914. [Google Scholar] [CrossRef]
- Zybura, A.S.; Baucum, A.J., 2nd; Rush, A.M.; Cummins, T.R.; Hudmon, A. CaMKII enhances voltage-gated sodium channel Nav1.6 activity and neuronal excitability. J. Biol. Chem. 2020, 295, 11845–11865. [Google Scholar] [CrossRef] [PubMed]
- Solé, L.; Tamkun, M.M. Trafficking mechanisms underlying Na(v) channel subcellular localization in neurons. Channels 2020, 14, 1–17. [Google Scholar] [CrossRef]
- Scheuer, T. Regulation of sodium channel activity by phosphorylation. Sem. Cell Dev. Biol. 2011, 22, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Forty Years of Sodium Channels: Structure, Function, Pharmacology, and Epilepsy. Neurochem. Res. 2017, 42, 2495–2504. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397. [Google Scholar] [CrossRef]
- Berendt, F.J.; Park, K.S.; Trimmer, J.S. Multisite phosphorylation of voltage-gated sodium channel alpha subunits from rat brain. J. Proteome Res. 2010, 9, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Cerda, O.; Baek, J.H.; Trimmer, J.S. Mining recent brain proteomic databases for ion channel phosphosite nuggets. J. Gen. Physiol. 2011, 137, 3–16. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [PubMed]
- Rossie, S.; Catterall, W.A. Phosphorylation of the alpha subunit of rat brain sodium channels by cAMP-dependent protein kinase at a new site containing Ser686 and Ser687. J. Biol. Chem. 1989, 264, 14220–14224. [Google Scholar] [CrossRef]
- Schaller, K.L.; Krzemien, D.M.; Yarowsky, P.J.; Krueger, B.K.; Caldwell, J.H. A novel, abundant sodium channel expressed in neurons and glia. J. Neurosci. 1995, 15, 3231–3242. [Google Scholar] [CrossRef]
- Burgess, D.L.; Kohrman, D.C.; Galt, J.; Plummer, N.W.; Jones, J.M.; Spear, B.; Meisler, M.H. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant motor endplate disease. Nat. Genet. 1995, 10, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Plummer, N.W.; Galt, J.; Jones, J.M.; Burgess, D.L.; Sprunger, L.K.; Kohrman, D.C.; Meisler, M.H. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics 1998, 54, 287–296. [Google Scholar] [CrossRef] [PubMed]
- García, K.D.; Sprunger, L.K.; Meisler, M.H.; Beam, K.G. The sodium channel Scn8a is the major contributor to the postnatal developmental increase of sodium current density in spinal motoneurons. J. Neurosci. 1998, 18, 5234–5239. [Google Scholar] [CrossRef]
- Smith, M.R.; Smith, R.D.; Plummer, N.W.; Meisler, M.H.; Goldin, A.L. Functional Analysis of the Mouse Scn8a Sodium Channel. J. Neurosci. 1998, 18, 6093. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.I.; Khaliq, Z.M.; Aman, T.K.; Grieco, T.M.; Kearney, J.A.; Raman, I.M.; Meisler, M.H. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J. Neurophysiol. 2006, 96, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, Z.M.; Gouwens, N.W.; Raman, I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: An experimental and modeling study. J. Neurosci. 2003, 23, 4899–4912. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E.; Meisler, M.H. Sodium channel SCN8A (Nav1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 2013, 4, 213. [Google Scholar] [CrossRef]
- Pappalardo, L.W.; Black, J.A.; Waxman, S.G. Sodium channels in astroglia and microglia. Glia 2016, 64, 1628–1645. [Google Scholar] [CrossRef]
- Rush, A.M.; Dib-Hajj, S.D.; Waxman, S.G. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J. Physiol. 2005, 564, 803–815. [Google Scholar] [CrossRef]
- Liang, R.; Liu, X.; Wei, L.; Wang, W.; Zheng, P.; Yan, X.; Zhao, Y.; Liu, L.; Cao, X. The modulation of the excitability of primary sensory neurons by Ca(2)(+)-CaM-CaMKII pathway. J. Neurosci. 2012, 33, 1083–1093. [Google Scholar] [CrossRef]
- Chen, L.; Huang, J.; Zhao, P.; Persson, A.K.; Dib-Hajj, F.B.; Cheng, X.; Tan, A.; Waxman, S.G.; Dib-Hajj, S.D. Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep. 2018, 8, 3845. [Google Scholar] [CrossRef]
- Noujaim, S.F.; Kaur, K.; Milstein, M.; Jones, J.M.; Furspan, P.; Jiang, D.; Auerbach, D.S.; Herron, T.; Meisler, M.H.; Jalife, J. A null mutation of the neuronal sodium channel NaV1.6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. J. FASEB 2012, 26, 63–72. [Google Scholar] [CrossRef]
- Struckman, H.L.; Baine, S.; Thomas, J.; Mezache, L.; Mykytyn, K.; Györke, S.; Radwański, P.B.; Veeraraghavan, R. Super-Resolution Imaging Using a Novel High-Fidelity Antibody Reveals Close Association of the Neuronal Sodium Channel Na(V)1.6 with Ryanodine Receptors in Cardiac Muscle. Microsc. Microanal. 2020, 26, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lv, Y.; Xu, J.; Mao, X.; Chen, Z.; Lu, W. Over-expression of Nav1.6 channels is associated with lymph node metastases in colorectal cancer. World J. Surg. Oncol. 2019, 17, 175. [Google Scholar] [CrossRef]
- Mao, W.; Zhang, J.; Körner, H.; Jiang, Y.; Ying, S. The Emerging Role of Voltage-Gated Sodium Channels in Tumor Biology. Front. Oncol. 2019, 9, 124. [Google Scholar] [CrossRef]
- Lopez-Charcas, O.; Espinosa, A.M.; Alfaro, A.; Herrera-Carrillo, Z.; Ramirez-Cordero, B.E.; Cortes-Reynosa, P.; Perez Salazar, E.; Berumen, J.; Gomora, J.C. The invasiveness of human cervical cancer associated to the function of Na(V)1.6 channels is mediated by MMP-2 activity. Sci. Rep. 2018, 8, 12995. [Google Scholar] [CrossRef]
- Duménieu, M.; Oulé, M.; Kreutz, M.R.; Lopez-Rojas, J. The Segregated Expression of Voltage-Gated Potassium and Sodium Channels in Neuronal Membranes: Functional Implications and Regulatory Mechanisms. Front. Cell Neurosci. 2017, 11, 115. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites and synapses. Proc. Nat. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.M.; Bennett, V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 2001, 155, 739–746. [Google Scholar] [CrossRef]
- Tzoumaka, E.; Tischler, A.C.; Sangameswaran, L.; Eglen, R.M.; Hunter, J.C.; Novakovic, S.D. Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J. Neurosci. Res. 2000, 60, 37–44. [Google Scholar] [CrossRef]
- Osorio, N.; Alcaraz, G.; Padilla, F.; Couraud, F.; Delmas, P.; Crest, M. Differential targeting and functional specialization of sodium channels in cultured cerebellar granule cells. J. Physiol. 2005, 569, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.; Nusser, Z. Molecular identity of dendritic voltage-gated sodium channels. Science 2010, 328, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tian, C.; Li, T.; Yang, M.; Hou, H.; Shu, Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci. 2009, 12, 996–1002. [Google Scholar] [CrossRef]
- Bender, K.J.; Trussell, L.O. The Physiology of the Axon Initial Segment. Neurosci. Annu. Rev. 2012, 35, 249–265. [Google Scholar] [CrossRef]
- Kole, M.H.; Stuart, G.J. Signal processing in the axon initial segment. Neuron 2012, 73, 235–247. [Google Scholar] [CrossRef]
- Grubb, M.S.; Shu, Y.; Kuba, H.; Rasband, M.N.; Wimmer, V.C.; Bender, K.J. Short- and long-term plasticity at the axon initial segment. J. Neurosci. 2011, 31, 16049–16055. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Rasband, M.N. Axon initial segments: Structure, function, and disease. Ann. N. Y. Acad. Sci. 2018, 1420, 46–61. [Google Scholar] [CrossRef]
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 2018, 38, 2135. [Google Scholar] [CrossRef]
- Royeck, M.; Horstmann, M.T.; Remy, S.; Reitze, M.; Yaari, Y.; Beck, H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J. Neurophysiol. 2008, 100, 2361–2380. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.; Nusser, Z. Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 2008, 28, 14329–14340. [Google Scholar] [CrossRef]
- Kaplan, M.R.; Cho, M.H.; Ullian, E.M.; Isom, L.L.; Levinson, S.R.; Barres, B.A. Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 2001, 30, 105–119. [Google Scholar] [CrossRef]
- Van Wart, A.; Matthews, G. Impaired firing and cell-specific compensation in neurons lacking Nav1.6 sodium channels. J. Neurosci. 2006, 26, 7172–7180. [Google Scholar] [CrossRef]
- Akin, E.J.; Solé, L.; Dib-Hajj, S.D.; Waxman, S.G.; Tamkun, M.M. Preferential targeting of Nav1.6 voltage-gated Na+ Channels to the axon initial segment during development. PLoS ONE 2015, 10, e0124397. [Google Scholar] [CrossRef] [PubMed]
- Kole, M.H.; Stuart, G.J. Is action potential threshold lowest in the axon? Nat. Neurosci. 2008, 11, 1253–1255. [Google Scholar] [CrossRef]
- Stuart, G.J.; Sakmann, B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature 1994, 367, 69–72. [Google Scholar] [CrossRef]
- Hedstrom, K.L.; Ogawa, Y.; Rasband, M.N. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell Biol. 2008, 183, 635–640. [Google Scholar] [CrossRef]
- Garrido, J.J.; Giraud, P.C.E.; Fernandes, F.; Moussif, A.; Fache, M.P.; Debanne, D.; Dargent, B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 2003, 300, 2091–2094. [Google Scholar] [CrossRef]
- Gasser, A.; Ho, T.S.; Cheng, X.; Chang, K.J.; Waxman, S.G.; Rasband, M.N.; Dib-Hajj, S.D. An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J. Neurosci. 2012, 32, 7232–7243. [Google Scholar] [CrossRef]
- Boiko, T.; Rasband, M.N.; Levinson, S.R.; Caldwell, J.H.; Mandel, G.; Trimmer, J.S.; Matthews, G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 2001, 30, 91–104. [Google Scholar] [CrossRef]
- Boiko, T. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J. Neurosci. 2003, 23, 2306–2313. [Google Scholar] [CrossRef]
- Lemaillet, G.; Walker, B.; Lambert, S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 2003, 278, 27333–27339. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.J.; Kleopa, K.A.; Cooper, E.C.; Scherer, S.S. KCNQ2 is a nodal K+ channel. J. Neurosci. 2004, 24, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef]
- Bréchet, A.; Fache, M.P.; Brachet, A.; Ferracci, G.; Baude, A.; Irondelle, M.; Pereira, S.; Leterrier, C.; Dargent, B. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J. Cell Biol. 2008, 183, 1101–1114. [Google Scholar] [CrossRef]
- Hien, Y.E.; Montersino, A.; Castets, F.; Leterrier, C.; Filhol, O.; Vacher, H.; Dargent, B. CK2 accumulation at the axon initial segment depends on sodium channel Nav1. FEBS Lett. 2014, 588, 3403–3408. [Google Scholar] [CrossRef] [PubMed]
- Akin, E.J.; Tamkun, M.M. Another piece to the intracellular FGF/Na+ channel puzzle. Proc. Nat. Aca. Sci. USA 2016, 113, 5147–5149. [Google Scholar] [CrossRef]
- Goaillard, J.-M.; Moubarak, E.; Tapia, M.; Tell, F. Diversity of Axonal and Dendritic Contributions to Neuronal Output. Front. Cell Neurosci. 2020, 13, 570. [Google Scholar] [CrossRef]
- Gasparini, S.; Magee, J.C. State-dependent dendritic computation in hippocampal CA1 pyramidal neurons. J. Neurosci. 2006, 26, 2088–2100. [Google Scholar] [CrossRef]
- Larkum, M.E.; Waters, J.; Sakmann, B.; Helmchen, F. Dendritic spikes in apical dendrites of neocortical layer 2/3 pyramidal neurons. J. Neurosci. 2007, 27, 8999–9008. [Google Scholar] [CrossRef] [PubMed]
- Kamondi, A.; Acsády, L.; Buzsáki, G. Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J. Neurosci. 1998, 18, 3919–3928. [Google Scholar] [CrossRef]
- Colbert, C.M.; Magee, J.C.; Hoffman, D.A.; Johnston, D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J. Neurosci. 1997, 17, 6512–6521. [Google Scholar] [CrossRef]
- Gasparini, S.; Magee, J.C. Phosphorylation-dependent differences in the activation properties of distal and proximal dendritic Na+ channels in rat CA1 hippocampal neurons. J. Physiol. 2002, 541, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Bywalez, W.G.; Patirniche, D.; Rupprecht, V.; Stemmler, M.; Herz, A.V.M.; Pálfi, D.; Rózsa, B.; Egger, V. Local Postsynaptic Voltage-Gated Sodium Channel Activation in Dendritic Spines of Olfactory Bulb Granule Cells. Neuron 2015, 85, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Araya, R.; Nikolenko, V.; Eisenthal, K.B.; Yuste, R. Sodium channels amplify spine potentials. Proc. Nat. Aca. Sci. USA 2007, 104, 12347. [Google Scholar] [CrossRef] [PubMed]
- Engel, D.; Jonas, P. Presynaptic action potential amplification by voltage-gated Na+ channels in hippocampal mossy fiber boutons. Neuron 2005, 45, 405–417. [Google Scholar] [CrossRef]
- Leão, R.M.; Kushmerick, C.; Pinaud, R.; Renden, R.; Li, G.-L.; Taschenberger, H.; Spirou, G.; Levinson, S.R.; von Gersdorff, H. Presynaptic Na+ channels: Locus, development, and recovery from inactivation at a high-fidelity synapse. J. Neurosci. 2005, 25, 3724–3738. [Google Scholar] [CrossRef]
- Stuart, G.; Sakmann, B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 1995, 15, 1065–1076. [Google Scholar] [CrossRef]
- Golding, N.L.; Spruston, N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron 1998, 21, 1189–1200. [Google Scholar] [CrossRef]
- Raman, I.M.; Bean, B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 1997, 17, 4517–4526. [Google Scholar] [CrossRef]
- Raman, I.M.; Sprunger, L.K.; Meisler, M.H.; Bean, B.P. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 1997, 19, 881–891. [Google Scholar] [CrossRef]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Crill, W.E. Persistent Sodium Current in Mammalian Central Neurons. Annu. Rev. Physiol. 1996, 58, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Osorio, N.; Cathala, L.; Meisler, M.H.; Crest, M.; Magistretti, J.; Delmas, P. Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J. Physiol. 2010, 588, 651–670. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, F.H.; Sharp, E.M.; Beacham, D.; Scheuer, T.; Catterall, W.A. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol. Cell Neurosci. 2008, 38, 607–615. [Google Scholar] [CrossRef]
- Taddese, A.; Bean, B.P. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron 2002, 33, 587–600. [Google Scholar] [CrossRef]
- van Drongelen, W.; Lee, H.C.; Stevens, R.L.; Hereld, M. Propagation of Seizure-like Activity in a Model of Neocortex. J. Clin. Neurophysiol. 2007, 24, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Veeramah, K.R.; O’Brien, J.E.; Meisler, M.H.; Cheng, X.; Dib-Hajj, S.D.; Waxman, S.G.; Talwar, D.; Girirajan, S.; Eichler, E.E.; Restifo, L.L.; et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 2012, 90, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Bant, J.S.; Raman, I.M. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc. Nat. Acad. Sci. USA 2010, 107, 12357–12362. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, C.; Marx, S.O.; Pitt, G.S. Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 2017, 149, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Meisler, M.H.; Plummer, N.W.; Burgess, D.L.; Buchner, D.A.; Sprunger, L.K. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica 2004, 122, 37–45. [Google Scholar] [CrossRef]
- Patel, R.R.; Barbosa, C.; Brustovetsky, T.; Brustovetsky, N.; Cummins, T.R. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 2016, 139, 2164–2181. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Santiago, L.F.; Yuan, Y.; Wagnon, J.L.; Hull, J.M.; Frasier, C.R.; O’Malley, H.A.; Meisler, M.H.; Isom, L.L. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Nat. Acad. Sci. USA 2017, 114, 2383–2388. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007, 7, 15–22. [Google Scholar] [CrossRef]
- Xie, W.; Strong, J.A.; Ye, L.; Mao, J.X.; Zhang, J.M. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 2013, 154, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Sittl, R.; Lampert, A.; Huth, T.; Schuy, E.T.; Link, A.S.; Fleckenstein, J.; Alzheimer, C.; Grafe, P.; Carr, R.W. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc. Nat. Acad. Sci. USA 2012, 109, 6704–6709. [Google Scholar] [CrossRef]
- Cummins, T.R.; Dib-Hajj, S.D.; Herzog, R.I.; Waxman, S.G. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005, 579, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Raman, I.M.; Bean, B.P. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: Evidence for two mechanisms. J. Biophys. 2001, 80, 729–737. [Google Scholar] [CrossRef]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef]
- Patel, R.R.; Barbosa, C.; Xiao, Y.; Cummins, T.R. Human Nav1.6 Channels Generate Larger Resurgent Currents than Human Nav1.1 Channels, but the Navβ4 Peptide Does Not Protect Either Isoform from Use-Dependent Reduction. PLoS ONE 2015, 10, e0133485. [Google Scholar] [CrossRef]
- Barbosa, C.; Tan, Z.Y.; Wang, R.; Xie, W.; Strong, J.A.; Patel, R.R.; Vasko, M.R.; Zhang, J.M.; Cummins, T.R. Navβ4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol. Pain 2015, 11, 60. [Google Scholar] [CrossRef]
- Barbosa, C.; Xiao, Y.; Johnson, A.J.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. FHF2 isoforms differentially regulate Nav1.6-mediated resurgent sodium currents in dorsal root ganglion neurons. Pflug. Arch. 2017, 469, 195–212. [Google Scholar] [CrossRef]
- Pan, Y.; Cummins, T.R. Distinct functional alterations in SCN8A epilepsy mutant channels. J. Physiol. 2019, 598, 381–401. [Google Scholar] [CrossRef] [PubMed]
- Wagnon, J.L.; Bunton-Stasyshyn, R.K.; Meisler, M.H. Chapter 10–Mutations of Sodium Channel SCN8A (Nav1.6) in Neurological Disease. In Ion Channels in Health and Disease; Pitt, G.S., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 239–264. [Google Scholar] [CrossRef]
- Meisler, M.H.; Helman, G.; Hammer, M.F.; Fureman, B.E.; Gaillard, W.D.; Goldin, A.L.; Hirose, S.; Ishii, A.; Kroner, B.L.; Lossin, C.; et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia 2016, 57, 1027–1035. [Google Scholar] [CrossRef]
- Butler, K.M.; da Silva, C.; Shafir, Y.; Weisfeld-Adams, J.D.; Alexander, J.J.; Hegde, M.; Escayg, A. De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis. Epilepsy Res. 2017, 129, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, M.M.; Dalton, J.C.; Day, J.W.; Ranum, L.P.; Meisler, M.H. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J. Med. Genet. 2006, 43, 527–530. [Google Scholar] [CrossRef]
- Sharkey, L.M.; Cheng, X.; Drews, V.; Buchner, D.A.; Jones, J.M.; Justice, M.J.; Waxman, S.G.; Dib-Hajj, S.D.; Meisler, M.H. The ataxia3 Mutation in the N-Terminal Cytoplasmic Domain of Sodium Channel Nav1.6 Disrupts Intracellular Trafficking. J. Neurosci. 2009, 29, 2733. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; O’Brien, J.E.; Conravey, A.; Hammer, M.F.; Waxman, S.G.; Dib-Hajj, S.D.; Meisler, M.H. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 2014, 69, 117–123. [Google Scholar] [CrossRef]
- Vaher, U.; Nõukas, M.; Nikopensius, T.; Kals, M.; Annilo, T.; Nelis, M.; Ounap, K.; Reimand, T.; Talvik, I.; Ilves, P.; et al. De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J. Child Neurol. 2014, 29, Np202–Np206. [Google Scholar] [CrossRef]
- Bialer, M.; Johannessen, S.I.; Koepp, M.J.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs in preclinical and early clinical development. Epilepsia 2018, 59, 1811–1841. [Google Scholar] [CrossRef]
- Beatch, G.; Namdari, R.; Cadieux, J.; Kato, H.; Aycardi, E. A Phase 1 Study to Assess the Safety, Tolerability and Pharmacokinetics of Two Formulations of a Novel Nav1.6 Sodium Channnel Blocker (XEN901) in Healthy Adult Subjects. (4757). Neurology 2020, 94, 4757. [Google Scholar]
- Wengert, E.R.; Saga, A.U.; Panchal, P.S.; Barker, B.S.; Patel, M.K. Prax330 reduces persistent and resurgent sodium channel currents and neuronal hyperexcitability of subiculum neurons in a mouse model of SCN8A epileptic encephalopathy. Neuropharmacology 2019, 158, 107699. [Google Scholar] [CrossRef]
- Mason, E.R.; Cummins, T.R. Differential Inhibition of Human Nav1.2 Resurgent and Persistent Sodium Currents by Cannabidiol and GS967. Int. J. Mol. Sci. 2020, 21, 2454. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.M.; Thompson, C.H.; Hawkins, N.A.; Wagnon, J.L.; Wengert, E.R.; Patel, M.K.; George, A.L., Jr.; Meisler, M.H.; Kearney, J.A. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 2018, 59, 1166–1176. [Google Scholar] [CrossRef]
- Weuring, W.J.; Singh, S.; Volkers, L.; Rook, M.B.; van’t Slot, R.H.; Bosma, M.; Inserra, M.; Vetter, I.; Verhoeven-Duif, N.M.; Braun, K.P.J.; et al. NaV1.1 and NaV1.6 selective compounds reduce the behavior phenotype and epileptiform activity in a novel zebrafish model for Dravet Syndrome. PLoS ONE 2020, 15, e0219106. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Strong, J.A.; Zhang, J.M. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 2015, 291, 317–330. [Google Scholar] [CrossRef]
- Laedermann, C.J.; Abriel, H.; Decosterd, I. Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front. Pharmacol. 2015, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, M.A.; Khan, M.R.; Alasmari, F.; Alshehri, A.O.; Ali, R.; Boudjelal, M.; Alhosaini, K.A.; Niazy, A.A.; Alshammari, T.K. Changes in the Fluorescence Tracking of NaV1.6 Protein Expression in a BTBR T+Itpr3tf/J Autistic Mouse Model. Neural Plast. 2019, 2019, 12. [Google Scholar] [CrossRef]
- Zhu, H.; Lin, W.; Zhao, Y.; Wang, Z.; Lao, W.; Kuang, P.; Zhou, H. Transient upregulation of Nav1.6 expression in the genu of corpus callosum following middle cerebral artery occlusion in the rats. Brain Res. Bull. 2017, 132, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Wittmack, E.K.; Rush, A.M.; Hudmon, A.; Waxman, S.G.; Dib-Hajj, S.D. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J. Neurosci. 2005, 25, 6621–6630. [Google Scholar] [CrossRef]
- Wu, J.-X.; Tong, L.; Hu, L.; Xia, C.-M.; Li, M.; Chen, Q.-H.; Chen, F.-X.; Du, D.-S. Upregulation of Nav1.6 expression in the rostral ventrolateral medulla of stress-induced hypertensive rats. Hypertens. Res. 2018, 41, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Hargus, N.J.; Nigam, A.; Bertram, E.H., 3rd; Patel, M.K. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J. Neurophysiol. 2013, 110, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, H.; Lampert, A.; Klein, J.P.; Mission, J.; Chen, M.C.; Rivera, M.; Dib-Hajj, S.; Brennan, A.R.; Hains, B.C.; Waxman, S.G. Role of hippocampal sodium channel Nav1.6 in kindling epileptogenesis. Epilepsia 2009, 50, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.S.; Tang, B.; Papale, L.A.; Yu, F.H.; Catterall, W.A.; Escayg, A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum. Mol. Genet. 2007, 16, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Makinson, C.D.; Tanaka, B.S.; Lamar, T.; Goldin, A.L.; Escayg, A. Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiol. Dis. 2014, 68, 16–25. [Google Scholar] [CrossRef]
- Wong, J.C.; Makinson, C.D.; Lamar, T.; Cheng, Q.; Wingard, J.C.; Terwilliger, E.F.; Escayg, A. Selective targeting of Scn8a prevents seizure development in a mouse model of mesial temporal lobe epilepsy. Sci. Rep. 2018, 8, 126. [Google Scholar] [CrossRef]
- Lenk, G.M.; Jafar-Nejad, P.; Hill, S.F.; Huffman, L.D.; Smolen, C.E.; Wagnon, J.L.; Petit, H.; Yu, W.; Ziobro, J.; Bhatia, K.; et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann. Neurol. 2020, 87, 339–346. [Google Scholar] [CrossRef]
- O’Malley, H.A.; Isom, L.L. Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Calhoun, J.D.; Chen, C.; Miyazaki, H.; Nukina, N.; Oyama, F.; Ranscht, B.; Isom, L.L. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc. Nat. Acad. Sci. USA 2010, 107, 2283–2288. [Google Scholar] [CrossRef]
- Aman, T.K.; Grieco-Calub, T.M.; Chen, C.; Rusconi, R.; Slat, E.A.; Isom, L.L.; Raman, I.M. Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J. Neurosci. 2009, 29, 2027–2042. [Google Scholar] [CrossRef]
- Grieco, T.M.; Afshari, F.S.; Raman, I.M. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar purkinje neurons. J. Neurosci. 2002, 22, 3100–3107. [Google Scholar] [CrossRef]
- Bouza, A.A.; Philippe, J.M.; Edokobi, N.; Pinsky, A.M.; Offord, J.; Calhoun, J.D.; Lopez-Florán, M.; Lopez-Santiago, L.F.; Jenkins, P.M.; Isom, L.L. Sodium channel β1 subunits are post-translationally modified by tyrosine phosphorylation, S-palmitoylation, and regulated intramembrane proteolysis. J. Biol. Chem. 2020, 295, 10380–10393. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236. [Google Scholar] [CrossRef]
- Schoorlemmer, J.; Goldfarb, M. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr. Biol. 2001, 11, 793–797. [Google Scholar] [CrossRef]
- Schoorlemmer, J.; Goldfarb, M. Fibroblast growth factor homologous factors and the islet brain-2 scaffold protein regulate activation of a stress-activated protein kinase. J. Biol. Chem. 2002, 277, 49111–49119. [Google Scholar] [CrossRef]
- Pablo, J.L.; Pitt, G.S. Fibroblast Growth Factor Homologous Factors: New Roles in Neuronal Health and Disease. Neuroscientist 2016, 22, 19–25. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Waxman, S.G. Isoform-specific and pan-channel partners regulate trafficking and plasma membrane stability; and alter sodium channel gating properties. Neurosci. Let. 2010, 486, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Dover, K.; Laezza, F.; Shtraizent, N.; Huang, X.; Tchetchik, D.; Eliseenkova, A.V.; Xu, C.F.; Neubert, T.A.; Ornitz, D.M.; et al. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J. Biol. Chem. 2009, 284, 17883–17896. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dib-Hajj, S.D.; Waxman, S.G. Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN). J. Biol. Chem. 2001, 276, 18925–18933. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, C.; Hoch, E.G.; Pitt, G.S. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J. Biol. Chem. 2011, 286, 24253–24263. [Google Scholar] [CrossRef]
- Wang, C.; Chung, B.C.; Yan, H.; Lee, S.Y.; Pitt, G.S. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
- Munoz-Sanjuan, I.; Smallwood, P.M.; Nathans, J. Isoform diversity among fibroblast growth factor homologous factors is generated by alternative promoter usage and differential splicing. J. Biol. Chem. 2000, 275, 2589–2597. [Google Scholar] [CrossRef]
- Wang, Q.; McEwen, D.G.; Ornitz, D.M. Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech. Dev. 2000, 90, 283–287. [Google Scholar] [CrossRef]
- Laezza, F.; Lampert, A.; Kozel, M.A.; Gerber, B.R.; Rush, A.M.; Nerbonne, J.M.; Waxman, S.G.; Dib-Hajj, S.D.; Ornitz, D.M. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol. Cell Neurosci. 2009, 42, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Bosch, M.K.; Nerbonne, J.M.; Ornitz, D.M. FGF14 localization and organization of the axon initial segment. Mol. Cell Neurosci. 2013, 56, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Wittmack, E.K.; Rush, A.M.; Craner, M.J.; Goldfarb, M.; Waxman, S.G.; Dib-Hajj, S.D. Fibroblast Growth Factor Homologous Factor 2B: Association with Nav1.6 and Selective Colocalization at Nodes of Ranvier of Dorsal Root Axons. J. Neurosci. 2004, 24, 6765. [Google Scholar] [CrossRef]
- Rush, A.M.; Wittmack, E.K.; Tyrrell, L.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Differential modulation of sodium channel Nav1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. Euro J. Neurosci. 2006, 23, 2551–2562. [Google Scholar] [CrossRef]
- Xia, Z.; Storm, D.R. The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 2005, 6, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Sack, J.S.; Greenhough, T.J.; Bugg, C.E.; Means, A.R.; Cook, W.J. Three-dimensional structure of calmodulin. Nature 1985, 315, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kretsinger, R.H.; Rudnick, S.E.; Weissman, L.J. Crystal structure of calmodulin. J. Inorg. Biochem. 1986, 28, 289–302. [Google Scholar] [CrossRef]
- Chattopadhyaya, R.; Meador, W.E.; Means, A.R.; Quiocho, F.A. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 1992, 228, 1177–1192. [Google Scholar] [CrossRef]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef]
- Linse, S.; Helmersson, A.; Forsén, S. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 1991, 266, 8050–8054. [Google Scholar] [CrossRef]
- O’Neil, K.T.; DeGrado, W.F. How calmodulin binds its targets: Sequence independent recognition of amphiphilic alpha-helices. Trend Biochem. Sci 1990, 15, 59–64. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. J. FEBS 2013, 280, 5551–5565. [Google Scholar] [CrossRef] [PubMed]
- Babitch, J. Channel hands. Nature 1990, 346, 321–322. [Google Scholar] [CrossRef]
- Mori, M.; Konno, T.; Ozawa, T.; Murata, M.; Imoto, K.; Nagayama, K. Novel Interaction of the Voltage-Dependent Sodium Channel (VDSC) with Calmodulin: Does VDSC Acquire Calmodulin-Mediated Ca2+-Sensitivity? Biochemistry 2000, 39, 1316–1323. [Google Scholar] [CrossRef]
- Bähler, M.; Rhoads, A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002, 513, 107–113. [Google Scholar] [CrossRef]
- Shah, V.N.; Chagot, B.; Chazin, W.J. Calcium-Dependent Regulation of Ion Channels. Calcium Bind. Proteins 2006, 1, 203–212. [Google Scholar]
- Shah, V.N.; Wingo, T.L.; Weiss, K.L.; Williams, C.K.; Balser, J.R.; Chazin, W.J. Calcium-dependent regulation of the voltage-gated sodium channel hH1: Intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. USA 2006, 103, 3592–3597. [Google Scholar] [CrossRef]
- Wingo, T.L.; Shah, V.N.; Anderson, M.E.; Lybrand, T.P.; Chazin, W.J.; Balser, J.R. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 2004, 11, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ghosh, S.; Liu, H.; Tateyama, M.; Kass, R.S.; Pitt, G.S. Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 2004, 279, 45004–45012. [Google Scholar] [CrossRef] [PubMed]
- Miloushev, V.Z.; Levine, J.A.; Arbing, M.A.; Hunt, J.F.; Pitt, G.S.; Palmer, A.G., 3rd. Solution structure of the NaV1.2 C-terminal EF-hand domain. J. Biol. Chem. 2009, 284, 6446–6454. [Google Scholar] [CrossRef] [PubMed]
- Gardill, B.R.; Rivera-Acevedo, R.E.; Tung, C.C.; Van Petegem, F. Crystal structures of Ca(2+)-calmodulin bound to Na(V) C-terminal regions suggest role for EF-hand domain in binding and inactivation. Proc. Nat. Acad. Sci. USA 2019, 116, 10763–10772. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, C. Calmodulin and calcium differentially regulate the neuronal Nav1.1 voltage-dependent sodium channel. Biochem. Biophys. Res. Commun. 2011, 411, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.I.; Liu, C.; Waxman, S.G.; Cummins, T.R. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J. Neurosci. 2003, 23, 8261–8270. [Google Scholar] [CrossRef]
- Sarhan, M.F.; Tung, C.C.; Van Petegem, F.; Ahern, C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Nat. Acad. Sci. USA 2012, 109, 3558–3563. [Google Scholar] [CrossRef]
- Deschenes, I. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 2002, 90, e49–e57. [Google Scholar] [CrossRef]
- Tan, H.L. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 2002, 415, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Young, K.A.; Caldwell, J.H. Modulation of skeletal and cardiac voltage-gated sodium channels by calmodulin. J. Physiol. 2005, 565, 349–370. [Google Scholar] [CrossRef]
- Biswas, S. Calmodulin regulation of Nav1.4 current: Role of binding to the carboxyl terminus. J. Gen. Physiol. 2008, 131, 197–209. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Xiao, Y.; Seetharaman, J.; Cummins, T.R.; Sivaraman, J. Structural basis for the modulation of the neuronal voltage-gated sodium channel NaV1.6 by calmodulin. Sci. Rep. 2013, 3, 2435. [Google Scholar] [CrossRef]
- Wang, Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.-S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Na(v)1.5. Channels 2020, 14, 268–286. [Google Scholar] [CrossRef]
- Burel, S.; Coyan, F.C.; Lorenzini, M.; Meyer, M.R.; Lichti, C.F.; Brown, J.H.; Loussouarn, G.; Charpentier, F.; Nerbonne, J.M.; Townsend, R.R.; et al. C-terminal phosphorylation of NaV1.5 impairs FGF13-dependent regulation of channel inactivation. J. Biol. Chem. 2017, 292, 17431–17448. [Google Scholar] [CrossRef] [PubMed]
- Waechter, C.J.; Schmidt, J.W.; Catterall, W.A. Glycosylation is required for maintenance of functional sodium channels in neuroblastoma cells. J. Biol. Chem. 1983, 258, 5117–5123. [Google Scholar] [CrossRef]
- Schmidt, J.W.; Catterall, W.A. Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J. Biol. Chem. 1987, 262, 13713–13723. [Google Scholar] [CrossRef]
- Ednie, A.R.; Bennett, E.S. Modulation of voltage-gated ion channels by sialylation. Compr. Physiol. 2012, 2, 1269–1301. [Google Scholar] [CrossRef]
- Baycin-Hizal, D.; Gottschalk, A.; Jacobson, E.; Mai, S.; Wolozny, D.; Zhang, H.; Krag, S.S.; Betenbaugh, M.J. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem. Biophys. Res. Commun. 2014, 453, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Lazniewska, J.; Weiss, N. The "sweet" side of ion channels. Rev. Physiol. 2014, 167, 67–114. [Google Scholar] [CrossRef]
- Mercier, A.; Clément, R.; Harnois, T.; Bourmeyster, N.; Bois, P.; Chatelier, A. Nav1.5 channels can reach the plasma membrane through distinct N-glycosylation states. Biochim. Biophys. Acta 2015, 1850, 1215–1223. [Google Scholar] [CrossRef]
- Jones, J.M.; Dionne, L.; Dell’Orco, J.; Parent, R.; Krueger, J.N.; Cheng, X.; Dib-Hajj, S.D.; Bunton-Stasyshyn, R.K.; Sharkey, L.M.; Dowling, J.J.; et al. Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiol. Dis. 2016, 89, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Recio-Pinto, E.; Thornhill, W.B.; Duch, D.S.; Levinson, S.R.; Urban, B.W. Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron 1990, 5, 675–684. [Google Scholar] [CrossRef]
- Bennett, E.; Urcan, M.S.; Tinkle, S.S.; Koszowski, A.G.; Levinson, S.R. Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. J. Gen. Physiol. 1997, 109, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hartmann, H.A.; Satin, J. Glycosylation Influences Voltage-Dependent Gating of Cardiac and Skeletal Muscle Sodium Channels. J. Membr. Biol. 1999, 171, 195–207. [Google Scholar] [CrossRef]
- Tyrrell, L.; Renganathan, M.; Dib-Hajj, S.D.; Waxman, S.G. Glycosylation Alters Steady-State Inactivation of Sodium Channel Nav1.9/NaN in Dorsal Root Ganglion Neurons and Is Developmentally Regulated. J. Neurosci. 2001, 21, 9629. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Abriel, H.; Staub, O. Ubiquitylation of Ion Channels. Physiology 2005, 20, 398–407. [Google Scholar] [CrossRef]
- Shih, S.C.; Sloper-Mould, K.E.; Hicke, L. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. J. EMBO 2000, 19, 187–198. [Google Scholar] [CrossRef]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fotia, A.B.; Ekberg, J.; Adams, D.J.; Cook, D.I.; Poronnik, P.; Kumar, S. Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J. Biol. Chem. 2004, 279, 28930–28935. [Google Scholar] [CrossRef]
- Rougier, J.S.; van Bemmelen, M.X.; Bruce, M.C.; Jespersen, T.; Gavillet, B.; Apothéloz, F.; Cordonier, S.; Staub, O.; Rotin, D.; Abriel, H. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. American journal of physiology. Cell Physiol. 2005, 288, C692–C701. [Google Scholar] [CrossRef] [PubMed]
- Gasser, A.; Cheng, X.; Gilmore, E.S.; Tyrrell, L.; Waxman, S.G.; Dib-Hajj, S.D. Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J. Biol. Chem. 2010, 285, 26149–26161. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Brown, G.E.; Yaffe, M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care. Med. 2000, 28, N67–N77. [Google Scholar] [CrossRef] [PubMed]
- Shipston, M.J. Ion channel regulation by protein palmitoylation. J. Biol. Chem. 2011, 286, 8709–8716. [Google Scholar] [CrossRef] [PubMed]
- Shipston, M.J. Ion channel regulation by protein S-acylation. J. Gen. Physiol. 2014, 143, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xiao, Y.; Pei, Z.; Cummins, T.R. S-palmitoylation of the sodium channel Nav1.6 regulates its activity and neuronal excitability. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Cantrell, A.R.; Catterall, W.A. Neuromodulation of Na+ channels: An unexpected form of cellular plasticity. Nat. Rev. Neurosci. 2001, 2, 397–407. [Google Scholar] [CrossRef]
- Numann, R.; Catterall, W.A.; Scheuer, T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science 1991, 254, 115–118. [Google Scholar] [CrossRef]
- Vijayaragavan, K.; Boutjdir, M.; Chahine, M. Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J. Neurophysiol. 2004, 91, 1556–1569. [Google Scholar] [CrossRef]
- Numann, R.; Hauschka, S.D.; Catterall, W.A.; Scheuer, T. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. J. Neurosci. 1994, 14, 4226–4236. [Google Scholar] [CrossRef]
- Bendahhou, S.; Cummins, T.R.; Potts, J.F.; Tong, J.; Agnew, W.S. Serine-1321-independent regulation of the mu 1 adult skeletal muscle Na+ channel by protein kinase C. Proc. Nat. Acad. Sci. USA 1995, 92, 12003–12007. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Rogers, J.; Tanada, T.; Scheuer, T.; Catterall, W.A. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 3289–3293. [Google Scholar] [CrossRef]
- Cantrell, A.R.; Tibbs, V.C.; Yu, F.H.; Murphy, B.J.; Sharp, E.M.; Qu, Y.; Catterall, W.A.; Scheuer, T. Molecular mechanism of convergent regulation of brain Na(+) channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol. Cell Neurosci. 2002, 21, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; West, J.W.; Lai, Y.; Scheuer, T.; Catterall, W.A. Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron 1992, 8, 1151–1159. [Google Scholar] [CrossRef]
- Smith, R.D.; Goldin, A.L. Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. J. Neurosci. 1997, 17, 6086–6093. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.; Goldin, A.L. Functional analysis of the rat I sodium channel in xenopus oocytes. J. Neurosci. 1998, 18, 811–820. [Google Scholar] [CrossRef]
- Ono, K.; Fozzard, H.A.; Hanck, D.A. Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ. Res. 1993, 72, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.J.; Lee, H.; Shibata, E.F. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ. Res. 1992, 70, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.M.; Okuse, K.; Wood, J.N.; Dolphin, A.C.; Moss, S.J. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J. Physiol. 1999, 516 Pt 2, 433–446. [Google Scholar] [CrossRef]
- Murphy, B.J.; Rossie, S.; De Jongh, K.S.; Catterall, W.A. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel alpha subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J. Biol. Chem. 1993, 268, 27355–27362. [Google Scholar] [CrossRef]
- Cantrell, A.R.; Smith, R.D.; Goldin, A.L.; Scheuer, T.; Catterall, W.A. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit. J. Neurosci. 1997, 17, 7330–7338. [Google Scholar] [CrossRef]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Herren, A.W.; Weber, D.M.; Rigor, R.R.; Margulies, K.B.; Phinney, B.S.; Bers, D.M. CaMKII Phosphorylation of Na(V)1.5: Novel in Vitro Sites Identified by Mass Spectrometry and Reduced S516 Phosphorylation in Human Heart Failure. J. Proteome Res. 2015, 14, 2298–2311. [Google Scholar] [CrossRef]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac Na channel: Contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Cerda, O.; Trimmer, J.S. Mass spectrometry-based phosphoproteomics reveals multisite phosphorylation on mammalian brain voltage-gated sodium and potassium channels. Semin. Cell Dev. Biol. 2011, 22, 153–159. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Ma, Q.-H.; Yang, W.-l.; Zhang, X.-G.; Dawe, G.S.; Xiao, Z.-C. Amyloid precursor protein modulates Nav1.6 sodium channel currents through a Go-coupled JNK pathway. Sci. Rep. 2016, 6, 39320. [Google Scholar] [CrossRef]
- Ciccone, R.; Franco, C.; Piccialli, I.; Boscia, F.; Casamassa, A.; de Rosa, V.; Cepparulo, P.; Cataldi, M.; Annunziato, L.; Pannaccione, A. Amyloid β-Induced Upregulation of Na(v)1.6 Underlies Neuronal Hyperactivity in Tg2576 Alzheimer’s Disease Mouse Model. Sci. Rep. 2019, 9, 13592. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.G.; Zhou, T.T.; Li, N.; Jang, C.Y.; Xiao, Z.C.; Ma, Q.H.; Li, S. Elevated Neuronal Excitability Due to Modulation of the Voltage-Gated Sodium Channel Nav1.6 by Abeta1-42. Front. Neurosci. 2016, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tan, F.C.; Xiao, Z.C.; Dawe, G.S. Amyloid precursor protein enhances Nav1.6 sodium channel cell surface expression. J. Biol. Chem. 2015, 290, 12048–12057. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-Y.; Snider, W. Functions of GSK-3 Signaling in Development of the Nervous System. Front. Mol. Neurosci. 2011, 4, 44. [Google Scholar] [CrossRef]
- Jaworski, T. Control of neuronal excitability by GSK-3beta: Epilepsy and beyond. Biochim. Biophys. Acta 2020, 1867, 118745. [Google Scholar] [CrossRef]
- Jaworski, T.; Banach-Kasper, E.; Gralec, K. GSK-3β at the Intersection of Neuronal Plasticity and Neurodegeneration. Neural. Plast. 2019, 2019, 4209475. [Google Scholar] [CrossRef] [PubMed]
- Scala, F.; Nenov, M.N.; Crofton, E.J.; Singh, A.K.; Folorunso, O.; Zhang, Y.; Chesson, B.C.; Wildburger, N.C.; James, T.F.; Alshammari, M.A.; et al. Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway. Cell Rep. 2018, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, P.A.; Folorunso, O.; Nguyen, N.; Singh, A.K.; D’Amico, D.; Powell, R.T.; Brunell, D.; Allen, J.; Stephan, C.; Laezza, F. High-throughput screening against protein:protein interaction interfaces reveals anti-cancer therapeutics as potent modulators of the voltage-gated Na+ channel complex. Sci. Rep. 2019, 9, 16890. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-C.; Nenov, M.N.; Shavkunov, A.; Panova, N.; Zhan, M.; Laezza, F. Identifying a kinase network regulating FGF14:Nav1.6 complex assembly using split-luciferase complementation. PLoS ONE 2015, 10, e0117246. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Liu, Z.; Nenov, M.N.; Folorunso, O.; Singh, A.; Scala, F.; Chen, H.; James, T.F.; Alshammari, M.; Panova-Elektronova, N.I.; et al. Functional Modulation of Voltage-Gated Sodium Channels by a FGF14-Based Peptidomimetic. ACS Chem. Neurosci. 2018, 9, 976–987. [Google Scholar] [CrossRef]
- Ali, S.R.; Singh, A.K.; Laezza, F. Identification of Amino Acid Residues in Fibroblast Growth Factor 14 (FGF14) Required for Structure-Function Interactions with Voltage-gated Sodium Channel Nav1.6. J. Biol. Chem. 2016, 291, 11268–11284. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zybura, A.; Hudmon, A.; Cummins, T.R. Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability. Cells 2021, 10, 1595. https://doi.org/10.3390/cells10071595
Zybura A, Hudmon A, Cummins TR. Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability. Cells. 2021; 10(7):1595. https://doi.org/10.3390/cells10071595
Chicago/Turabian StyleZybura, Agnes, Andy Hudmon, and Theodore R. Cummins. 2021. "Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability" Cells 10, no. 7: 1595. https://doi.org/10.3390/cells10071595
APA StyleZybura, A., Hudmon, A., & Cummins, T. R. (2021). Distinctive Properties and Powerful Neuromodulation of Nav1.6 Sodium Channels Regulates Neuronal Excitability. Cells, 10(7), 1595. https://doi.org/10.3390/cells10071595