
Impact of Progerin Expression on Adipogenesis in Hutchinson—Gilford Progeria Skin-Derived Precursor Cells



Abstract
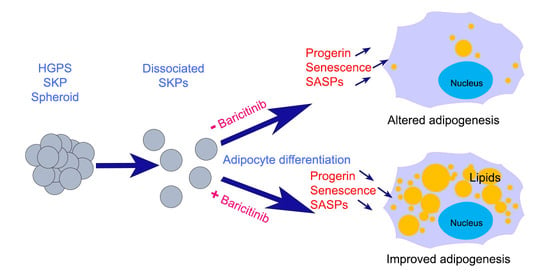
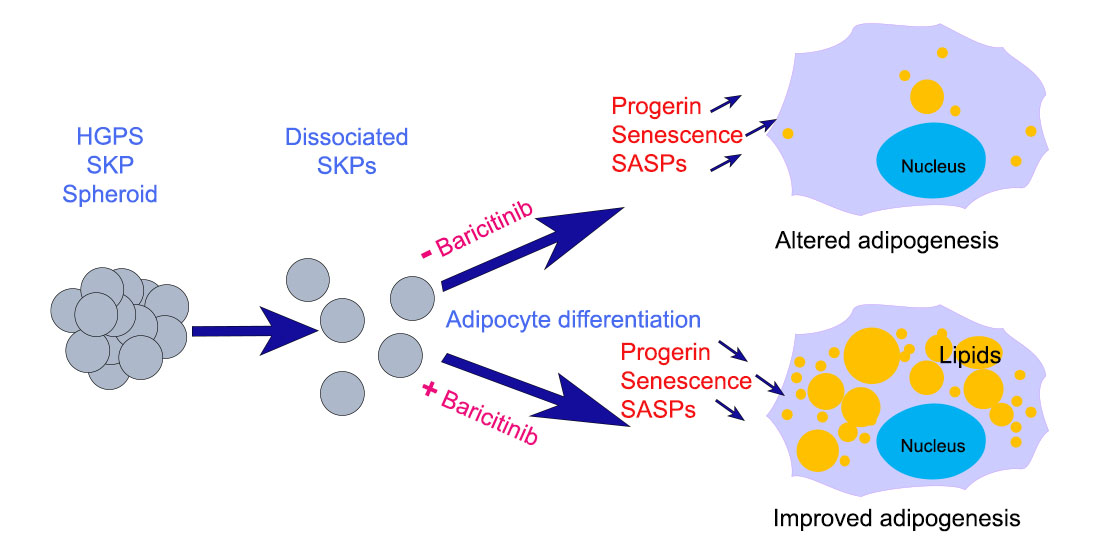{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Low-pH SKP Isolation and Culture
2.3. Differentiation of SKPs into Adipocytes
2.4. Differentiation of 3T3-L1 Cells
2.5. Senescence Associated Beta-Galactosidase (SA-β-Gal)
2.6. Oil Red O (ORO) Staining
2.7. Bodipy Staining
2.8. Immunocytochemistry
2.9. Image Analysis
2.10. Statistical Evaluation and Graphics
3. Results
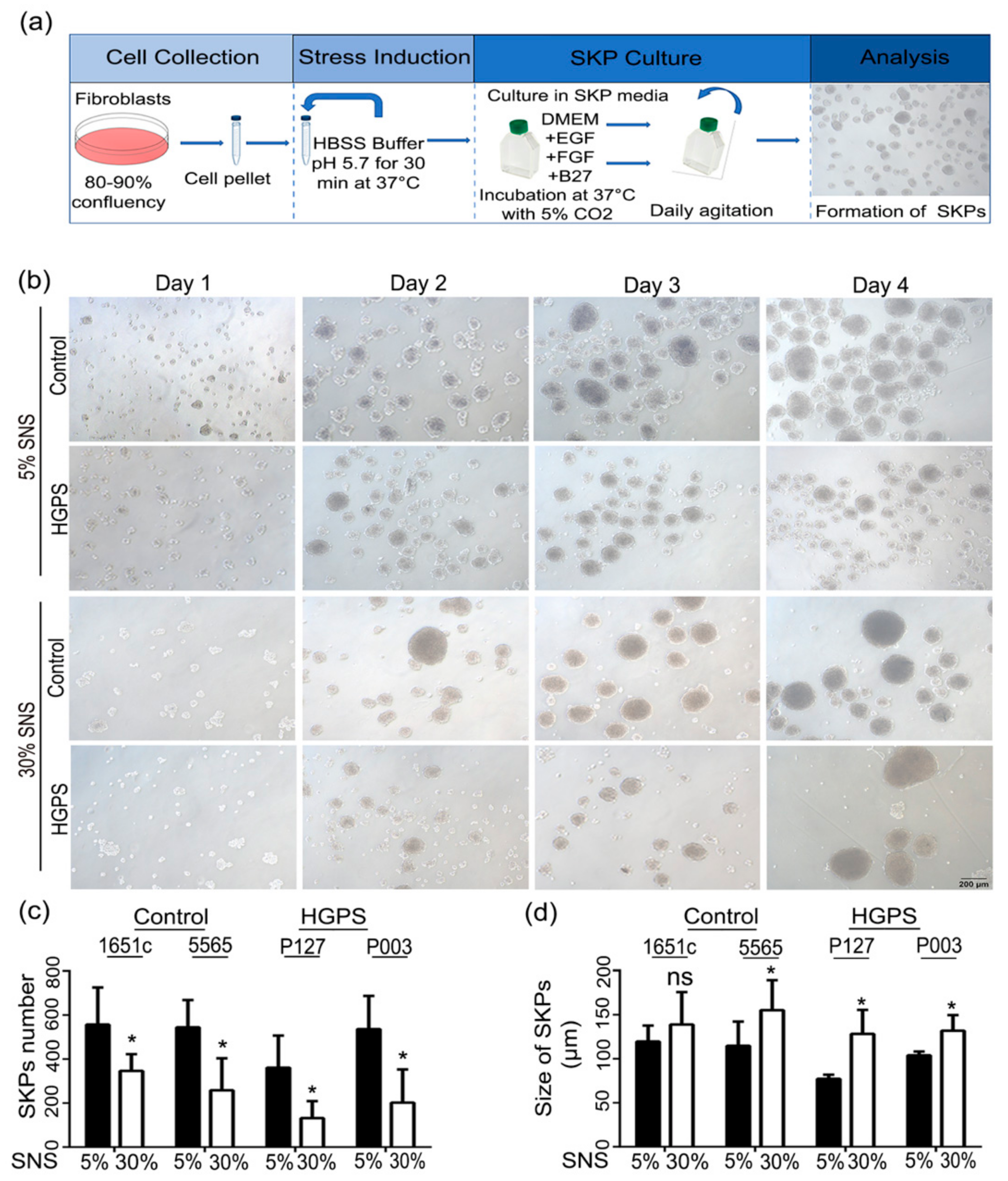
3.1. Characterization of HGPS SKPs
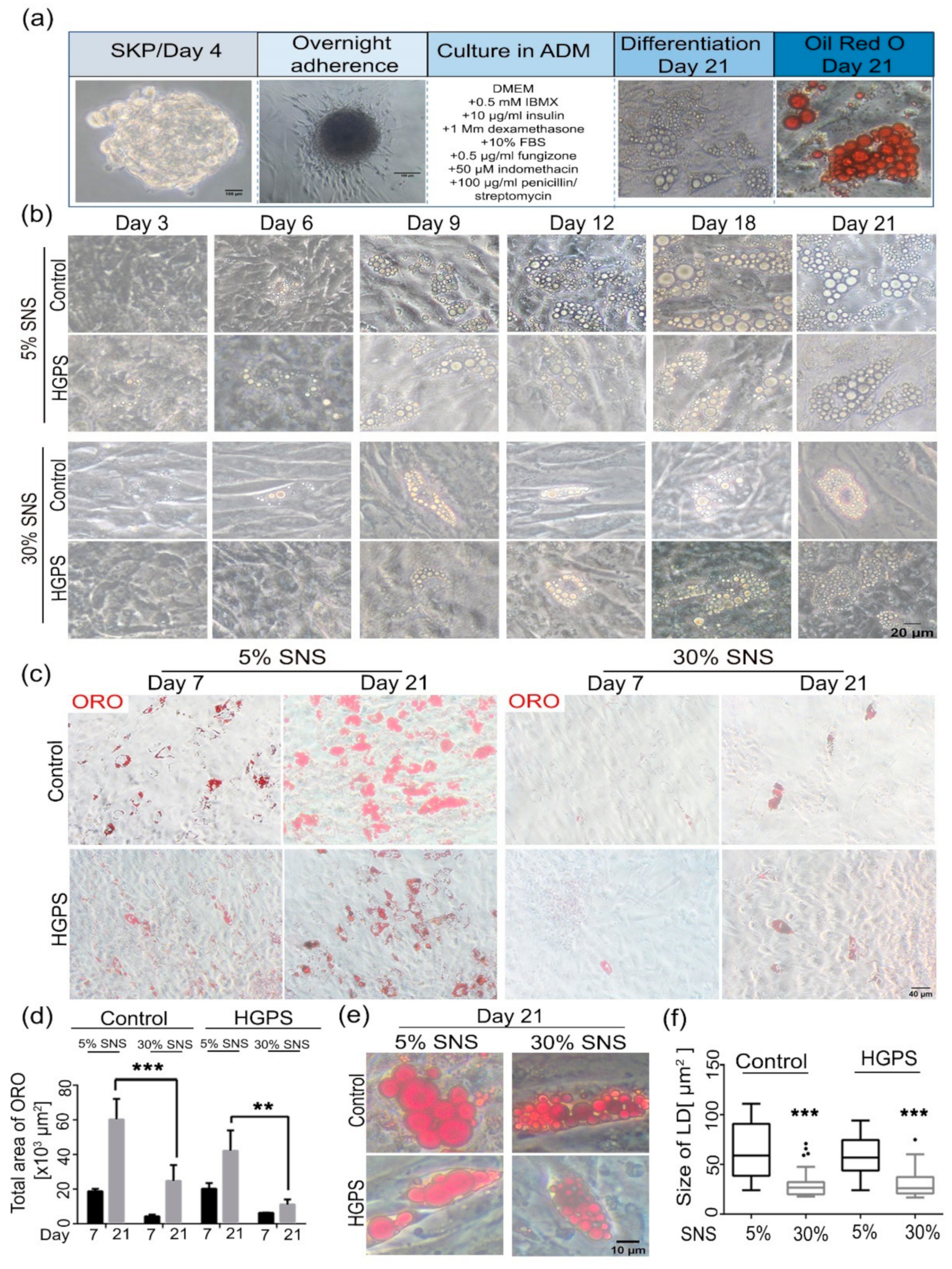
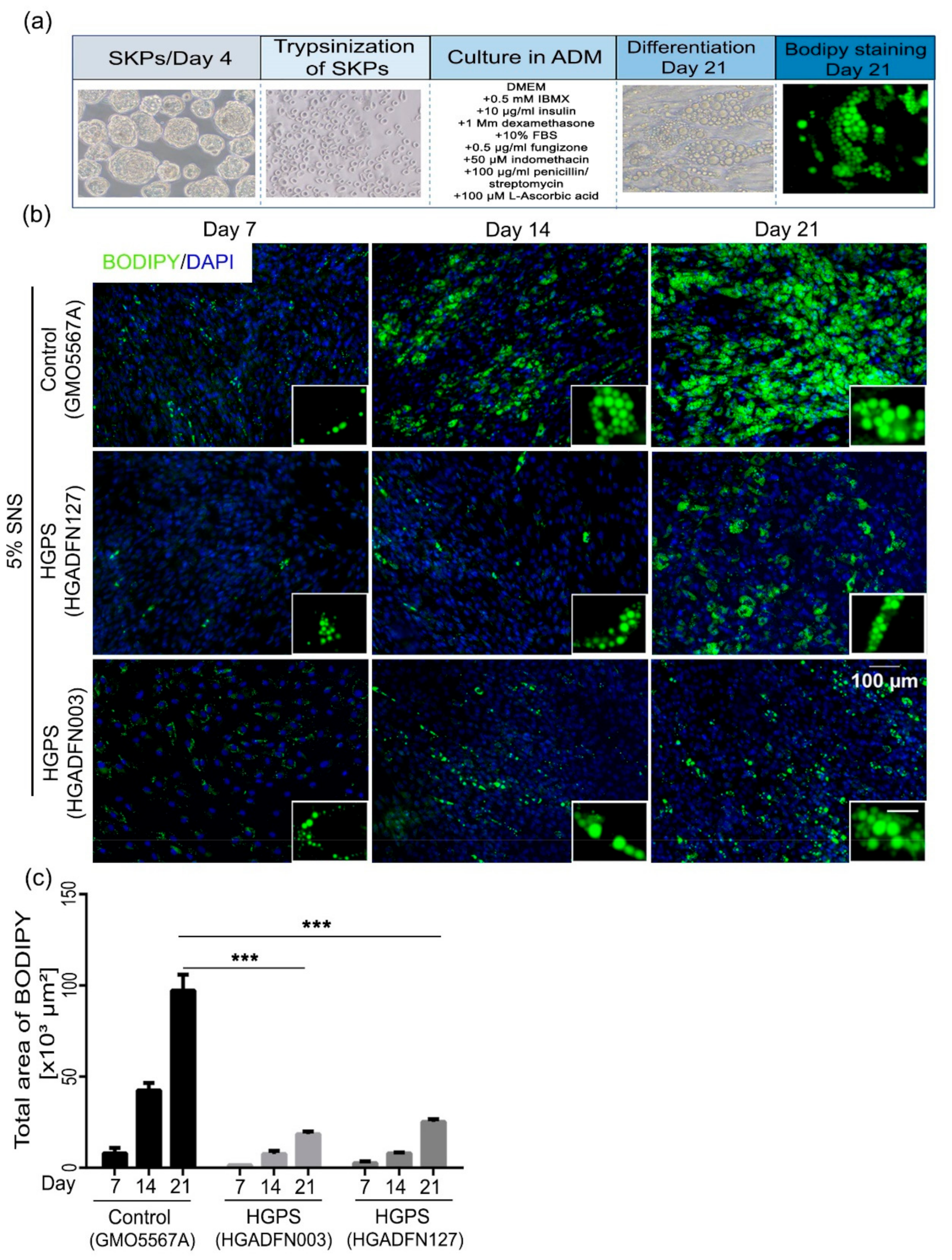
3.2. Differentiation of SKP Spheroids into Adipocytes
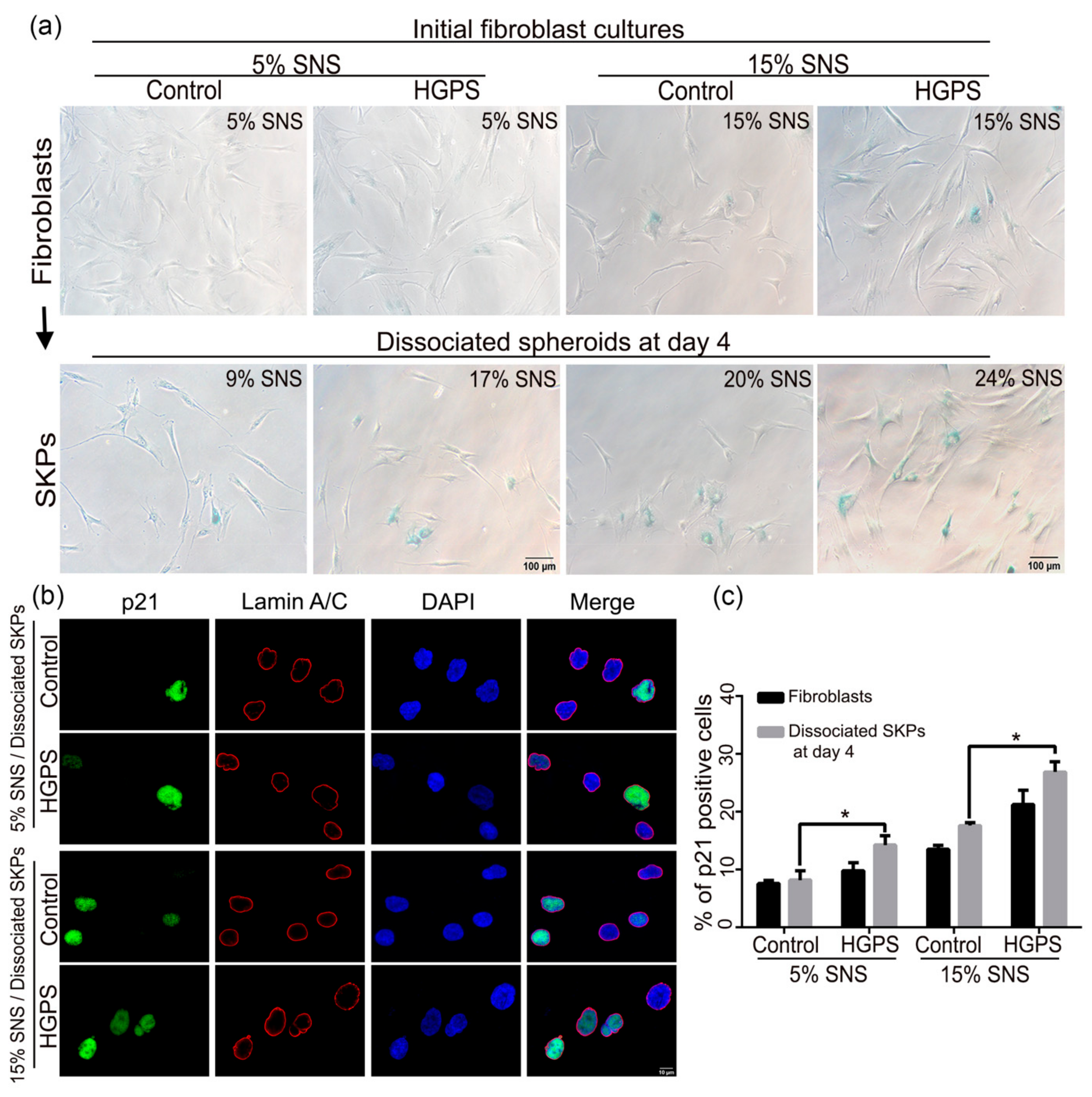
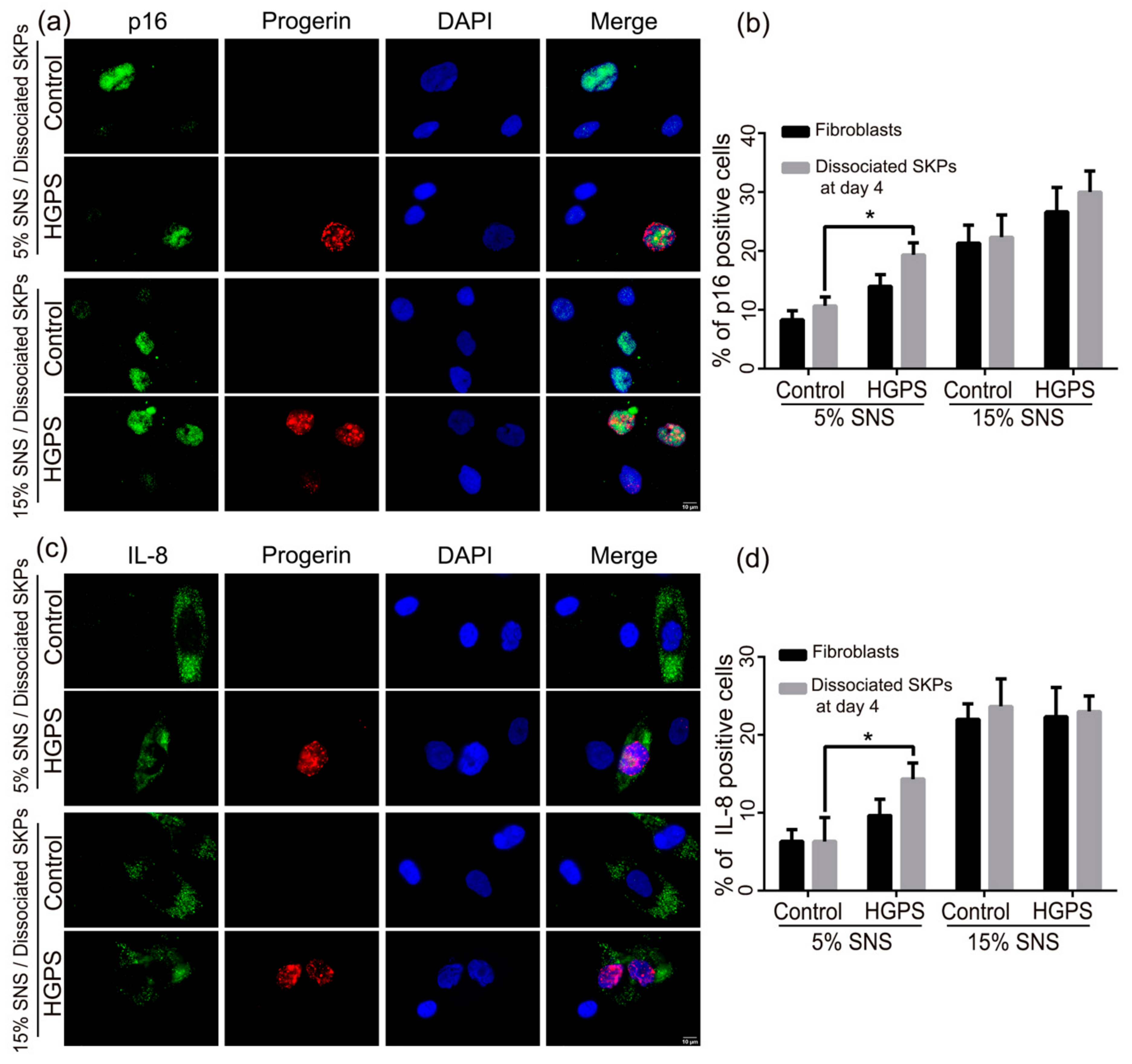
3.3. Characterization of the Senescence Index of SKP Spheroids
3.4. Adipogenesis of Dissociated SKP Spheroids Derived from Young Fibroblast Cultures
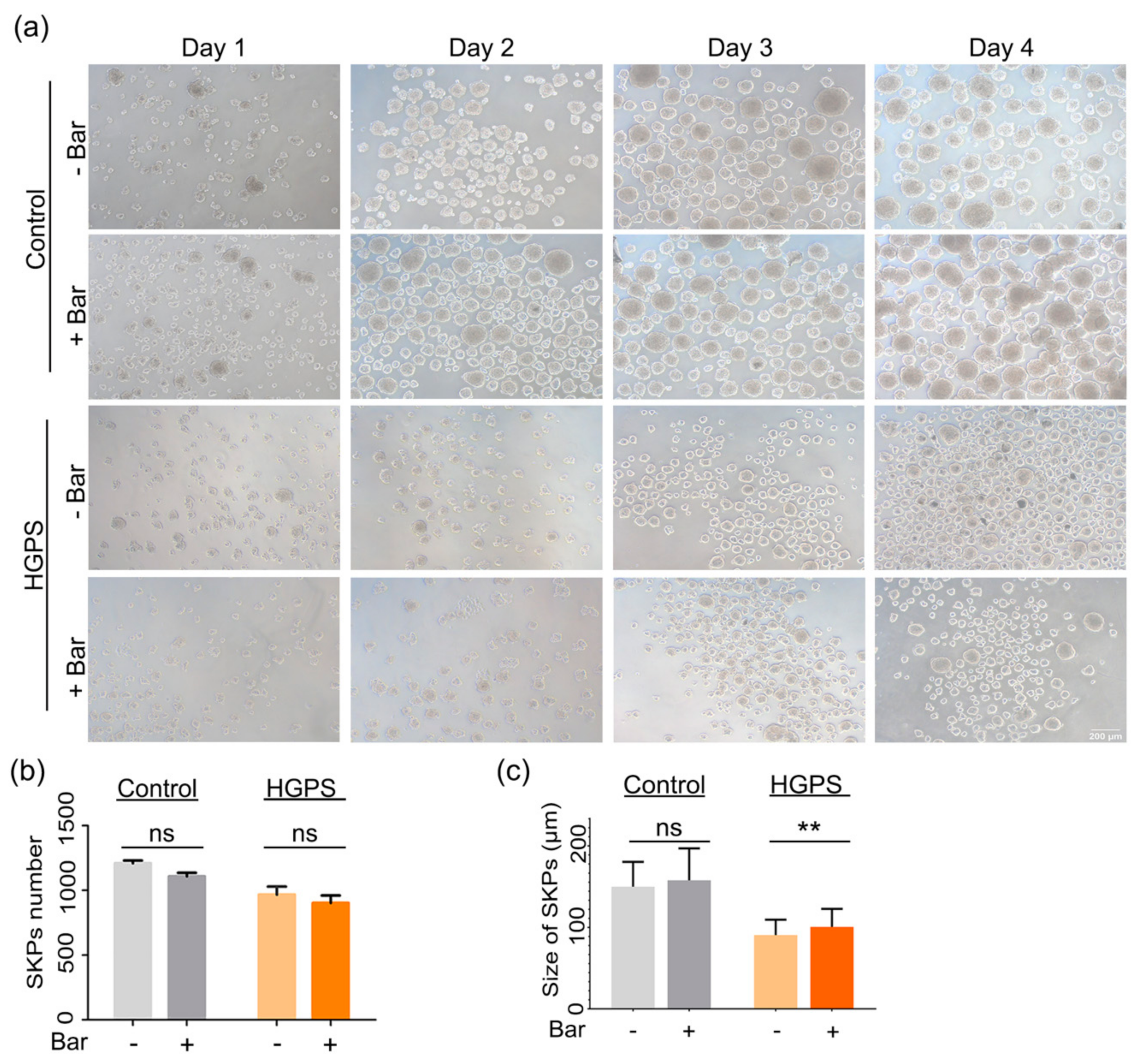
3.5. Effect of Baricitinib, a Specific JAK1/2 Inhibitor, on Spheroid Formation and Adipogenesis
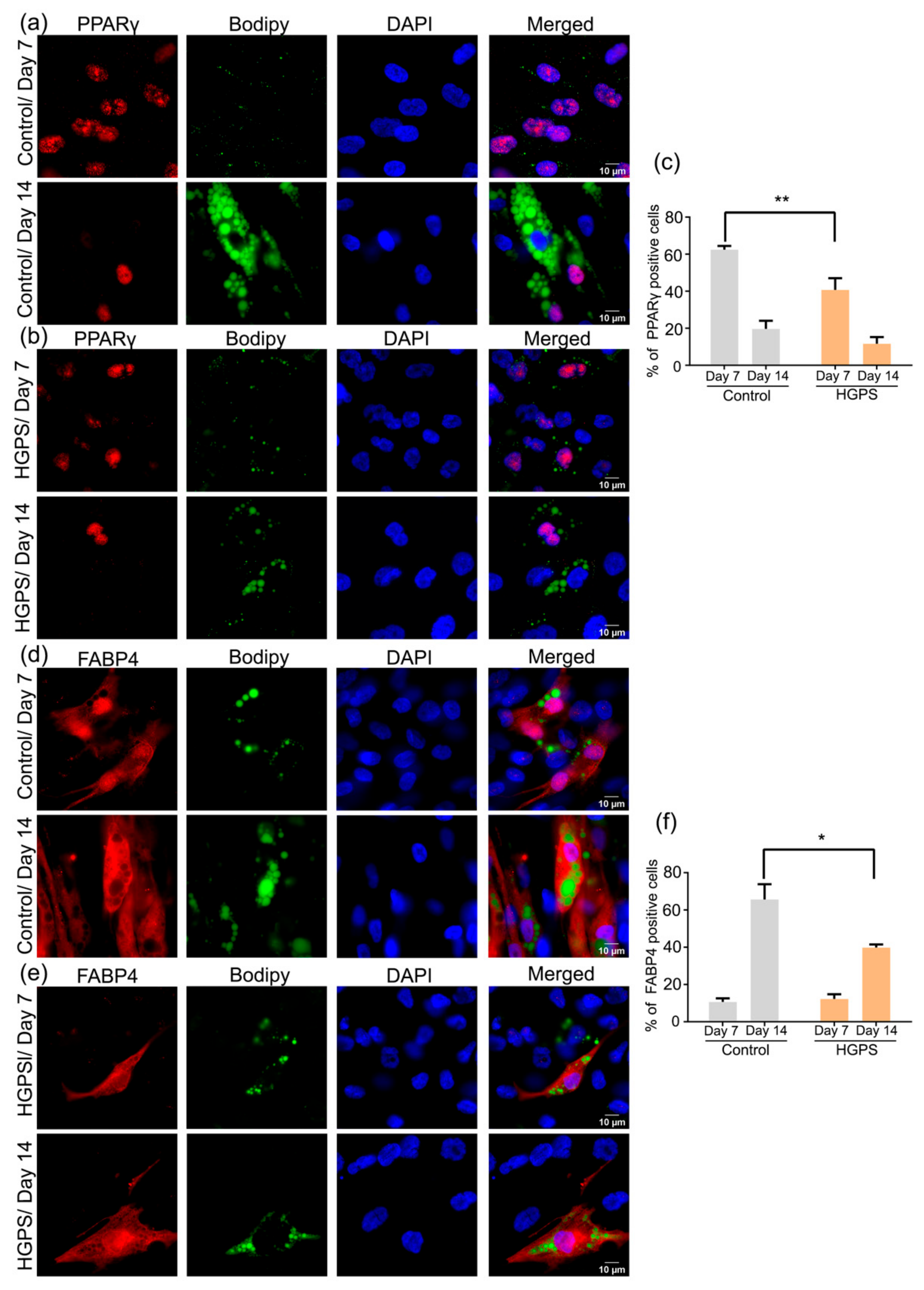
3.6. Expression of PPARγ and FABP4 in Control and HGPS Adipocytes
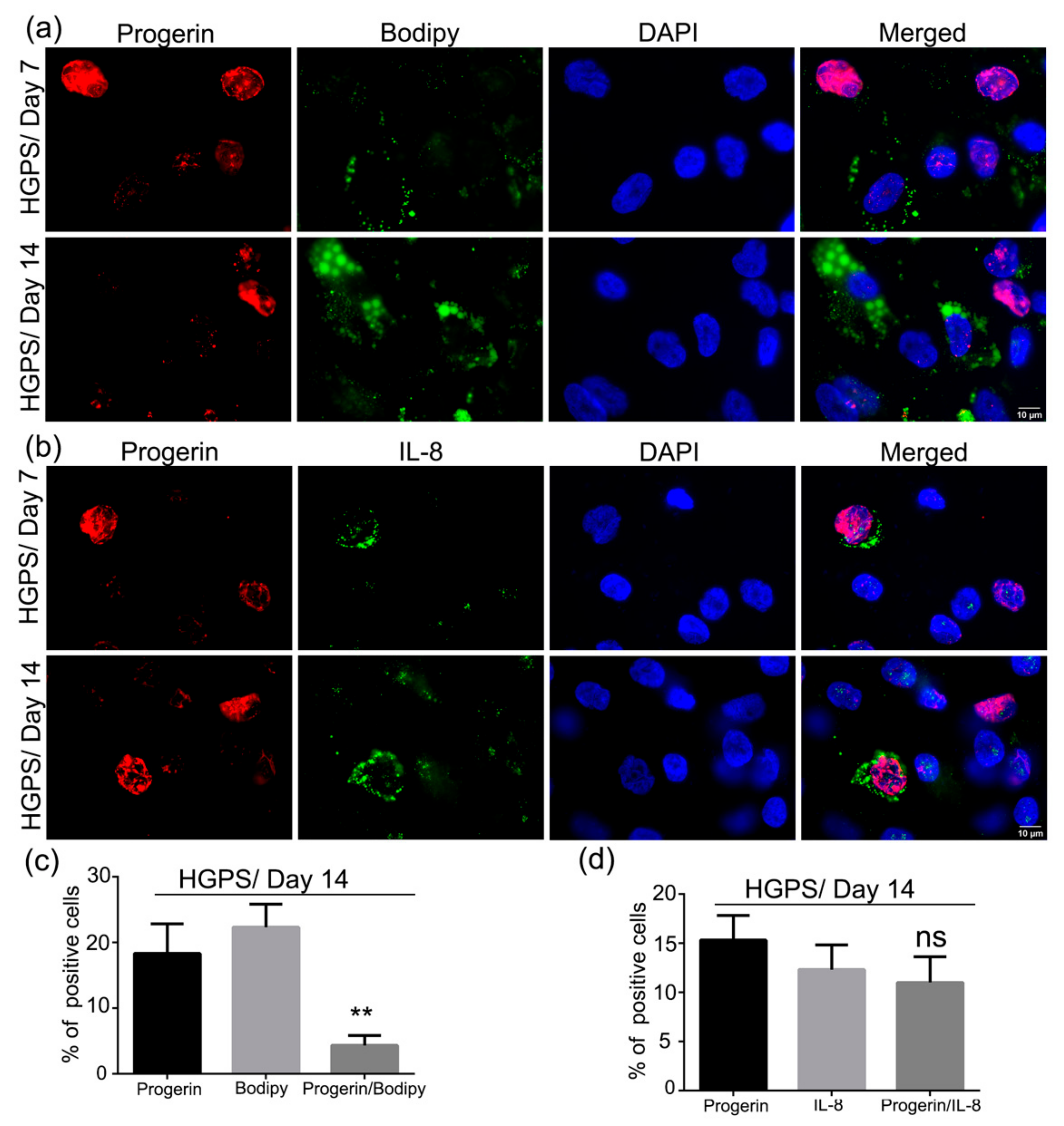
3.7. Accumulation of Progerin and Increased Senescence Underlied HGPS Deffective Adipogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Progeria Research Foundation. PRF BY THE NUMBERS. Available online: https://www.progeriaresearch.org/quick-facts/ (accessed on 31 March 2021).
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef]
- Gordon, L.B.; McCarten, K.M.; Giobbie-Hurder, A.; Machan, J.T.; Campbell, S.E.; Berns, S.D.; Kieran, M.W. Disease progression in Hutchinson-Gilford progeria syndrome: Impact on growth and development. Pediatrics 2007, 120, 824–833. [Google Scholar] [CrossRef]
- Ackerman, J.; Gilbert-Barness, E. Hutchinson-Gilford progeria syndrome: A pathologic study. Pediatr. Pathol. Mol. Med. 2002, 21, 1–13. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Rusinol, A.E.; Sinensky, M.S. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J. Cell Sci. 2006, 119, 3265–3272. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Reddel, C.J.; Weiss, A.S. Lamin A expression levels are unperturbed at the normal and mutant alleles but display partial splice site selection in Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2004, 41, 715–717. [Google Scholar] [CrossRef][Green Version]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef]
- Marji, J.; O’Donoghue, S.I.; McClintock, D.; Satagopam, V.P.; Schneider, R.; Ratner, D.; Worman, H.J.; Gordon, L.B.; Djabali, K. Defective lamin A-Rb signaling in Hutchinson-Gilford Progeria Syndrome and reversal by farnesyltransferase inhibition. PLoS ONE 2010, 5, e11132. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef]
- Wozniak, S.E.; Gee, L.L.; Wachtel, M.S.; Frezza, E.E. Adipose tissue: The new endocrine organ? A review article. Dig. Dis. Sci. 2009, 54, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Garg, A. Genetic disorders of adipose tissue development, differentiation, and death. Annu. Rev. Genom. Hum. Genet. 2006, 7, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Lowe, C.E.; O’Rahilly, S.; Rochford, J.J. Adipogenesis at a glance. J. Cell Sci. 2011, 124, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.L.; Savage, D.B. The genetics of lipid storage and human lipodystrophies. Trends Mol. Med. 2015, 21, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. The physiology of cellular liporegulation. Annu. Rev. Physiol. 2003, 65, 333–347. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Mazereeuw-Hautier, J.; Wilson, L.C.; Mohammed, S.; Smallwood, D.; Shackleton, S.; Atherton, D.J.; Harper, J.I. Hutchinson-Gilford progeria syndrome: Clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br. J. Dermatol. 2007, 156, 1308–1314. [Google Scholar] [CrossRef]
- DeBusk, F.L. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J. Pediatr. 1972, 80, 697–724. [Google Scholar] [CrossRef]
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [CrossRef]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin A inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bidault, G.; Vatier, C.; Capeau, J.; Vigouroux, C.; Bereziat, V. LMNA-linked lipodystrophies: From altered fat distribution to cellular alterations. Biochem. Soc. Trans. 2011, 39, 1752–1757. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Lee, S.J.; Jung, Y.S.; Yoon, M.H.; Kang, S.M.; Oh, A.Y.; Lee, J.H.; Jun, S.Y.; Woo, T.G.; Chun, H.Y.; Kim, S.K.; et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J. Clin. Investig. 2016, 126, 3879–3893. [Google Scholar] [CrossRef]
- Revechon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtacnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.M.; LaDana, C.; Wu, D.; Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging 2013, 5, 288–303. [Google Scholar] [CrossRef]
- Wenzel, V.; Roedl, D.; Gabriel, D.; Gordon, L.B.; Herlyn, M.; Schneider, R.; Ring, J.; Djabali, K. Naive adult stem cells from patients with Hutchinson-Gilford progeria syndrome express low levels of progerin in vivo. Biol. Open 2012, 1, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.J.; McKenzie, I.A.; Mill, P.; Smith, K.M.; Akhavan, M.; Barnabe-Heider, F.; Biernaskie, J.; Junek, A.; Kobayashi, N.R.; Toma, J.G.; et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat. Cell Biol. 2004, 6, 1082–1093. [Google Scholar] [CrossRef]
- Kumar, R.; Sinha, S.; Hagner, A.; Stykel, M.; Raharjo, E.; Singh, K.K.; Midha, R.; Biernaskie, J. Adult skin-derived precursor Schwann cells exhibit superior myelination and regeneration supportive properties compared to chronically denervated nerve-derived Schwann cells. Exp. Neurol. 2016, 278, 127–142. [Google Scholar] [CrossRef]
- Mao, D.; Yao, X.; Feng, G.; Yang, X.; Mao, L.; Wang, X.; Ke, T.; Che, Y.; Kong, D. Skin-derived precursor cells promote angiogenesis and stimulate proliferation of endogenous neural stem cells after cerebral infarction. Biomed. Res. Int. 2015, 2015, 945846. [Google Scholar] [CrossRef]
- Willis, M.A.; Fox, R.J. Progressive Multiple Sclerosis. Continuum 2016, 22, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Toma, J.G.; Akhavan, M.; Fernandes, K.J.; Barnabe-Heider, F.; Sadikot, A.; Kaplan, D.R.; Miller, F.D. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat. Cell Biol. 2001, 3, 778–784. [Google Scholar] [CrossRef]
- Toma, J.G.; McKenzie, I.A.; Bagli, D.; Miller, F.D. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells 2005, 23, 727–737. [Google Scholar] [CrossRef]
- Budel, L.; Djabali, K. Rapid isolation and expansion of skin-derived precursor cells from human primary fibroblast cultures. Biol. Open 2017, 6, 1745–1755. [Google Scholar] [CrossRef]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs—Mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Meuth, M. An established pre-adipose cell line and its differentiation in culture. Cell 1974, 3, 127–133. [Google Scholar] [CrossRef]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, J.M.; Arnold, R.; Djabali, K. Nuclear Pore Complexes Cluster in Dysmorphic Nuclei of Normal and Progeria Cells during Replicative Senescence. Cells 2021, 10, 153. [Google Scholar] [CrossRef]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Candelario, J.; Chen, L.Y.; Marjoram, P.; Reddy, S.; Comai, L. A filtering strategy identifies FOXQ1 as a potential effector of lamin A dysfunction. Aging 2012, 4, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef]
- Hegele, R.A. Molecular basis of partial lipodystrophy and prospects for therapy. Trends Mol. Med. 2001, 7, 121–126. [Google Scholar] [CrossRef]
- Maraldi, N.M.; Capanni, C.; Lattanzi, G.; Camozzi, D.; Facchini, A.; Manzoli, F.A. SREBP1 interaction with prelamin A forms: A pathogenic mechanism for lipodystrophic laminopathies. Adv. Enzyme. Regul. 2008, 48, 209–223. [Google Scholar] [CrossRef]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Levy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Lee, Y.S.; Kim, W.; Kim, S.J.; Shin, K.O.; Yu, J.Y.; Lee, M.K.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J. Nutr. Biochem. 2014, 25, 201–207. [Google Scholar] [CrossRef]
- Osorio, F.G.; Barcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Gordon, L.B.; Campbell, S.E.; Massaro, J.M.; D’Agostino, R.B., Sr.; Kleinman, M.E.; Kieran, M.W.; Moses, M.A. Survey of plasma proteins in children with progeria pre-therapy and on-therapy with lonafarnib. Pediatr. Res. 2018, 83, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Billon, C.; Bedia-Diaz, G.; Albert, C.J.; Toth, Z.; Butler, A.A.; McBride-Gagyi, S.; Ford, D.A.; Baldan, A.; Burris, T.P.; et al. Doubled lifespan and patient-like pathologies in progeria mice fed high-fat diet. Aging Cell 2019, 18, e12852. [Google Scholar] [CrossRef] [PubMed]
- Heizer, P.J.; Yang, Y.; Tu, Y.; Kim, P.H.; Chen, N.Y.; Hu, Y.; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Morales, J.E.; et al. Deficiency in ZMPSTE24 and resulting farnesyl-prelamin A accumulation only modestly affect mouse adipose tissue stores. J. Lipid. Res. 2020, 61, 413–421. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najdi, F.; Krüger, P.; Djabali, K. Impact of Progerin Expression on Adipogenesis in Hutchinson—Gilford Progeria Skin-Derived Precursor Cells. Cells 2021, 10, 1598. https://doi.org/10.3390/cells10071598
Najdi F, Krüger P, Djabali K. Impact of Progerin Expression on Adipogenesis in Hutchinson—Gilford Progeria Skin-Derived Precursor Cells. Cells. 2021; 10(7):1598. https://doi.org/10.3390/cells10071598
Chicago/Turabian StyleNajdi, Farah, Peter Krüger, and Karima Djabali. 2021. "Impact of Progerin Expression on Adipogenesis in Hutchinson—Gilford Progeria Skin-Derived Precursor Cells" Cells 10, no. 7: 1598. https://doi.org/10.3390/cells10071598
APA StyleNajdi, F., Krüger, P., & Djabali, K. (2021). Impact of Progerin Expression on Adipogenesis in Hutchinson—Gilford Progeria Skin-Derived Precursor Cells. Cells, 10(7), 1598. https://doi.org/10.3390/cells10071598