
Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Combined and Sequential Treatment of Cells with Epirubicin, Cyclophosphamide and Paclitaxel
2.3. Cell Growth Evaluation
2.3.1. Cell Growth in 2D Culture
2.3.2. Clonogenic Assay in 2D Culture
2.3.3. Cell Growth in 3D Culture
2.4. Invasion Assay
2.5. In Vivo Invasive Assay Using Zebrafish Xenograft Model
2.6. Sphere Forming Assay
2.7. Flow Cytometry Analysis
2.8. RNA Extraction and Real Time-PCR (qRT-PCR)
2.9. Western Blot Analysis
2.10. Vimentin Knock down and Overexpression
2.10.1. siRNA Transfection
2.10.2. Transient Vimentin Overexpression
2.11. Statistical Analysis
3. Results
3.1. TNBC Cells Surviving Chemotherapeutic Drug Treatment Exhibit Enhanced Agressiveness in 3D Culture
3.2. Persistent Cells Display Enhanced Invasion in Zebrafish
3.3. Persistent Cells Present Enhanced Expression of Cancer Stem Cell Markers and Vimentin
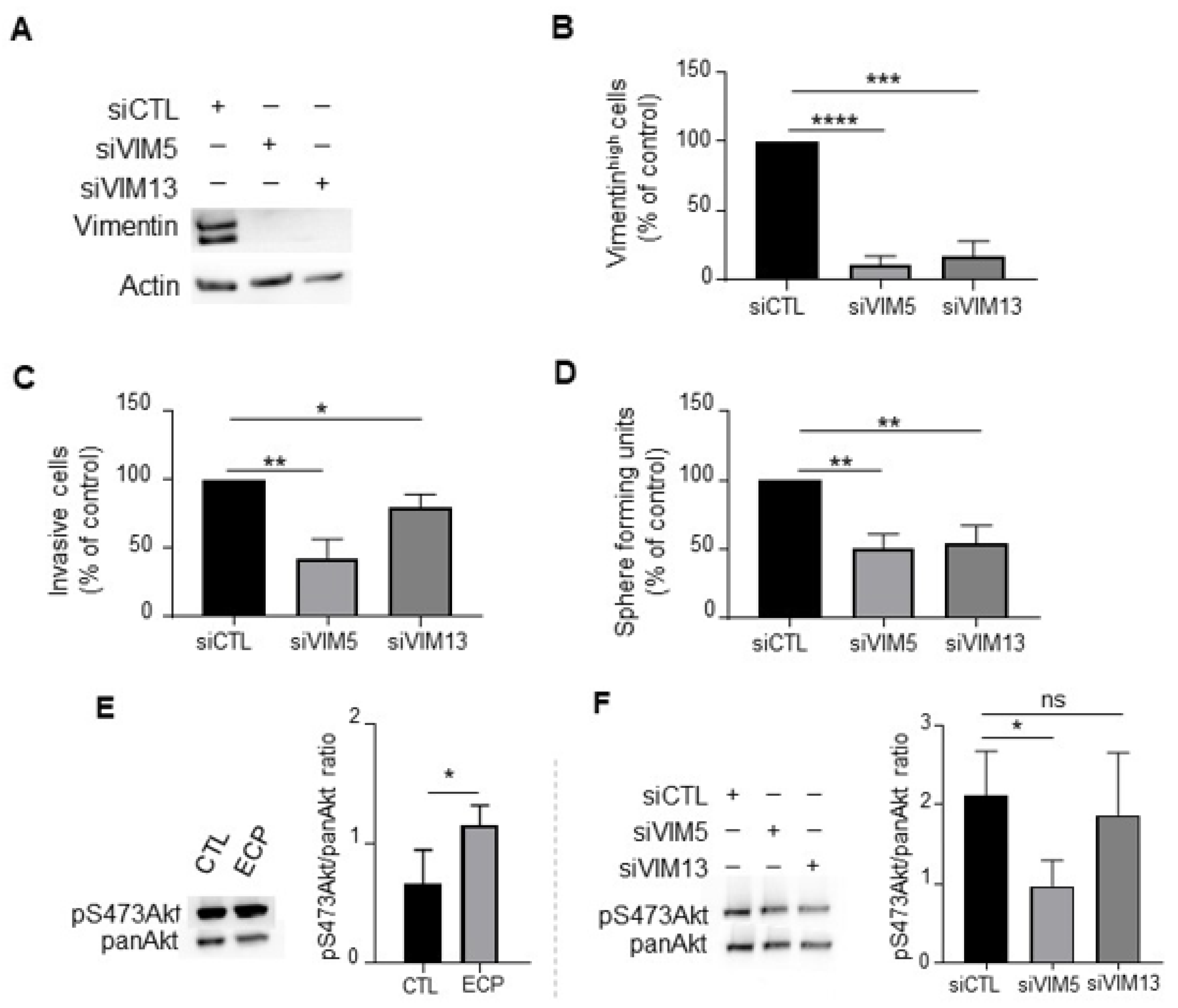
3.4. Vimentin Down-Regulation Reduces Invasion and Sphere Formation as Well as Akt Phosphorylation in MDA-MB-231 Persistent Cells
3.5. Vimentin Promotes Aggressive Phenotype and Drug Resistance of Native MDA-MB-231 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickey, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Siddahart, S.; Sharma, D. Racial disparity and triple-negative breast cancer in African-american women: A multifaceted affair between obesity, biology, and socioeconomic determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Fraser Symmans, W.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Mukherjee, P.; Gupta, A.; Chattopadhyay, D.; Chatterji, U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci. Rep. 2017, 7, 9170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef]
- Volk-draper, L.; Hall, K.; Griggs, C.; Rajput, S.; Kohio, P.; DeNardo, D.; Ran, S. Paclitaxel therapy promotes breast cancer metastasis in TLR4-dependent manner. Cancer Res. 2014, 74, 5421–5434. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Damjanović, A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-How we can rise to the challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Tomao, F.; Papa, A.; Zaccarelli, E.; Rossi, L.; Caruso, D.; Minozzi, M.; Vici, P.; Frati, L.; Tomao, S. Triple-negative breast cancer: New perspectives for targeted therapies. OncoTarget Ther. 2015, 8, 177–193. [Google Scholar] [CrossRef]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef]
- Ichinose, I.; Hamada, Y.; Mitsuyama, S.; Ishikawa, E.; Ikeda, T.; Kobayashi, S.; Horikoshi, N.; Tamura, K. Dose escalation study of epirubicin and docetaxel in patients with advanced or recurrent breast cancer. Chemotherapy 2008, 54, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Mielke, S.; Sparreboom, A.; Steinberg, S.M.; Gelderblom, H.; Unger, C.; Behringer, D.; Mross, K. Association of paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin. Cancer Res. 2005, 11, 4843–4850. [Google Scholar] [CrossRef]
- Tran, T.A.; Gillet, L.; Roger, S.; Besson, P.; White, E.; Le Guennec, J.V. Non-anti-mitotic concentrations of taxol reduce breast cancer cell invasiveness. Biochem. Biophys. Res. Commun. 2009, 379, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.A.; El-Sokkary, G.; Saber, S.H. Low doses of paclitaxel repress breast cancer invasion through DJ-1/KLF17 signalling pathway. Clin. Exp. Pharm. Physiol. 2018, 45, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ma, Z.; Liu, Y.; Kan, X.; Wang, C.; Su, B.; Li, Y.; Zhang, Y.; Wang, P.; Luo, Y.; et al. Low doses of paclitaxel enhance liver metastasis of breast cancer cells in the mouse model. FEBS J. 2016, 283, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Chen, M.J.; Lin, J.C.; Lin, C.H.; Huang, W.C.; Cheng, S.P.; Chen, S.N.; Chang, Y.H. Doxorubicin promotes migration and invasion of breast cancer cells through the upregulation of the RhoA/MLC pathway. J. Breast Cancer 2019, 22, 185–195. [Google Scholar] [CrossRef]
- Lévêque, R.; Corbet, C.; Aubert, L.; Guilbert, M.; Lagadec, C.; Adriaenssens, E.; Duval, J.; Finetti, P.; Birnbaum, D.; Magné, N.; et al. ProNGF increases breast tumor aggressiveness through functional association of TrkA with EphA2. Cancer Lett. 2019, 449, 196–206. [Google Scholar] [CrossRef]
- Lagadec, C.; Meignan, S.; Adriaenssens, E.; Foveau, B.; Vanhecke, E.; Romon, R.; Toillon, R.A.; Oxombre, B.; Hondermarck, H.; Le Bourhis, X. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 2009, 28, 1960–1970. [Google Scholar] [CrossRef]
- Grolez, G.P.; Hammadi, M.; Barras, A.; Gordienko, D.; Slomianny, C.; Völkel, P.; Angrand, P.O.; Pinault, M.; Guimaraes, C.; Potier-Cartereau, M.; et al. Encapsulation of TRPM8 agonist, WS12, in lipid nanocapsules potentiates PC3 prostate cancer cell migration inhibition through channel activation. Sci. Rep. 2019, 9, 7926. [Google Scholar] [CrossRef]
- Bidan, N.; Bailleul-Dubois, J.; Duval, J.; Winter, M.; Denoulet, M.; Hannebicque, K.; El-Sayed, I.Y.; Ginestier, C.; Forissier, V.; Matsunaga, Y.; et al. Transcriptomic analysis of breast cancer stem cells and development of a pALDH1A1:mNeptune reporter system for live tracking. Proteomics 2019, 19, 21–22. [Google Scholar] [CrossRef]
- Aubert, L.; Guilbert, M.; Corbet, C.; Génot, E.; Adriaenssens, E.; Chassat, T.; Bertucci, F.; Daubon, T.; Magné, N.; Le Bourhis, X.; et al. NGF-induced TrkA/CD44 association is involved in tumor aggressiveness and resistance to lestaurtinib. Oncotarget 2015, 6, 9807–9819. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.; Patterson, G.; Ting, L.M.; Wang, Y.; Condeelis, J.; Verkhusha, V. A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat. Methods 2011, 8, 771–777. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 2018, 53, 156–167. [Google Scholar] [CrossRef]
- Geng, S.Q.; Alexandrou, A.T.; Li, J.J. Breast cancer stem cells: Multiple capacities in tumor metastasis. Cancer Lett. 2014, 349, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Miragaya, J.; Palafox, M.; Paré, L.; Yoldi, G.; Ferrer, I.; Vila, S.; Galván, P.; Pellegrini, P.; Pérez-Montoyo, H.; Igea, A.; et al. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of CD49f+ tumor-initiating population. Stem Cell Rep. 2017, 19, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptative resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef]
- Danielson, F.; Peterson, M.K.; Caldeira Araújo, H.; Lautenschläger, F.; Gad, A.K.B. Vimentin diversity in health and disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Morata-Tarifa, C.; Jiménez, G.; García, M.A.; Entrena, J.M.; Griñán-Lisón, C.; Aguilera, M.; Picon-Ruiz, M.; Marchal, J.A. Low adherent cancer cell subpopulation are enriched in tumorigenic and metastatic epithelial-to-mesenchymal transition-induced cancer stem-like cells. Sci. Rep. 2016, 6, 187772. [Google Scholar] [CrossRef]
- Hinz, N.; Jücker, M. Distinct function of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCarffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Wei, X.; Zhang, S.; Song, Z.; Chen, X.; Zhang, J. Role of inhibitor of yes-associated protein 1 in triple negative breast cancer with taxol-based chemoresistance. Cancer Sci. 2019, 110, 561–567. [Google Scholar] [CrossRef]
- Blanchard, Z.; Paul, B.T.; Craft, B.; ElShamy, W.M. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast Cancer Res. 2015, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feeback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef]
- Liu, P.; Kumar, I.S.; Brown, S.; Kannappan, V.; Tawari, P.E.; Tang, J.Z.; Jiang, W.; Armesilla, A.L.; Darling, J.L.; Wang, W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistance triple-negative breast cancer cells. Brit. J. Cancer 2013, 109, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Nagata, T.; Shimada, Y.; Sekine, S.; Hori, R.; Matsui, K.; Okumura, T.; Sawada, S.; Fukuoka, J.; Tsukada, K. Prognostic significance of NANOG and KFL4 for breast cancer. Breast Cancer 2014, 21, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mazur, S.J.; Lin, T.; Appella, E.; Xu, Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 2014, 33, 2655–2664. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [CrossRef]
- Friedrichs, K.; Ruiz, P.; Franke, F.; Gille, I.; Terpe, H.J.; Imhof, B.A. High expression level of α6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995, 55, 901–906. [Google Scholar] [PubMed]
- Ye, F.; Qiu, Y.; Li, L.; Yang, L.; Cheng, F.; Zhang, H.; Wei, B.; Zhang, Z.; Sun, L.; Bu, H. The presence of EpCAM−/CD49f+ cells in breast cancer is associated with a poor clinical outcome. J. Breast Cancer 2015, 18, 242–248. [Google Scholar] [CrossRef]
- Brooks, D.; Schwab, L.; Krutilina, R.; Park, D.; Sethuraman, A.; Hoogewijs, D.; Schörg, A.; Gotwald, L.; Fan, M.; Wenger, R.; et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol. Cancer 2016, 15. [Google Scholar] [CrossRef]
- Strouhalova, K.; Přechová, M.; Gandalovičová, A.; Brábek, J.; Gregor, M.; Rosel, D. Vimentin intermediate filaments as potential target for cancer treatment. Cancers 2020, 12, 184. [Google Scholar] [CrossRef] [PubMed]
- Peuhu, E.; Virtakoivu, R.; Mai, A.; Wärri, A.; Ivaska, J. Epithelial vimentin plays a functional role in mammary gland development. Development 2017, 144, 4103–41113. [Google Scholar] [CrossRef]
- Yamashita, N.; Tokunanga, E.; Kitao, H.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; Aishima, S.; Morita, M.; Maehara, Y. Vimentin as a poor prognostic factor for triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Javir, G.; Joshi, K.; Khedkar, V.; Rojatkar, S. 6 α-Hydroxy-4[14], 10[15]-guainadien-8β, 12-olide induced cell cycle arrest via modulation of EMT and Wnt/β-catenin pathway in HER-2 positive breast cancer cells. J. Steroid Biochem. Mol. Biol. 2020, 197, 105514. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef] [PubMed]
- Leduc, C.; Etienne-Manneville, S. Regulation of microtubule-associated motors drives intermediate filament network polarization. J. Cell Biol. 2017, 216, 1689–1703. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Jiu, Y.; Lehtimäki, J.; Tojkander, S.; Cheng, F.; Jäälinoja, H.; Liu, X.; Varjosalo, M.; Eriksson, J.E.; Lappalainen, P. Bidirectional interplay between vimentin intermediate filaments and contractile actin stress fibers. Cell Rep. 2015, 11, 1511–1518. [Google Scholar] [CrossRef]
- Jiu, Y.; Peränen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and Rhoa. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef]
- Ivaska, J.; Vuoriluoto, K.; Huovinen, T.; Izawa, I.; Inagaki, M.; Parker, P.J. PKCε-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005, 24, 3834–3845. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin-ERK signaling uncouples slug gene regulatory function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef]
- Zelenko, Z.; Gallagher, E.J.; Tobin-Hess, A.; Belardi, V.; Rostoker, R.; Blank, J.; Dina, Y.; LeRoith, D. Silencing vimentin expression decreases pulmonary metastases in pre-diabetic mouse model of mammary tumor progression. Oncogene 2017, 36, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, J.; Zhang, Y.; Liu, Y.; Zuo, S.; Li, R. MAP2K4 interacts with vimentin to activate the PI3K/AKT pathway and promotes breast cancer pathogenesis. Aging 2019, 11, 10697–10710. [Google Scholar] [CrossRef] [PubMed]
- Matte, B.F.; Kumar, A.; Placone, J.K.; Zanella, V.G.; Martins, M.D.; Engler, A.J.; Lamers, M.L. Matrix stiffness mechanically conditions EMT and migratory behavior of oral squamous cell carcinoma. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Antfolk, D.; Sjöqvist, M.; Cheng, F.; Isoniemi, K.; Duran, C.; Rivero-Müller, A.; Antila, C.; Niemi, R.; Landor, S.; Bouten, C.; et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. USA 2017, 114, E4574–E4581. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, N.; Rodriguez, F.; Rivero-Müller, A.; Ristori, T.; Duran, C.; Stassen, O.; Antfolk, D.; Driessen, R.; Ruohonen, S.; Ruohonen, S.; et al. Vimentin regulates Notch signaling strength and arterial remodeling in response to hemodynamic stress. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Wang, W.; Yi, M.; Zhang, R.; Li, J.; Chen, S.; Cai, J.; Zeng, Z.; Li, X.; Xiong, W.; Wang, L.; et al. Vimentin is a crucial target for anti-metastasis therapy of nasopharyngeal carcinoma. Mol. Cell Biochem. 2018, 438, 47–57. [Google Scholar] [CrossRef]
- Trogden, K.; Battaglia, R.; Kabiraj, P.; Madden, V.; Herrmann, H.; Snider, N. An image-based small-molecules screen identifies vimentin as a pharmacologically relevant target of simvastatin in cancer cells. FASEB J. 2018, 32, 2841–2854. [Google Scholar] [CrossRef] [PubMed]
- Stan, S.; Hahm, E.R.; Warin, R.; Singh, S. Withaferin A causes FOXO3a- and Bim-Dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008, 15, 7661–7769. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hahm, E.R.; Marcus, A.; Singh, S. Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors. Mol. Carcinog. 2015, 54, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Bargagna-Mohan, P.; Hamza, A.; Kim, Y.; Ho, Y.; Mor-Vaknin, N.; Wendschlag, N.; Liu, J.; Evans, R.; Markovitz, D.; Zhan, C.; et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 2007, 14, 623–634. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| Nanog | 5′GTG-ATT-TGT-GGG-CCT-GAA-GA3′ | 5′ACA-CAG-CTG-GGT-GGA-GA3′ |
| Oct4 | 5′GAA-GGA-TGT-GGT-CCG-AGT-GT3′ | 5′GTG-AAG-TGA-GGG-CTC-CCA-TA3′ |
| Sox2 | 5′AAC-CCC-AAG-ATG-CAC-AAC-TC3′ | 5′CGG-GGC-CGG-TAT-TTA-TAA-TC3′ |
| Vimentin | 5′TCT-AGG-AGG-AGA-TGC-GG3′ | 5′GGT-CAA-GAC-GTG-CCA-GAG-AC3′ |
| RPLP0 | 5′GCG-ACC-TGG-AAG-TCC-AAC-TA3′ | 5′TGT-CTG-CTC-CCA-TGA-AG3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winter, M.; Meignan, S.; Völkel, P.; Angrand, P.-O.; Chopin, V.; Bidan, N.; Toillon, R.-A.; Adriaenssens, E.; Lagadec, C.; Le Bourhis, X. Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment. Cells 2021, 10, 1504. https://doi.org/10.3390/cells10061504
Winter M, Meignan S, Völkel P, Angrand P-O, Chopin V, Bidan N, Toillon R-A, Adriaenssens E, Lagadec C, Le Bourhis X. Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment. Cells. 2021; 10(6):1504. https://doi.org/10.3390/cells10061504
Chicago/Turabian StyleWinter, Marie, Samuel Meignan, Pamela Völkel, Pierre-Olivier Angrand, Valérie Chopin, Nadège Bidan, Robert-Alain Toillon, Eric Adriaenssens, Chann Lagadec, and Xuefen Le Bourhis. 2021. "Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment" Cells 10, no. 6: 1504. https://doi.org/10.3390/cells10061504
APA StyleWinter, M., Meignan, S., Völkel, P., Angrand, P.-O., Chopin, V., Bidan, N., Toillon, R.-A., Adriaenssens, E., Lagadec, C., & Le Bourhis, X. (2021). Vimentin Promotes the Aggressiveness of Triple Negative Breast Cancer Cells Surviving Chemotherapeutic Treatment. Cells, 10(6), 1504. https://doi.org/10.3390/cells10061504