
Efficacy and Selectivity of FGF2-Saporin Cytosolically Delivered by PCI in Cells Overexpressing FGFR1

 ,
,  and
and
Abstract
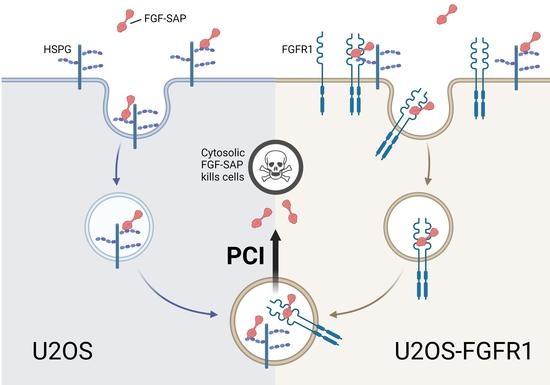
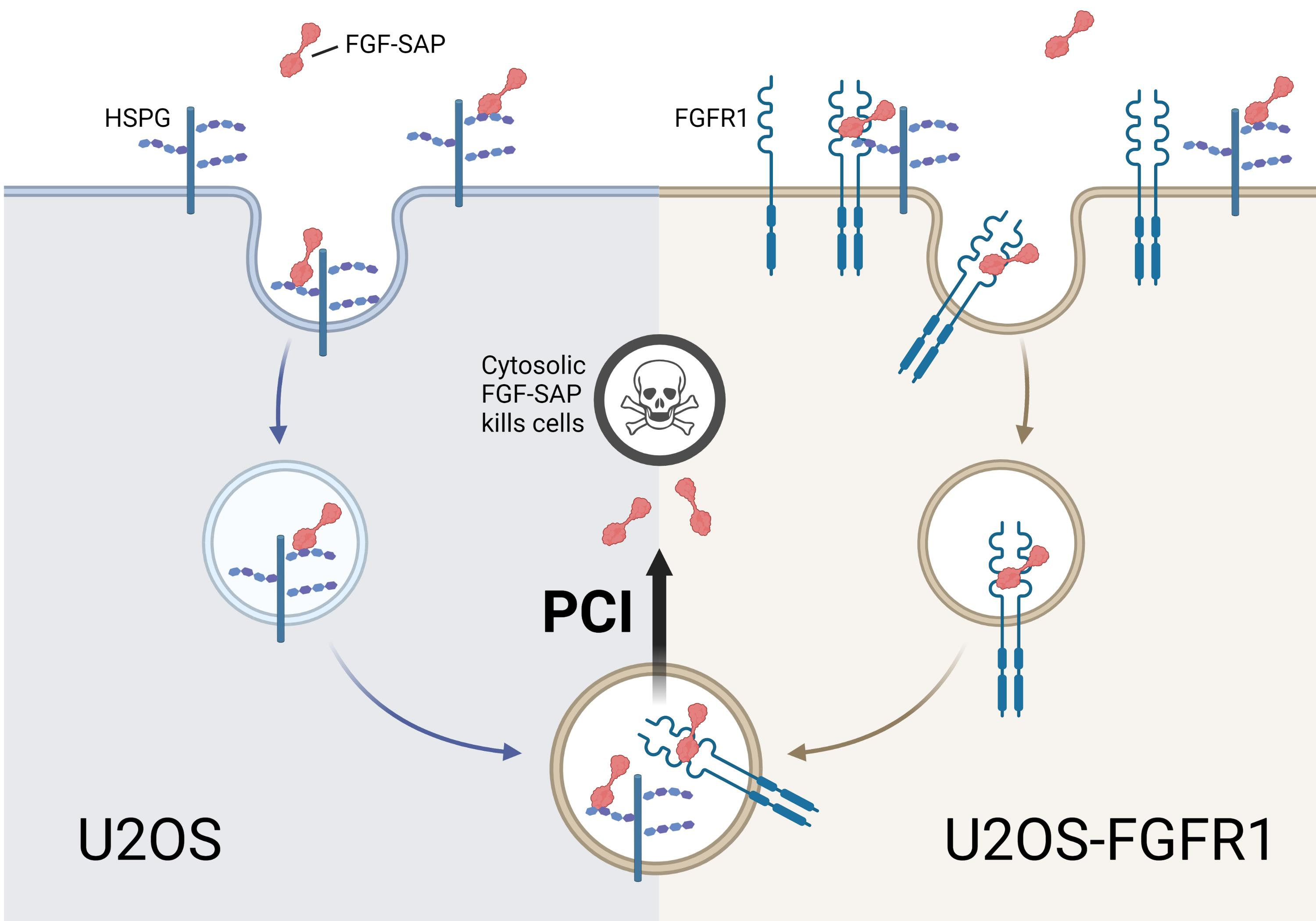{kind=link}
{kind=link}
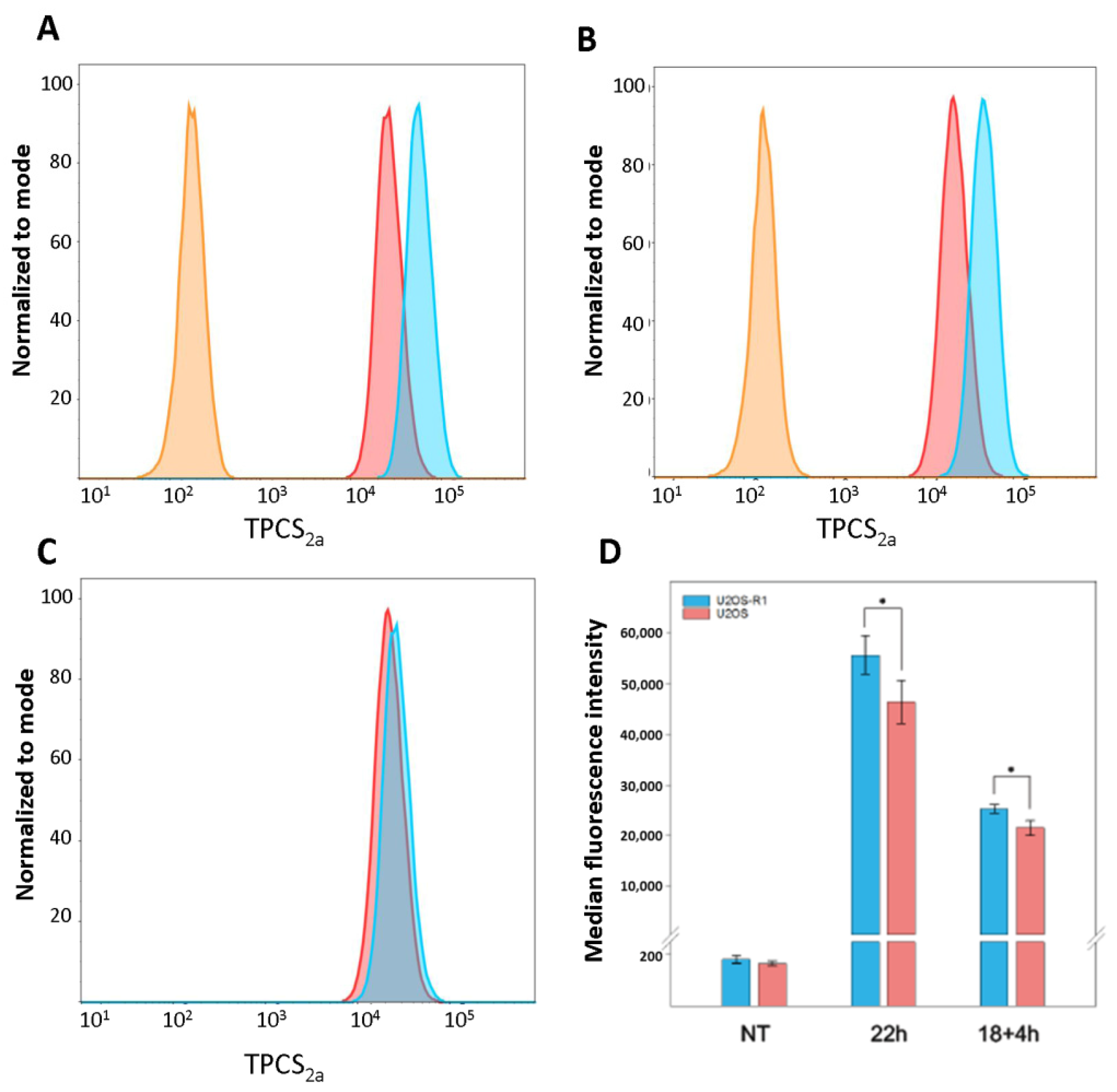{kind=link}
{kind=link}
{kind=link}
{kind=link}
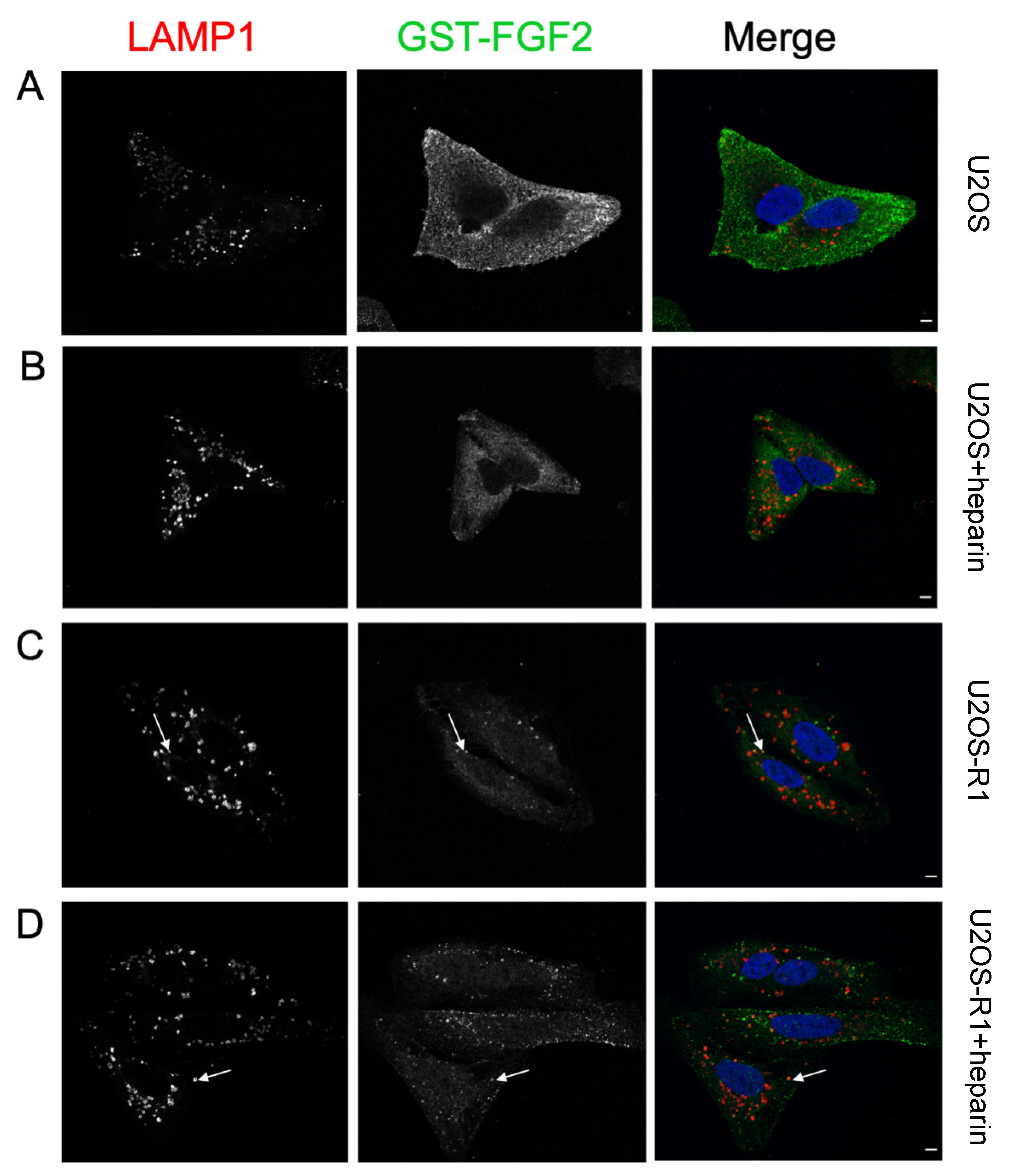{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Materials, Antibodies, and Compounds
2.3. PDT and PCI Treatment
2.4. Viability Assay
2.5. Flow Cytometry
2.6. Immunofluorescence Confocal Microscopy
2.7. Western Blotting
3. Results
3.1. Optimization of PCI Parameters
3.2. Cellular Uptake of TPCS2a
3.3. PCI of FGF-SAP
3.3.1. Cytotoxic Effects of PCI of SAP and FGF-SAP
3.3.2. Cytotoxic Effect of PCI after a Short (Four Hours) FGF-SAP Incubation
3.3.3. Co-Incubation of FGF-SAP and Heparin
3.4. Association of FGF-SAP to U2OS Cells Assessed by Immunofluorescence
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release Off. J. Control. Release Soc. 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Auguste, D.T. Cancer targeted therapeutics: From molecules to drug delivery vehicles. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Delehedde, M.; Lyon, M.; Gallagher, J.T.; Rudland, P.S.; Fernig, D.G. Fibroblast growth factor-2 binds to small heparin-derived oligosaccharides and stimulates a sustained phosphorylation of p42/44 mitogen-activated protein kinase and proliferation of rat mammary fibroblasts. Biochem. J. 2002, 366, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Wilkinson, M.C.; Fernig, D.G. The heparanome and regulation of cell function: Structures, functions and challenges. Front Biosci. 2008, 13, 4309–4338. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. MCR 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjølsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Yip, W.L.; Weyergang, A.; Berg, K.; Tønnesen, H.H.; Selbo, P.K. Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positive cancer cells. Mol. Pharm. 2007, 4, 241–251. [Google Scholar] [CrossRef]
- Berstad, M.B.; Cheung, L.H.; Berg, K.; Peng, Q.; Fremstedal, A.S.; Patzke, S.; Rosenblum, M.G.; Weyergang, A. Design of an EGFR-targeting toxin for photochemical delivery: In vitro and in vivo selectivity and efficacy. Oncogene 2015, 34, 5582–5592. [Google Scholar] [CrossRef]
- Weyergang, A.; Fremstedal, A.S.; Skarpen, E.; Peng, Q.; Mohamedali, K.A.; Eng, M.S.; Cheung, L.H.; Rosenblum, M.G.; Waltenberger, J.; Berg, K. Light-enhanced VEGF(121)/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Control. Release Off. J. Control. Release Soc. 2018, 288, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Eng, M.S.; Kaur, J.; Prasmickaite, L.; Engesæter, B.; Weyergang, A.; Skarpen, E.; Berg, K.; Rosenblum, M.G.; Mælandsmo, G.M.; Høgset, A.; et al. Enhanced targeting of triple-negative breast carcinoma and malignant melanoma by photochemical internalization of CSPG4-targeting immunotoxins. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2018, 17, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Selbo, P.K.; Rosenblum, M.G.; Cheung, L.H.; Zhang, W.; Berg, K. Multi-modality therapeutics with potent anti-tumor effects: Photochemical internalization enhances delivery of the fusion toxin scFvMEL/rGel. PLoS ONE 2009, 4, e6691. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Bostad, M.; Castro, R.; Skarpen, E.; Berg, K.; Høgset, A.; Myklebost, O.; Selbo, P.K. Photochemical internalization of CD133-targeting immunotoxins efficiently depletes sarcoma cells with stem-like properties and reduces tumorigenicity. Biochim. Biophys. Acta 2013, 1830, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Bostad, M.; Olsen, C.E.; Peng, Q.; Berg, K.; Høgset, A.; Selbo, P.K. Light-controlled endosomal escape of the novel CD133-targeting immunotoxin AC133-saporin by photochemical internalization - A minimally invasive cancer stem cell-targeting strategy. J. Control. Release Off. J. Control. Release Soc. 2015, 206, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Bull-Hansen, B.; Cao, Y.; Berg, K.; Skarpen, E.; Rosenblum, M.G.; Weyergang, A. Photochemical activation of the recombinant HER2-targeted fusion toxin MH3-B1/rGel; Impact of HER2 expression on treatment outcome. J. Control. Release Off. J. Control. Release Soc. 2014, 182, 58–66. [Google Scholar] [CrossRef]
- Lappi, D.A.; Martineau, D.; Baird, A. Biological and chemical characterization of basic FGF-saporin mitotoxin. Biochem. Biophys. Res. Commun. 1989, 160, 917–923. [Google Scholar] [CrossRef]
- Beattie, G.M.; Lappi, D.A.; Baird, A.; Hayek, A. Selective elimination of fibroblasts from pancreatic islet monolayers by basic fibroblast growth factor-saporin mitotoxin. Diabetes 1990, 39, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.G.; Davol, P.; Clark, J.W.; Kato, J.; Medina, M.; Frackelton, A.R., Jr.; Lappi, D.A.; Baird, A.; Calabresi, P. Antitumor activity of basic fibroblast growth factor-saporin mitotoxin in vitro and in vivo. Cancer Res. 1992, 52, 227–230. [Google Scholar] [PubMed]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [PubMed]
- Sletten, T.; Kostas, M.; Bober, J.; Sorensen, V.; Yadollahi, M.; Olsnes, S.; Tomala, J.; Otlewski, J.; Zakrzewska, M.; Wiedlocha, A. Nucleolin regulates phosphorylation and nuclear export of fibroblast growth factor 1 (FGF1). PLoS ONE 2014, 9, e90687. [Google Scholar] [CrossRef]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Høgset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2011, 10, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Zakrzewska, M.; Brech, A.; Pust, S.; Olsnes, S.; Sandvig, K.; Wesche, J. Clathrin- and dynamin-independent endocytosis of FGFR3--implications for signalling. PLoS ONE 2011, 6, e21708. [Google Scholar] [CrossRef]
- Berg, K.; Weyergang, A.; Prasmickaite, L.; Bonsted, A.; Høgset, A.; Strand, M.T.; Wagner, E.; Selbo, P.K. Photochemical internalization (PCI): A technology for drug delivery. Methods Mol. Biol. 2010, 635, 133–145. [Google Scholar] [CrossRef]
- Zhen, Y.; Haugsten, E.M.; Singh, S.K.; Wesche, J. Proximity labeling by a recombinant APEX2-FGF1 fusion protein reveals interaction of FGF1 with the proteoglycans CD44 and CSPG4. Biochemistry 2018, 57, 3807–3816. [Google Scholar] [CrossRef]
- Nugent, M.A.; Iozzo, R.V. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 115–120. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef]
- Quarto, N.; Amalric, F. Heparan sulfate proteoglycans as transducers of FGF-2 signalling. J. Cell Sci. 1994, 107 Pt 11, 3201–3212. [Google Scholar] [CrossRef]
- Roghani, M.; Moscatelli, D. Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. J. Biol. Chem. 1992, 267, 22156–22162. [Google Scholar] [CrossRef]
- Colin, S.; Jeanny, J.C.; Mascarelli, F.; Vienet, R.; Al-Mahmood, S.; Courtois, Y.; Labarre, J. In vivo involvement of heparan sulfate proteoglycan in the bioavailability, internalization, and catabolism of exogenous basic fibroblast growth factor. Mol. Pharmacol. 1999, 55, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, E.; Lutgens, E.; Stan, R.V.; Simons, M. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J. Cell Sci. 2004, 117, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Urbinati, C.; Presta, M. Internalization of basic fibroblast growth factor (bFGF) in cultured endothelial cells: Role of the low affinity heparin-like bFGF receptors. J. Cell Physiol. 1993, 154, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Gleizes, P.E.; Noaillac-Depeyre, J.; Amalric, F.; Gas, N. Basic fibroblast growth factor (FGF-2) internalization through the heparan sulfate proteoglycans-mediated pathway: An ultrastructural approach. Eur. J. Cell Biol. 1995, 66, 47–59. [Google Scholar]
- Crowther, M.A.; Warkentin, T.E. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: Focus on new anticoagulant agents. Blood 2008, 111, 4871–4879. [Google Scholar] [CrossRef]
- Davol, P.; Beitz, J.G.; Mohler, M.; Ying, W.; Cook, J.; Lappi, D.A.; Frackelton, A.R., Jr. Saporin toxins directed to basic fibroblast growth factor receptors effectively target human ovarian teratocarcinoma in an animal model. Cancer 1995, 76, 79–85. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vikan, A.K.; Kostas, M.; Haugsten, E.M.; Selbo, P.K.; Wesche, J. Efficacy and Selectivity of FGF2-Saporin Cytosolically Delivered by PCI in Cells Overexpressing FGFR1. Cells 2021, 10, 1476. https://doi.org/10.3390/cells10061476
Vikan AK, Kostas M, Haugsten EM, Selbo PK, Wesche J. Efficacy and Selectivity of FGF2-Saporin Cytosolically Delivered by PCI in Cells Overexpressing FGFR1. Cells. 2021; 10(6):1476. https://doi.org/10.3390/cells10061476
Chicago/Turabian StyleVikan, Aurora K., Michal Kostas, Ellen Margrethe Haugsten, Pål K. Selbo, and Jørgen Wesche. 2021. "Efficacy and Selectivity of FGF2-Saporin Cytosolically Delivered by PCI in Cells Overexpressing FGFR1" Cells 10, no. 6: 1476. https://doi.org/10.3390/cells10061476
APA StyleVikan, A. K., Kostas, M., Haugsten, E. M., Selbo, P. K., & Wesche, J. (2021). Efficacy and Selectivity of FGF2-Saporin Cytosolically Delivered by PCI in Cells Overexpressing FGFR1. Cells, 10(6), 1476. https://doi.org/10.3390/cells10061476