
Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets

 , and
, and
Abstract
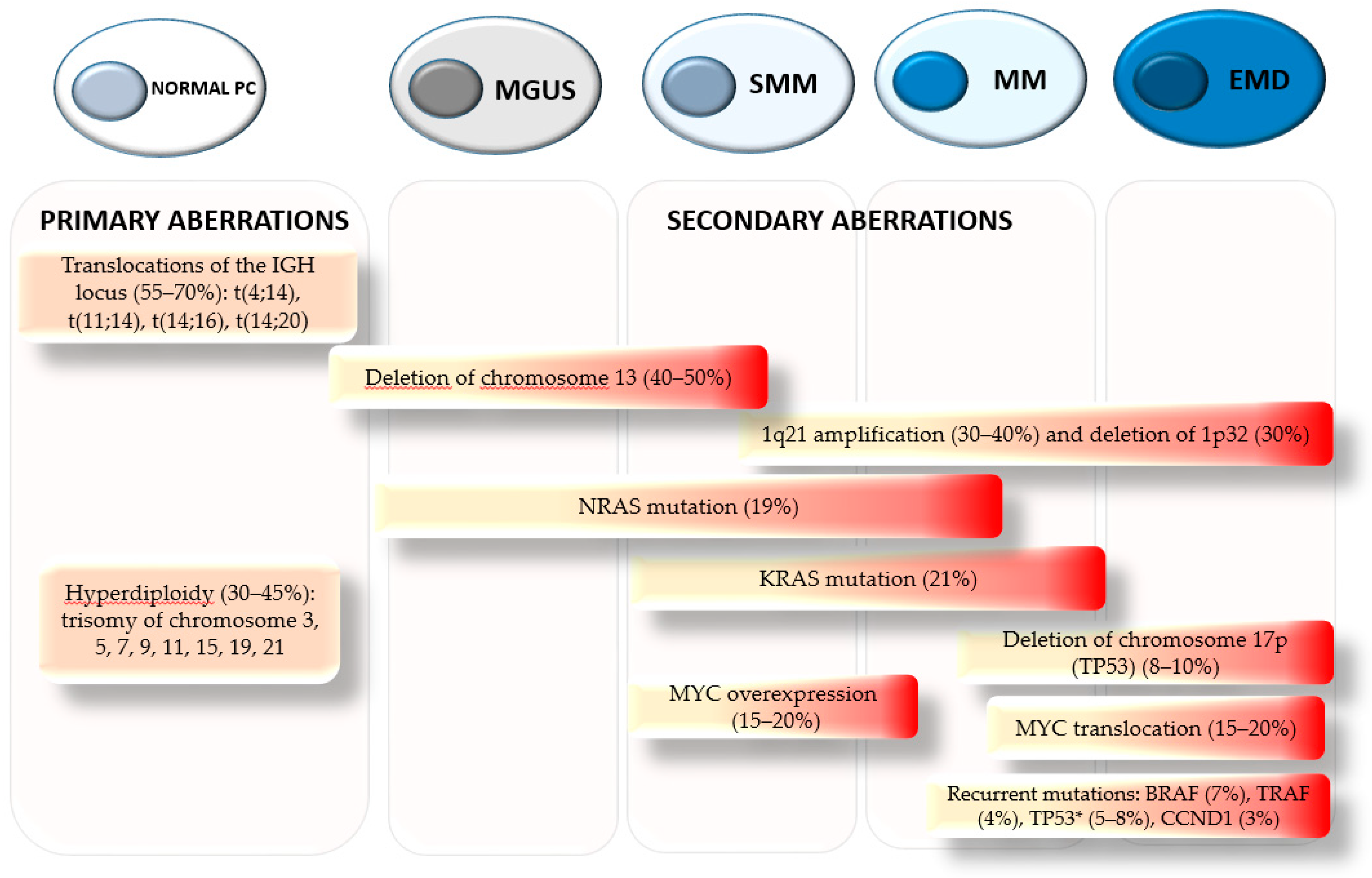
1. Introduction
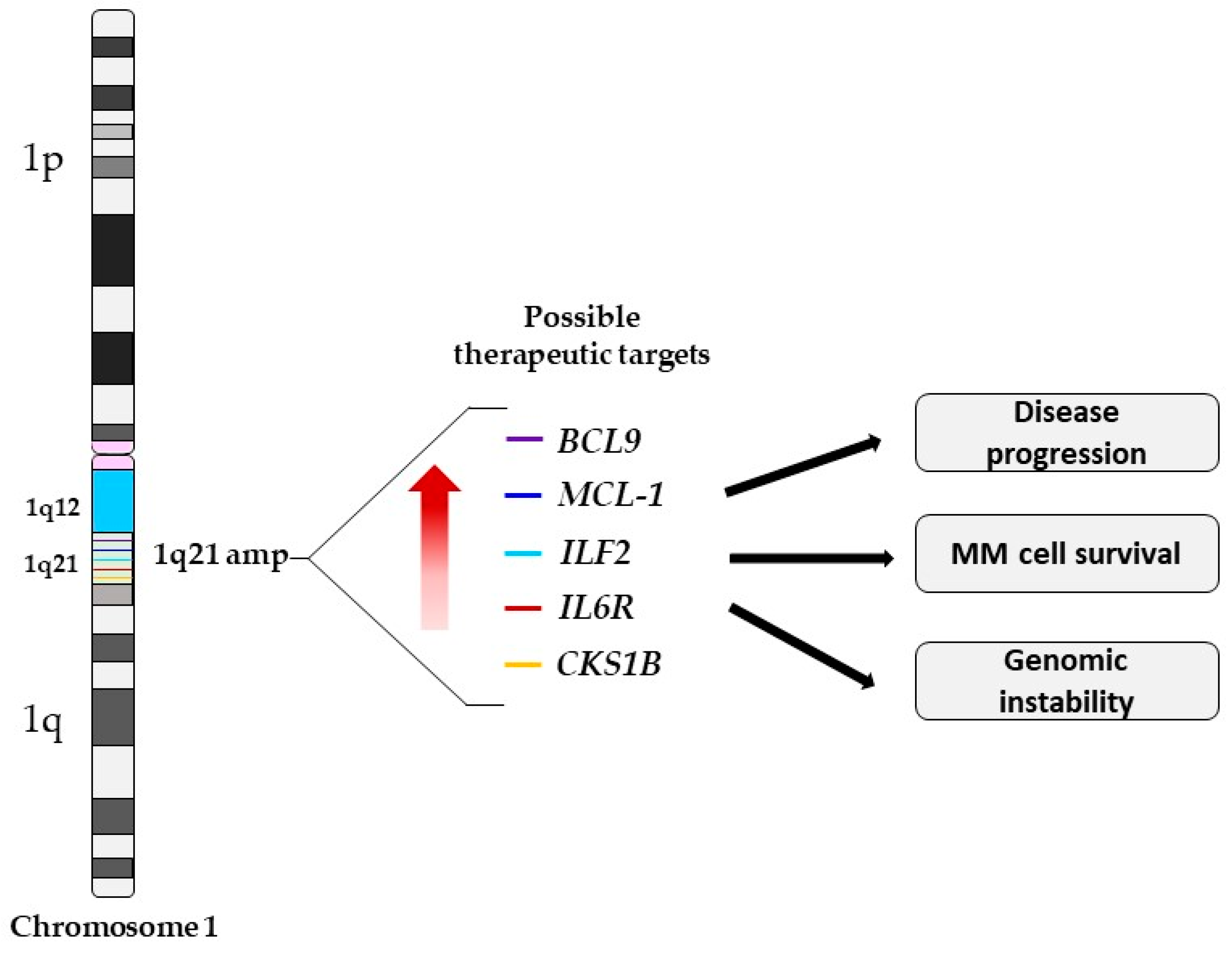
2. Pathogenesis and Clinical Implications of 1q21 Amplification
3. 1q21 Amplification and Potential Druggable Targets Genes
3.1. MCL-1
3.2. CKS1B
3.3. ILF2
3.4. IL6R
3.5. BCL9
4. Possible 1q21 Targets Genes Outside of Chromosome 1q
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavo, M.; Tacchetti, P.; Patriarca, F.; Petrucci, M.T.; Pantani, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Zamagni, E.; Palumbo, A.; et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: A randomised phase 3 study. Lancet 2010, 376, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Hillengass, J.; Usmani, S.; Rajkumar, S.V.; Durie, B.G.M.; Mateos, M.V.; Lonial, S.; Joao, C.; Anderson, K.C.; Garcia-Sanz, R.; Riva, E.; et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019, 20, e302–e312. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Attal, M.; Campion, L.; Caillot, D.; Hulin, C.; Marit, G.; Stoppa, A.M.; Voillat, L.; Wetterwald, M.; Pegourie, B.; et al. Long-term analysis of the IFM 99 trials for myeloma: Cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J. Clin. Oncol. 2012, 30, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and Validation of a Cytogenetic Prognostic Index Predicting Survival in Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Pawlyn, C.; Cairns, D.; Kaiser, M.; Striha, A.; Jones, J.; Shah, V.; Jenner, M.; Drayson, M.; Owen, R.; Gregory, W.; et al. The relative importance of factors predicting outcome for myeloma patients at different ages: Results from 3894 patients in the Myeloma XI trial. Leukemia 2020, 34, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Gerson, F.; Magrangeas, F.; Minvielle, S.; Harousseau, J.L.; Bataille, R.; Intergroupe Francophone du, M. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001, 98, 3082–3086. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Brioli, A.; Boyle, E.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Chng, W.J.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.V.; Oken, M.M.; Kyle, R.A.; Henderson, K.J.; Van Wier, S.; Greipp, P.; et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef]
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Baldini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An Italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353–21361. [Google Scholar] [CrossRef]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Baldini, L.; Trecca, D.; Cro, L.; Polli, E.; Maiolo, A.T. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood 1993, 81, 128–135. [Google Scholar] [CrossRef]
- D’Agostino, M.; Zaccaria, G.M.; Ziccheddu, B.; Rustad, E.H.; Genuardi, E.; Capra, A.; Oliva, S.; Auclair, D.; Yesil, J.; Colucci, P.; et al. Early Relapse Risk in Patients with Newly Diagnosed Multiple Myeloma Characterized by Next-generation Sequencing. Clin. Cancer Res. 2020, 26, 4832–4841. [Google Scholar] [CrossRef]
- Agnelli, L.; Bicciato, S.; Fabris, S.; Baldini, L.; Morabito, F.; Intini, D.; Verdelli, D.; Callegaro, A.; Bertoni, F.; Lambertenghi-Deliliers, G.; et al. Integrative genomic analysis reveals distinct transcriptional and genetic features associated with chromosome 13 deletion in multiple myeloma. Haematologica 2007, 92, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology 2005, 10 (Suppl. 1), 117–126. [Google Scholar] [CrossRef]
- Hanamura, I.; Stewart, J.P.; Huang, Y.; Zhan, F.; Santra, M.; Sawyer, J.R.; Hollmig, K.; Zangarri, M.; Pineda-Roman, M.; van Rhee, F.; et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: Incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood 2006, 108, 1724–1732. [Google Scholar] [CrossRef]
- Wu, K.L.; Beverloo, B.; Lokhorst, H.M.; Segeren, C.M.; van der Holt, B.; Steijaert, M.M.; Westveer, P.H.; Poddighe, P.J.; Verhoef, G.E.; Sonneveld, P.; et al. Abnormalities of chromosome 1p/q are highly associated with chromosome 13/13q deletions and are an adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Br. J. Haematol. 2007, 136, 615–623. [Google Scholar] [CrossRef]
- Barlogie, B.; Tricot, G.; Anaissie, E.; Shaughnessy, J.; Rasmussen, E.; van Rhee, F.; Fassas, A.; Zangari, M.; Hollmig, K.; Pineda-Roman, M.; et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N. Engl. J. Med. 2006, 354, 1021–1030. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Tricot, G.; Lukacs, J.L.; Binz, R.L.; Tian, E.; Barlogie, B.; Shaughnessy, J., Jr. Genomic instability in multiple myeloma: Evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer 2005, 42, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tricot, G.; Mattox, S.; Jagannath, S.; Barlogie, B. Jumping translocations of chromosome 1q in multiple myeloma: Evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood 1998, 91, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tian, E.; Heuck, C.J.; Epstein, J.; Johann, D.J.; Swanson, C.M.; Lukacs, J.L.; Johnson, M.; Binz, R.; Boast, A.; et al. Jumping translocations of 1q12 in multiple myeloma: A novel mechanism for deletion of 17p in cytogenetically defined high-risk disease. Blood 2014, 123, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tian, E.; Thomas, E.; Koller, M.; Stangeby, C.; Sammartino, G.; Goosen, L.; Swanson, C.; Binz, R.L.; Barlogie, B.; et al. Evidence for a novel mechanism for gene amplification in multiple myeloma: 1q12 pericentromeric heterochromatin mediates breakage-fusion-bridge cycles of a 1q12 approximately 23 amplicon. Br. J. Haematol. 2009, 147, 484–494. [Google Scholar] [CrossRef]
- An, G.; Li, Z.; Tai, Y.T.; Acharya, C.; Li, Q.; Qin, X.; Yi, S.; Xu, Y.; Feng, X.; Li, C.; et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin. Cancer Res. 2015, 21, 2148–2156. [Google Scholar] [CrossRef]
- Merz, M.; Hielscher, T.; Hoffmann, K.; Seckinger, A.; Hose, D.; Raab, M.S.; Hillengass, J.; Jauch, A.; Goldschmidt, H. Cytogenetic abnormalities in monoclonal gammopathy of undetermined significance. Leukemia 2018, 32, 2717–2719. [Google Scholar] [CrossRef]
- Cavo, M.; Pantani, L.; Petrucci, M.T.; Patriarca, F.; Zamagni, E.; Donnarumma, D.; Crippa, C.; Boccadoro, M.; Perrone, G.; Falcone, A.; et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood 2012, 120, 9–19. [Google Scholar] [CrossRef]
- Shaughnessy, J.D., Jr.; Qu, P.; Usmani, S.; Heuck, C.J.; Zhang, Q.; Zhou, Y.; Tian, E.; Hanamura, I.; van Rhee, F.; Anaissie, E.; et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3. Blood 2011, 118, 3512–3524. [Google Scholar] [CrossRef]
- Schmidt, T.M.; Barwick, B.G.; Joseph, N.; Heffner, L.T.; Hofmeister, C.C.; Bernal, L.; Dhodapkar, M.V.; Gupta, V.A.; Jaye, D.L.; Wu, J.; et al. Gain of Chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019, 9, 94. [Google Scholar] [CrossRef]
- D’Agostino, M.; Ruggeri, M.; Aquino, S.; Giuliani, N.; Arigoni, M.; Gentile, M.; Olivero, M.; Vincelli, I.D.; Capra, A.; Mussatto, C. Impact of Gain and Amplification of 1q in Newly Diagnosed Multiple Myeloma Patients Receiving Carfilzomib Based Treatment in the Forte Trial. Blood 2020, 136, 38–40. [Google Scholar] [CrossRef]
- Abdallah, N.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Clinical characteristics and treatment outcomes of newly diagnosed multiple myeloma with chromosome 1q abnormalities. Blood Adv. 2020, 4, 3509–3519. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yu, X.; Du, J.; Li, H.; Tang, W.; Jia, C.; Zan, Y.; Chen, M.; Zhang, Y.; Yu, M.; et al. The combination of C-Myc rearrangement and 1q21 gain is associated with poor prognosis in multiple myeloma. Ann. Hematol. 2021, 100, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Chung, T.H.; Chng, P.Y.Z.; Toh, S.H.M.; Chng, W.J. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica 2020, 105, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, M.; Ogoti, Y.; Fiorini, E.; Aktas Samur, A.; Nezi, L.; D’Anca, M.; Storti, P.; Samur, M.K.; Ganan-Gomez, I.; Fulciniti, M.T.; et al. ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell 2017, 32, 88–100. [Google Scholar] [CrossRef]
- Fonseca, R.; Van Wier, S.A.; Chng, W.J.; Ketterling, R.; Lacy, M.Q.; Dispenzieri, A.; Bergsagel, P.L.; Rajkumar, S.V.; Greipp, P.R.; Litzow, M.R.; et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006, 20, 2034–2040. [Google Scholar] [CrossRef]
- Le Gouill, S.; Podar, K.; Harousseau, J.L.; Anderson, K.C. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004, 3, 1259–1262. [Google Scholar] [CrossRef]
- Ganoth, D.; Bornstein, G.; Ko, T.K.; Larsen, B.; Tyers, M.; Pagano, M.; Hershko, A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell Biol. 2001, 3, 321–324. [Google Scholar] [CrossRef]
- Spruck, C.; Strohmaier, H.; Watson, M.; Smith, A.P.; Ryan, A.; Krek, T.W.; Reed, S.I. A CDK-independent function of mammalian Cks1: Targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol. Cell 2001, 7, 639–650. [Google Scholar] [CrossRef]
- Jones, S.A.; Horiuchi, S.; Topley, N.; Yamamoto, N.; Fuller, G.M. The soluble interleukin 6 receptor: Mechanisms of production and implications in disease. FASEB J. 2001, 15, 43–58. [Google Scholar] [CrossRef]
- Nourreddine, S.; Lavoie, G.; Paradis, J.; Ben El Kadhi, K.; Meant, A.; Aubert, L.; Grondin, B.; Gendron, P.; Chabot, B.; Bouvier, M.; et al. NF45 and NF90 Regulate Mitotic Gene Expression by Competing with Staufen-Mediated mRNA Decay. Cell Rep. 2020, 31, 107660. [Google Scholar] [CrossRef]
- Guan, D.; Altan-Bonnet, N.; Parrott, A.M.; Arrigo, C.J.; Li, Q.; Khaleduzzaman, M.; Li, H.; Lee, C.G.; Pe’ery, T.; Mathews, M.B. Nuclear factor 45 (NF45) is a regulatory subunit of complexes with NF90/110 involved in mitotic control. Mol. Cell. Biol. 2008, 28, 4629–4641. [Google Scholar] [CrossRef]
- Sustmann, C.; Flach, H.; Ebert, H.; Eastman, Q.; Grosschedl, R. Cell-type-specific function of BCL9 involves a transcriptional activation domain that synergizes with beta-catenin. Mol. Cell. Biol. 2008, 28, 3526–3537. [Google Scholar] [CrossRef]
- Mani, M.; Carrasco, D.E.; Zhang, Y.; Takada, K.; Gatt, M.E.; Dutta-Simmons, J.; Ikeda, H.; Diaz-Griffero, F.; Pena-Cruz, V.; Bertagnolli, M.; et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Bodrug, S.; Krajewska, M.; Shabaik, A.; Gascoyne, R.; Berean, K.; Reed, J.C. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am. J. Pathol. 1995, 146, 1309–1319. [Google Scholar]
- Zhuang, L.; Lee, C.S.; Scolyer, R.A.; McCarthy, S.W.; Zhang, X.D.; Thompson, J.F.; Hersey, P. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Modern Pathol. 2007, 20, 416–426. [Google Scholar] [CrossRef]
- Itoh, T.; Itoh, A.; Pleasure, D. Bcl-2-related protein family gene expression during oligodendroglial differentiation. J. Neurochem. 2003, 85, 1500–1512. [Google Scholar] [CrossRef]
- Belmar, J.; Fesik, S.W. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol. Ther. 2015, 145, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; De Vos, J.; Mechti, N.; Klein, B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: Interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. 2000, 7, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Derenne, S.; Monia, B.; Dean, N.M.; Taylor, J.K.; Rapp, M.J.; Harousseau, J.L.; Bataille, R.; Amiot, M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002, 100, 194–199. [Google Scholar] [CrossRef]
- Peperzak, V.; Vikstrom, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef]
- Slomp, A.; Moesbergen, L.M.; Gong, J.N.; Cuenca, M.; von dem Borne, P.A.; Sonneveld, P.; Huang, D.C.S.; Minnema, M.C.; Peperzak, V. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019, 3, 4202–4214. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Almasan, A. Development of venetoclax for therapy of lymphoid malignancies. Drug Des. Dev. Ther. 2017, 11, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W. Protein destruction: Adapting roles for Cks proteins. Curr. Biol. 2001, 11, R431–R435. [Google Scholar] [CrossRef]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D., Jr.; Barlogie, B. Using genomics to identify high-risk myeloma after autologous stem cell transplantation. Biol. Blood Marrow Transplant. 2006, 12, 77–80. [Google Scholar] [CrossRef]
- Chen, M.H.; Qi, C.; Reece, D.; Chang, H. Cyclin kinase subunit 1B nuclear expression predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with bortezomib. Hum. Pathol. 2012, 43, 858–864. [Google Scholar] [CrossRef]
- Shi, L.; Wang, S.; Zangari, M.; Xu, H.; Cao, T.M.; Xu, C.; Wu, Y.; Xiao, F.; Liu, Y.; Yang, Y.; et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010, 1, 22–33. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, Y.; Thomas, G.S.; Gu, Z.; Yang, Y.; Xu, H.; Tricot, G.; Zhan, F. NEDD8 Inhibition Overcomes CKS1B-Induced Drug Resistance by Upregulation of p21 in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 5532–5542. [Google Scholar] [CrossRef]
- Malek, E.; Abdel-Malek, M.A.; Jagannathan, S.; Vad, N.; Karns, R.; Jegga, A.G.; Broyl, A.; van Duin, M.; Sonneveld, P.; Cottini, F.; et al. Pharmacogenomics and chemical library screens reveal a novel SCF(SKP2) inhibitor that overcomes Bortezomib resistance in multiple myeloma. Leukemia 2017, 31, 645–653. [Google Scholar] [CrossRef]
- Tian, E.; Landowski, T.H.; Stephens, O.W.; Yaccoby, S.; Barlogie, B.; Shaughnessy, J.D., Jr. Ellipticine derivative NSC 338258 represents a potential new antineoplastic agent for the treatment of multiple myeloma. Mol. Cancer Ther. 2008, 7, 500–509. [Google Scholar] [CrossRef]
- Barille, S.; Bataille, R.; Amiot, M. The role of interleukin-6 and interleukin-6/interleukin-6 receptor-alpha complex in the pathogenesis of multiple myeloma. Eur. Cytokine Netw. 2000, 11, 546–551. [Google Scholar] [PubMed]
- Pulkki, K.; Pelliniemi, T.T.; Rajamaki, A.; Tienhaara, A.; Laakso, M.; Lahtinen, R. Soluble interleukin-6 receptor as a prognostic factor in multiple myeloma. Finnish Leukaemia Group. Br. J. Haematol. 1996, 92, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Ninomiya, H.; Hasegawa, Y.; Kobayashi, T.; Kojima, H.; Nagasawa, T.; Abe, T. Clinical significance of elevated soluble interleukin-6 receptor levels in the sera of patients with plasma cell dyscrasias. Br. J. Haematol. 1995, 91, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Min, H.J.; Park, H.K.; Oh, B.; Kim, T.Y.; She, C.J.; Hwang, S.M.; Kim, M.; Kim, H.K.; Kim, I.; et al. Increased copy number of the interleukin-6 receptor gene is associated with adverse survival in multiple myeloma patients treated with autologous stem cell transplantation. Biol. Blood Marrow Transplant. 2011, 17, 810–820. [Google Scholar] [CrossRef]
- Klein, B. Cytokine, cytokine receptors, transduction signals, and oncogenes in human multiple myeloma. Semin. Hematol. 1995, 32, 4–19. [Google Scholar] [PubMed]
- Gado, K.; Domjan, G.; Hegyesi, H.; Falus, A. Role of INTERLEUKIN-6 in the pathogenesis of multiple myeloma. Cell Biol. Int. 2000, 24, 195–209. [Google Scholar] [CrossRef]
- Matthes, T.; Manfroi, B.; Huard, B. Revisiting IL-6 antagonism in multiple myeloma. Crit. Rev. Oncol. Hematol. 2016, 105, 1–4. [Google Scholar] [CrossRef]
- Bataille, R.; Jourdan, M.; Zhang, X.G.; Klein, B. Serum levels of interleukin 6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J. Clin. Investig. 1989, 84, 2008–2011. [Google Scholar] [CrossRef]
- Juge-Morineau, N.; Francois, S.; Puthier, D.; Godard, A.; Bataille, R.; Amiot, M. The gp 130 family cytokines IL-6, LIF and OSM but not IL-11 can reverse the anti-proliferative effect of dexamethasone on human myeloma cells. Br. J. Haematol. 1995, 90, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Urashima, M.; Teoh, G.; Chauhan, D.; Hoshi, Y.; Ogata, A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. Interleukin-6 overcomes p21WAF1 upregulation and G1 growth arrest induced by dexamethasone and interferon-gamma in multiple myeloma cells. Blood 1997, 90, 279–289. [Google Scholar] [CrossRef]
- Chauhan, D.; Pandey, P.; Hideshima, T.; Treon, S.; Raje, N.; Davies, F.E.; Shima, Y.; Tai, Y.T.; Rosen, S.; Avraham, S.; et al. SHP2 mediates the protective effect of interleukin-6 against dexamethasone-induced apoptosis in multiple myeloma cells. J. Biol. Chem. 2000, 275, 27845–27850. [Google Scholar] [CrossRef]
- Song, Z.; Ren, D.; Xu, X.; Wang, Y. Molecular cross-talk of IL-6 in tumors and new progress in combined therapy. Thorac. Cancer 2018, 9, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9, 531. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Kremer, M.; Specht, K.; Calzada-Wack, J.; Nathrath, M.; Schaich, R.; Hofler, H.; Fend, F. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am. J. Pathol. 2003, 162, 1449–1461. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Demartis, A.; Bernassola, F.; Savino, R.; Melino, G.; Ciliberto, G. Interleukin 6 receptor superantagonists are potent inducers of human multiple myeloma cell death. Cancer Res. 1996, 56, 4213–4218. [Google Scholar]
- De Vos, J.; Jourdan, M.; Tarte, K.; Jasmin, C.; Klein, B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br. J. Haematol. 2000, 109, 823–828. [Google Scholar] [CrossRef]
- Lin, L.; Benson, D.M., Jr.; DeAngelis, S.; Bakan, C.E.; Li, P.K.; Li, C.; Lin, J. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 2012, 130, 1459–1469. [Google Scholar] [CrossRef]
- Scuto, A.; Krejci, P.; Popplewell, L.; Wu, J.; Wang, Y.; Kujawski, M.; Kowolik, C.; Xin, H.; Chen, L.; Wang, Y.; et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia 2011, 25, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, E.; Mondala, P.K.; Santos, N.D.; Miller, A.C.; Pineda, G.; Jiang, Q.; Leu, H.; Ali, S.A.; Ganesan, A.P.; Wu, C.N.; et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 2017, 8, 1922. [Google Scholar] [CrossRef]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt signaling in multiple myeloma: Molecular mechanisms and targeting options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted disruption of the BCL9/beta-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Pichiorri, F.; Suh, S.S.; Rocci, A.; De Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M., Jr.; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010, 18, 367–381. [Google Scholar] [CrossRef]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef]
- Lionetti, M.; Agnelli, L.; Lombardi, L.; Tassone, P.; Neri, A. MicroRNAs in the pathobiology of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Lin, J.; Zhu, D.; Wang, X.; Brooks, D.; Chen, M.; Chu, Z.B.; Takada, K.; Ciccarelli, B.; Admin, S.; et al. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/beta-catenin/BCL9 pathway. Cancer Res. 2014, 74, 1801–1813. [Google Scholar] [CrossRef]
- Zhan, F.; Colla, S.; Wu, X.; Chen, B.; Stewart, J.P.; Kuehl, W.M.; Barlogie, B.; Shaughnessy, J.D., Jr. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood 2007, 109, 4995–5001. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Jiang, N.; Jiang, H.; Saha, M.N.; Qi, C.; Xu, W.; Reece, D. CKS1B nuclear expression is inversely correlated with p27Kip1 expression and is predictive of an adverse survival in patients with multiple myeloma. Haematologica 2010, 95, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.A.; Ackley, J.; Kaufman, J.L.; Boise, L.H. BCL2 Family Inhibitors in the Biology and Treatment of Multiple Myeloma. Blood Lymphat. Cancer Targets Ther. 2021, 11, 11–24. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, R.; He, X.; Liu, X.; Zhang, Y.; Yang, Z.; Yang, P.; Wang, J.; Hu, K.; Yuan, X.; et al. Clinical prognostic implications of EPB41L4A expression in multiple myeloma. J. Cancer 2020, 11, 619–629. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas—Current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Mohty, B.; El-Cheikh, J.; Yakoub-Agha, I.; Avet-Loiseau, H.; Moreau, P.; Mohty, M. Treatment strategies in relapsed and refractory multiple myeloma: A focus on drug sequencing and ‘retreatment’ approaches in the era of novel agents. Leukemia 2012, 26, 73–85. [Google Scholar] [CrossRef]
- D’Agostino Mattia, M.; Lahuerta, J.-J.; Wester, R.; Waage, A.; Bertsch, U.; Zamagni, E.; Mateos, M.-V.; Larocca, A.; Dall’Olio, D.; van de Donk, N.W.C.J.; et al. A New Risk Stratification Model (R2-ISS) in Newly Diagnosed Multiple Myeloma: Analysis of Mature Data from 7077 Patients Collected By European Myeloma Network within Harmony Big Data Platform. Blood 2020, 136, 34–37. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Candidate Genes | Chromosomal Location | Biological Function | Function of Overexpressed Gene in MM Cells with 1q21 | References |
|---|---|---|---|---|
| MCL-1 | 1q21.2 | Anti-apoptotic | Apoptosis resistance, disease progression, and shorter OS | [35,38] |
| CSK1B | 1q21.3 | Cell division | Regulation of the p27 proteasome degradation | [39,40] |
| IL6R | 1q21.3 | IL6 regulator | Increased IL6 sensitivity and hyper-activation of the STAT3 pathway | [34,41] |
| ILF2 | 1q21.3 | mRNA stability, mRNA post-transcriptional regulation, and in mitotic control. | Promotes tolerance of genomic instability, enhancing MM cell survival and drug resistance | [36,42,43] |
| BCL9 | 1q21.2 | T ranscriptional co-activator of the Wnt/β-catenin pathway | Promotes cell proliferation? | [44,45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burroughs Garcìa, J.; Eufemiese, R.A.; Storti, P.; Sammarelli, G.; Craviotto, L.; Todaro, G.; Toscani, D.; Marchica, V.; Giuliani, N. Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets. Cells 2021, 10, 1360. https://doi.org/10.3390/cells10061360
Burroughs Garcìa J, Eufemiese RA, Storti P, Sammarelli G, Craviotto L, Todaro G, Toscani D, Marchica V, Giuliani N. Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets. Cells. 2021; 10(6):1360. https://doi.org/10.3390/cells10061360
Chicago/Turabian StyleBurroughs Garcìa, Jessica, Rosa Alba Eufemiese, Paola Storti, Gabriella Sammarelli, Luisa Craviotto, Giannalisa Todaro, Denise Toscani, Valentina Marchica, and Nicola Giuliani. 2021. "Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets" Cells 10, no. 6: 1360. https://doi.org/10.3390/cells10061360
APA StyleBurroughs Garcìa, J., Eufemiese, R. A., Storti, P., Sammarelli, G., Craviotto, L., Todaro, G., Toscani, D., Marchica, V., & Giuliani, N. (2021). Role of 1q21 in Multiple Myeloma: From Pathogenesis to Possible Therapeutic Targets. Cells, 10(6), 1360. https://doi.org/10.3390/cells10061360