
Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
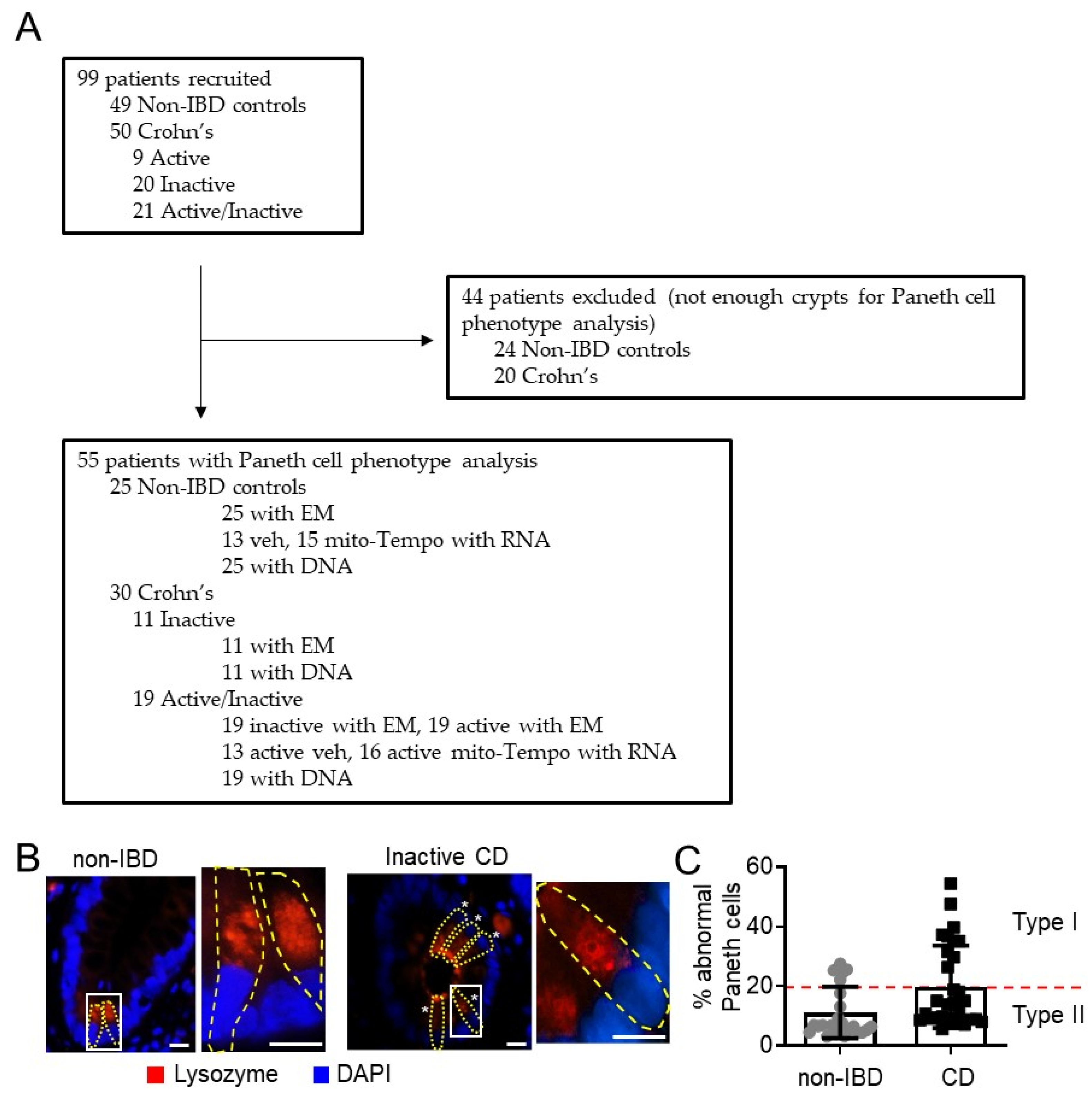
2.2. Paneth Cell Phenotype Analysis—Immunofluorescent Staining of Lysozyme and Quantitation of Lysozyme Granule Allocation Pattern
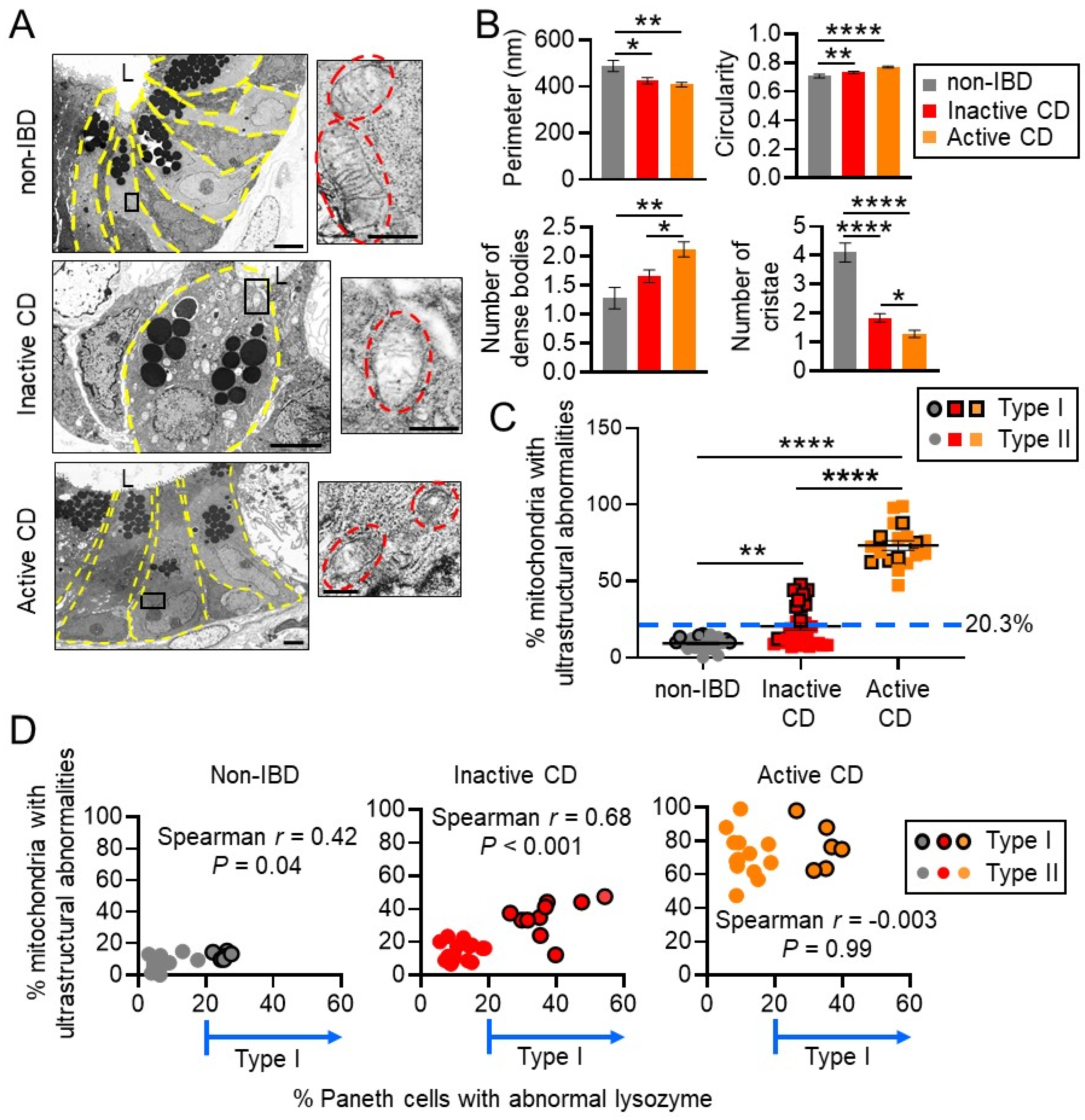
2.3. Transmission Electron Microscopy (TEM)
2.4. CD Risk Allele Analysis
2.5. Ex Vivo Mito-Tempo Treatment
2.6. Global RNA Sequencing (RNA-Seq)
2.7. Quantitative Real-Time PCR Analysis
2.8. Statistics
3. Results
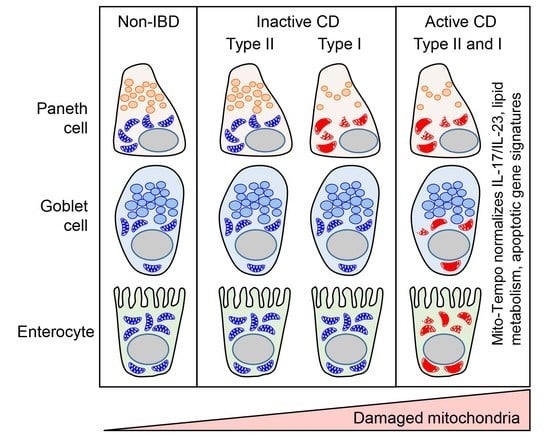
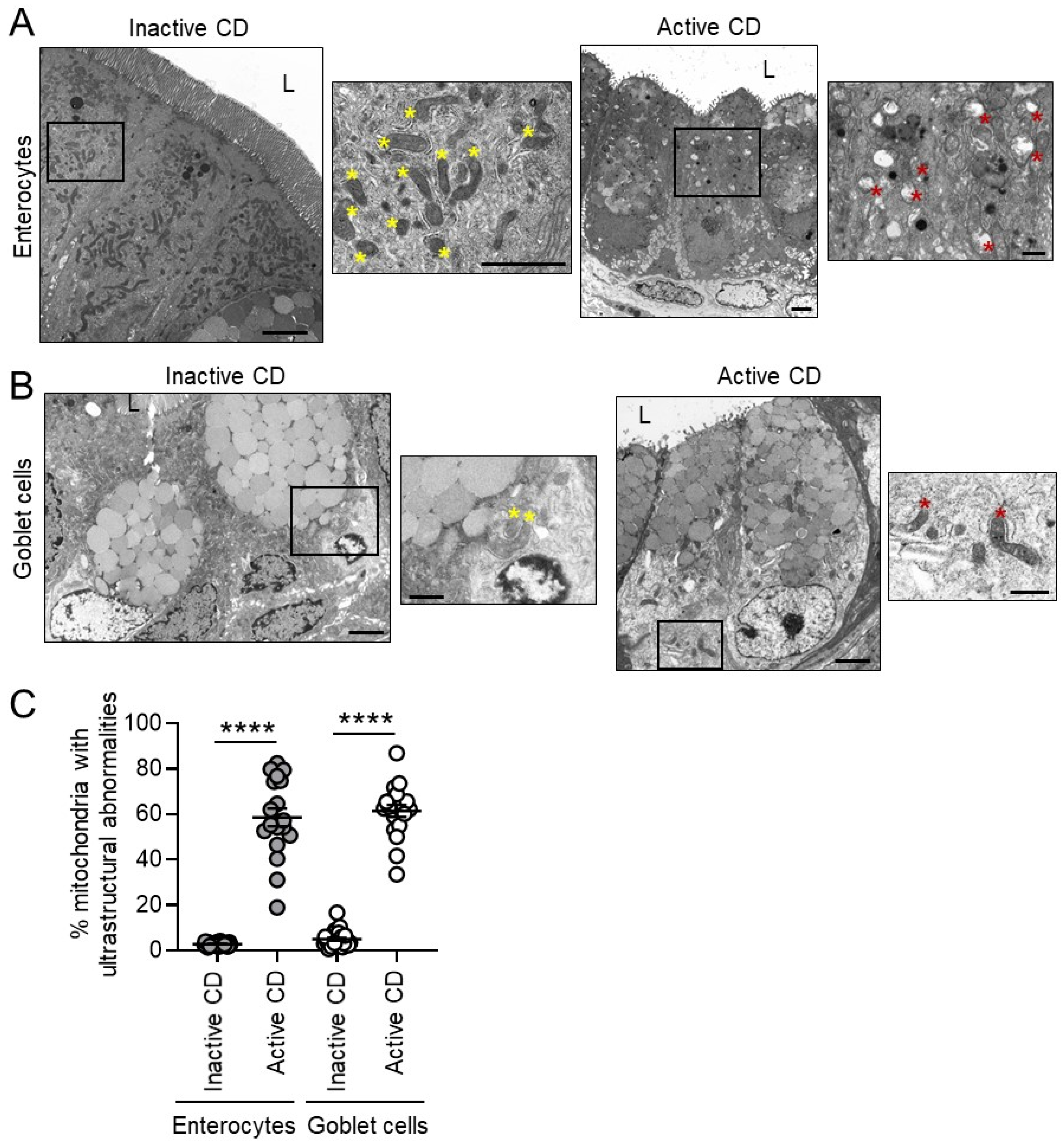
3.1. Mitochondria Exhibit Damage in Paneth Cells, Goblet Cells, and Enterocytes during Active CD and in Paneth Cells of Type I CD Patients Independent of Inflammation
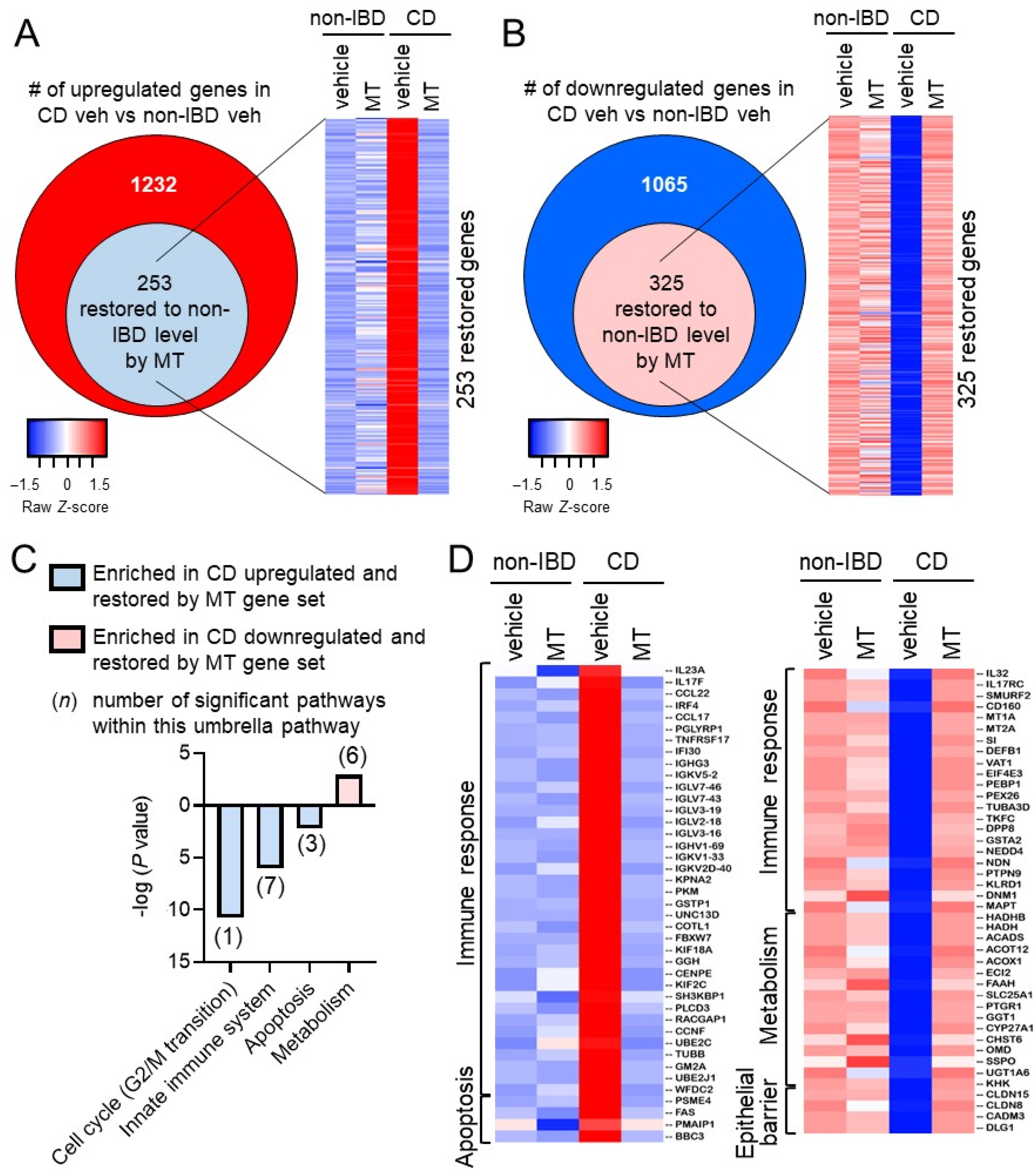
3.2. Mito-Tempo, a Mitochondrial-Targeted Therapeutic, Restores Altered CD Genes Involved in Immune Response, Apoptotic, and Metabolism Pathways to Non-IBD Expression Levels
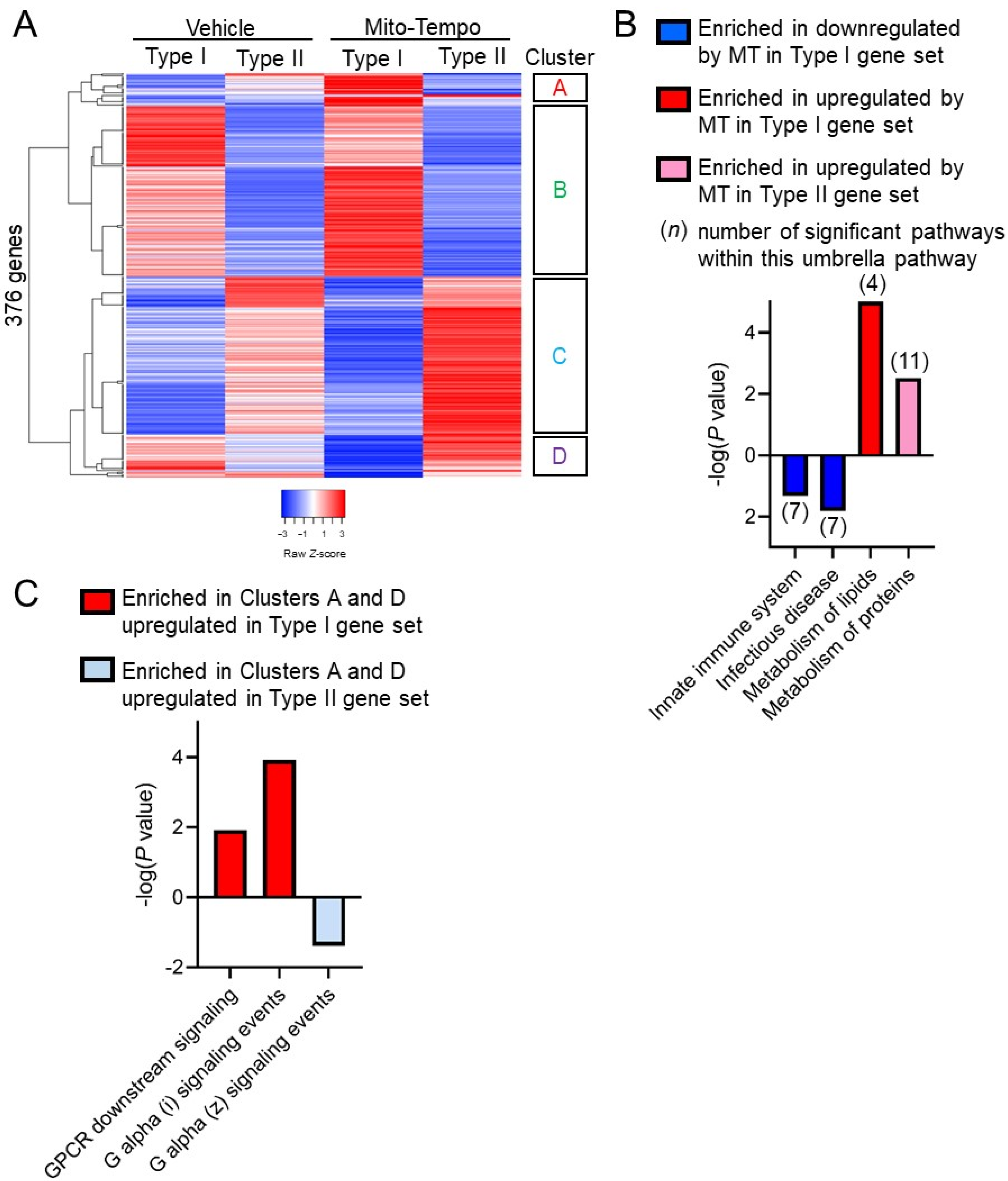
3.3. Paneth Cell Phenotype Is Associated with Unique Mito-Tempo-Induced Gene Signatures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. Mon. 2018, 64, 20–57. [Google Scholar] [CrossRef]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Liu, T.C.; Li, D.; Towfic, F.; Modiano, N.; Winter, R.; Haritunians, T.; Taylor, K.D.; Dhall, D.; Targan, S.R.; et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 2014, 146, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Liu, T.C.; Gao, F.; McGovern, D.P.; Stappenbeck, T.S. Spatial and temporal stability of paneth cell phenotypes in Crohn’s disease: Implications for prognostic cellular biomarker development. Inflamm. Bowel Dis. 2014, 20, 646–651. [Google Scholar] [CrossRef]
- Liu, T.C.; Gurram, B.; Baldridge, M.T.; Head, R.; Lam, V.; Luo, C.; Cao, Y.; Simpson, P.; Hayward, M.; Holtz, M.L.; et al. Paneth cell defects in Crohn’s disease patients promote dysbiosis. JCI Insight 2016, 1, e86907. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Naito, T.; Liu, Z.; VanDussen, K.L.; Haritunians, T.; Li, D.; Endo, K.; Kawai, Y.; Nagasaki, M.; Kinouchi, Y.; et al. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight 2017, 2, e91917. [Google Scholar] [CrossRef]
- Stappenbeck, T.S.; McGovern, D.P.B. Paneth Cell Alterations in the Development and Phenotype of Crohn’s Disease. Gastroenterology 2017, 152, 322–326. [Google Scholar] [CrossRef]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020, 69, 1928–1938. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef]
- Rath, E.; Haller, D. Mitochondria at the interface between danger signaling and metabolism: Role of unfolded protein responses in chronic inflammation. Inflamm. Bowel Dis. 2012, 18, 1364–1377. [Google Scholar] [CrossRef]
- Ho, G.T.; Aird, R.E.; Liu, B.; Boyapati, R.K.; Kennedy, N.A.; Dorward, D.A.; Noble, C.L.; Shimizu, T.; Carter, R.N.; Chew, E.T.S.; et al. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 2018, 11, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Daperno, M.; D’Haens, G.; Van Assche, G.; Baert, F.; Bulois, P.; Maunoury, V.; Sostegni, R.; Rocca, R.; Pera, A.; Gevers, A.; et al. Development and validation of a new, simplified endoscopic activity score for Crohn’s disease: The SES-CD. Gastrointest Endosc. 2004, 60, 505–512. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef]
- Liu, T.C.; Kern, J.T.; VanDussen, K.L.; Xiong, S.; Kaiko, G.E.; Wilen, C.B.; Rajala, M.W.; Caruso, R.; Holtzman, M.J.; Gao, F.; et al. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J. Clin. Investig. 2018, 128, 5110–5122. [Google Scholar] [CrossRef]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef]
- Fuller, M.K.; Faulk, D.M.; Sundaram, N.; Mahe, M.M.; Stout, K.M.; von Furstenberg, R.J.; Smith, B.J.; McNaughton, K.K.; Shroyer, N.F.; Helmrath, M.A.; et al. Intestinal stem cells remain viable after prolonged tissue storage. Cell Tissue Res. 2013, 354, 441–450. [Google Scholar] [CrossRef][Green Version]
- Heiseke, A.F.; Faul, A.C.; Lehr, H.A.; Forster, I.; Schmid, R.M.; Krug, A.B.; Reindl, W. CCL17 promotes intestinal inflammation in mice and counteracts regulatory T cell-mediated protection from colitis. Gastroenterology 2012, 142, 335–345. [Google Scholar] [CrossRef]
- Dornhoff, H.; Becker, C.; Wirtz, S.; Strand, D.; Tenzer, S.; Rosfa, S.; Neufert, C.; Mudter, J.; Markl, J.; Siebler, J.; et al. A variant of Smurf2 protects mice against colitis-associated colon cancer by inducing transforming growth factor beta signaling. Gastroenterology 2012, 142, 1183–1194.e4. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.-Y.; Shui, J.-R.; Mikulski, R.; Wang, Q.; Takahashi, D.; Giles, D.A.; Iwaya, H.; Sethi, A.; Kim, P.-H.; Cheroutre, H.; et al. CD160-HVEM signaling in intestinal epithelial cells modulates gut microbial homeostasis. J. Immunol 2019, 202, 191.11. [Google Scholar]
- Stange, E.F.; Schroeder, B.O. Microbiota and mucosal defense in IBD: An update. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Waeytens, A.; De Vos, M.; Laukens, D. Evidence for a potential role of metallothioneins in inflammatory bowel diseases. Mediators Inflamm. 2009, 2009, 729172. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Shu-Hong, Y.; Qiao, M. Expression of DLG1 and DLG5 in the Intestinal Epithelium of Patients with CD. Inflamm. Bowel Dis 2016, 22, E43–E44. [Google Scholar] [CrossRef] [PubMed]
- Darsigny, M.; Babeu, J.P.; Dupuis, A.A.; Furth, E.E.; Seidman, E.G.; Levy, E.; Verdu, E.F.; Gendron, F.P.; Boudreau, F. Loss of hepatocyte-nuclear-factor-4alpha affects colonic ion transport and causes chronic inflammation resembling inflammatory bowel disease in mice. PLoS ONE 2009, 4, e7609. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, K.; Kishi, H.; Komatsu, W.; Nagao, M.; Ohhira, S.; Kobashi, G. Lipid and Bile Acid Dysmetabolism in Crohn’s Disease. J. Immunol. Res. 2018, 2018, 7270486. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Chu, F.F.; Esworthy, R.S.; Doroshow, J.H.; Grasberger, H.; Donko, A.; Leto, T.L.; Gao, Q.; Shen, B. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 2017, 11, 144–156. [Google Scholar] [CrossRef]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef]
- Wang, A.; Keita, A.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.E.; Vincent, G.; Sodhi, C.P.; Novak, E.A.; Ranganathan, S.; Egan, C.E.; Stolz, D.B.; Rogers, M.B.; Firek, B.; Morowitz, M.J.; et al. Peroxisome Proliferator-activated Receptor-gamma Coactivator 1-alpha (PGC1alpha) Protects against Experimental Murine Colitis. J. Biol. Chem. 2016, 291, 10184–10200. [Google Scholar] [CrossRef]
- Matsue, H.; Edelbaum, D.; Shalhevet, D.; Mizumoto, N.; Yang, C.; Mummert, M.E.; Oeda, J.; Masayasu, H.; Takashima, A. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J. Immunol. 2003, 171, 3010–3018. [Google Scholar] [CrossRef]
- Kotsias, F.; Hoffmann, E.; Amigorena, S.; Savina, A. Reactive oxygen species production in the phagosome: Impact on antigen presentation in dendritic cells. Antioxid Redox Signal 2013, 18, 714–729. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, B.H.; Kim, K.S.; Jang, Y.S. Complement C5a promotes antigen cross-presentation by Peyer’s patch monocyte-derived dendritic cells and drives a protective CD8(+) T cell response. Cell Rep. 2021, 35, 108995. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.; Diehl, L. Dendritic cells in IBD pathogenesis: An area of therapeutic opportunity? J. Pathol. 2014, 232, 112–120. [Google Scholar] [CrossRef]
- Giuffrida, P.; Di Sabatino, A. Targeting T cells in inflammatory bowel disease. Pharmacol. Res. 2020, 159, 105040. [Google Scholar] [CrossRef]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef]
- A SPECIAL MEETING REVIEW EDITION: Highlights in Inflammatory Bowel Disease From the 14th Congress of ECCO: A Review of Selected Presentations From the 14th Congress of the European Crohn’s and Colitis Organisation (ECCO) March 6-9, 2019 Copenhagen, Denmark Special Reporting on: VARSITY: A Double-Blind, Double-Dummy, Randomized Controlled Trial of Vedolizumab Versus Adalimumab in Patients With Active Ulcerative Colitis Analyses of Data From the VISIBLE 1 and 2 Trials: Vedolizumab in Patients With Ulcerative Colitis or Crohn’s Disease Improved Endoscopic Outcomes and Mucosal Healing of Upadacitinib as an Induction Therapy in Adults With Moderately to Severely Active Ulcerative Colitis: Data From the U-ACHIEVE Study Long-Term Safety of Vedolizumab in Ulcerative Colitis and Crohn’s Disease: Final Results From the GEMINI LTS Study Pediatric Crohn’s Disease Adalimumab Level-Based Optimization Treatment (PAILOT) Randomized Controlled Trial Maintenance Treatment With Mirikizumab, a P19-Directed IL-23 Antibody: 52-Week Results in Patients With Moderately to Severely Active Ulcerative Colitis Real-World Effectiveness and Safety of Vedolizumab and Anti-TNF Therapy in Biologic-Naive Patients With Ulcerative Colitis or Crohn’s Disease: Results From the EVOLVE Study A Randomized, Multicenter, Double-Blind, Placebo-Controlled Study of a Targeted-Release Oral Cyclosporine Formulation in the Treatment of Mild to Moderate Ulcerative Colitis: Efficacy Results Real-World Analyses of Patients With IBD Treated With Vedolizumab PLUS Meeting Abstract Summaries With Expert Commentary by: Edward V. Loftus Jr, MD Professor of MedicineDivision of Gastroenterology and HepatologyMayo ClinicRochester, Minnesota. Gastroenterol. Hepatol. 2019, 15, 1–24.
- Medan, D.; Wang, L.; Toledo, D.; Lu, B.; Stehlik, C.; Jiang, B.H.; Shi, X.; Rojanasakul, Y. Regulation of Fas (CD95)-induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J. Cell Physiol. 2005, 203, 78–84. [Google Scholar] [CrossRef]
- Guikema, J.E.; Amiot, M.; Eldering, E. Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer. Expert Opin. Ther. Targets 2017, 21, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Low, I.C.; Pervaiz, S. Regulation of mitochondrial metabolism: Yet another facet in the biology of the oncoprotein Bcl-2. Biochem. J. 2011, 435, 545–551. [Google Scholar] [CrossRef]
- Macaluso, F.S.; Orlando, A.; Cottone, M. Anti-interleukin-12 and anti-interleukin-23 agents in Crohn’s disease. Expert Opin. Biol. Ther. 2019, 19, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 1089–1090. [Google Scholar] [CrossRef]
- Zhao, C.; Gu, Y.; Zeng, X.; Wang, J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin. Immunol. 2018, 197, 154–160. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.M.; Riestra, A.M.; Ali, S.R.; Fong, J.J.; Liu, J.Z.; Hughes, G.; Varki, A.; Nizet, V. Siglec-14 Enhances NLRP3-Inflammasome Activation in Macrophages. J. Innate Immun. 2020, 12, 333–343. [Google Scholar] [CrossRef]
- Melhem, H.; Kaya, B.; Ayata, C.K.; Hruz, P.; Niess, J.H. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells 2019, 8, 450. [Google Scholar] [CrossRef]
- Mashima, H.; Watanabe, N.; Sekine, M.; Matsumoto, S.; Asano, T.; Yuhashi, K.; Sagihara, N.; Urayoshi, S.; Uehara, T.; Fujiwara, J.; et al. The role of Galphaq/Galpha11 signaling in intestinal epithelial cells. Biochem. Biophys. Rep. 2018, 13, 93–98. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W.t.; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Non-IBD Control | CD | p-Value CD vs. Non-IBD |
|---|---|---|---|
| Number of patients | 25 | 30 | |
| Gender, n (%) | |||
| Male | 11 (44) | 19 (63) | 0.11 |
| Female | 14 (56) | 11 (37) | |
| Median age (range) | 61 (25–80) | 49 (20–71) | 0.0002 |
| Race, n (%) | |||
| Caucasian | 17 (68) | 24 (80) | 0.36 |
| African American | 8 (32) | 3 (10) | 0.09 |
| Asian | 0 | 1 (3) | 0.99 |
| Hispanic | 0 | 2 (6) | 0.50 |
| Harvey Bradshaw Index Score | N/A | 0–22 | |
| IBD treatment at biopsy, n (%) | |||
| 5-ASA | 5 (17) | ||
| 6-MP/AZA/MTX | 6 (20) | ||
| Biologics a | 17 (56) | ||
| Steroids | 6 (20) | ||
| Antibiotics | 0 | ||
| NSAIDS | 4 (13) | ||
| No treatment | 5 (17) | ||
| Tobacco use b, n (%) | |||
| Smoker | 1 (4) | 3 (10) | 0.99 |
| Non-smoker | 17 (68) | 26 (87) | 0.07 |
| Other conditions, n (%) | |||
| Coronary artery disease | 2 (8) | 1 (3) | 0.59 |
| Diabetes mellitus | 3 (12) | 4 (13) | 0.99 |
| Hypertension | 10 (40) | 9 (30) | 0.78 |
| COPD | 1 (4) | 0 | 0.45 |
| Congestive heart failure | 2 (8) | 1 (3) | 0.59 |
| Cerebrovascular event | 0 | 3 (10) | 0.24 |
| Peripheral vascular dis. | 1 (4) | 0 | 0.45 |
| Ankylosing spondylitis | 0 | 4 (13) | 0.12 |
| Rheumatoid Arthritis | 2 (8) | 1 (3) | 0.59 |
| Psoriasis | 0 | 1 (3) | 0.99 |
| Spondyloarthropathy | 0 | 2 (7) | 0.49 |
| Characteristic | Normal Mitochondria | Abnormal Mitochondria | p-Value Normal vs. Abnormal Mitochondria |
|---|---|---|---|
| Number of patients | 19 | 11 | |
| Gender, n (%) | |||
| Male | 14 (74) | 6 (54) | 0.24 |
| Female | 5 (26) | 5 (46) | |
| Age, n (%) | |||
| <40 | 8 (42) | 3 (27) | 0.47 |
| <50 | 10 (53) | 6 (54) | 0.99 |
| <60 | 14 (74) | 8 (72) | 0.99 |
| Race, n (%) | |||
| Caucasian | 16 (84) | 8 (72) | 0.64 |
| African American | 2 (11) | 1 (9) | 0.99 |
| Asian | 0 | 1 (9) | 0.37 |
| Hispanic | 1 (5) | 1 (9) | 0.99 |
| Harvey Bradshaw Index Score, n (%) | |||
| <5 | 11 (58) | 9 (82) | 0.25 |
| 5 to 7 | 5 (26) | 2 (18) | 0.99 |
| 8 to 16 | 1 (5) | 0 | 0.99 |
| >16 | 1 (5) | 0 | 0.99 |
| Endoscopically active disease present, n (%) | 11 (58) | 5 (46) | 0.71 |
| IBD treatment at biopsy, n (%) | |||
| 5-ASA | 3 (16) | 2 (18) | 0.99 |
| 6MP/AZA/MTX | 4 (21) | 2 (18) | 0.99 |
| Biologics | 10 (53) | 7 (64) | 0.71 |
| Steroids | 2 (11) | 4 (36) | 0.16 |
| NSAIDS | 1 (5) | 3 (27) | 0.13 |
| No treatment | 3 (16) | 2 (18) | 0.99 |
| Smoking, n (%) | 1 (5) | 2 (18) | 0.54 |
| Type I Paneth cell phenotype, n (%) | 1 (5) | 9 (82) | <0.0001 |
| Other conditions, n (%) a | |||
| Diabetes mellitus | 1 (5) | 3 (27) | 0.13 |
| Hypertension | 4 (21) | 5 (46) | 0.23 |
| Cerebrovascular event | 1 (5) | 2 (18) | 0.54 |
| Ankylosing spondylitis | 2 (11) | 2 (18) | 0.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alula, K.M.; Jackson, D.N.; Smith, A.D.; Kim, D.S.; Turner, K.; Odstrcil, E.; Kaipparettu, B.A.; Dassopoulos, T.; Venuprasad, K.; Feagins, L.A.; et al. Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease. Cells 2021, 10, 1349. https://doi.org/10.3390/cells10061349
Alula KM, Jackson DN, Smith AD, Kim DS, Turner K, Odstrcil E, Kaipparettu BA, Dassopoulos T, Venuprasad K, Feagins LA, et al. Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease. Cells. 2021; 10(6):1349. https://doi.org/10.3390/cells10061349
Chicago/Turabian StyleAlula, Kibrom M., Dakota N. Jackson, Andrew D. Smith, Daniel S. Kim, Kevin Turner, Elizabeth Odstrcil, Benny A. Kaipparettu, Themistocles Dassopoulos, K. Venuprasad, Linda A. Feagins, and et al. 2021. "Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease" Cells 10, no. 6: 1349. https://doi.org/10.3390/cells10061349
APA StyleAlula, K. M., Jackson, D. N., Smith, A. D., Kim, D. S., Turner, K., Odstrcil, E., Kaipparettu, B. A., Dassopoulos, T., Venuprasad, K., Feagins, L. A., & Theiss, A. L. (2021). Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease. Cells, 10(6), 1349. https://doi.org/10.3390/cells10061349