
Phagocytic Glial Cells in Brain Homeostasis

{kind=link}
{kind=link}
Abstract
1. Introduction
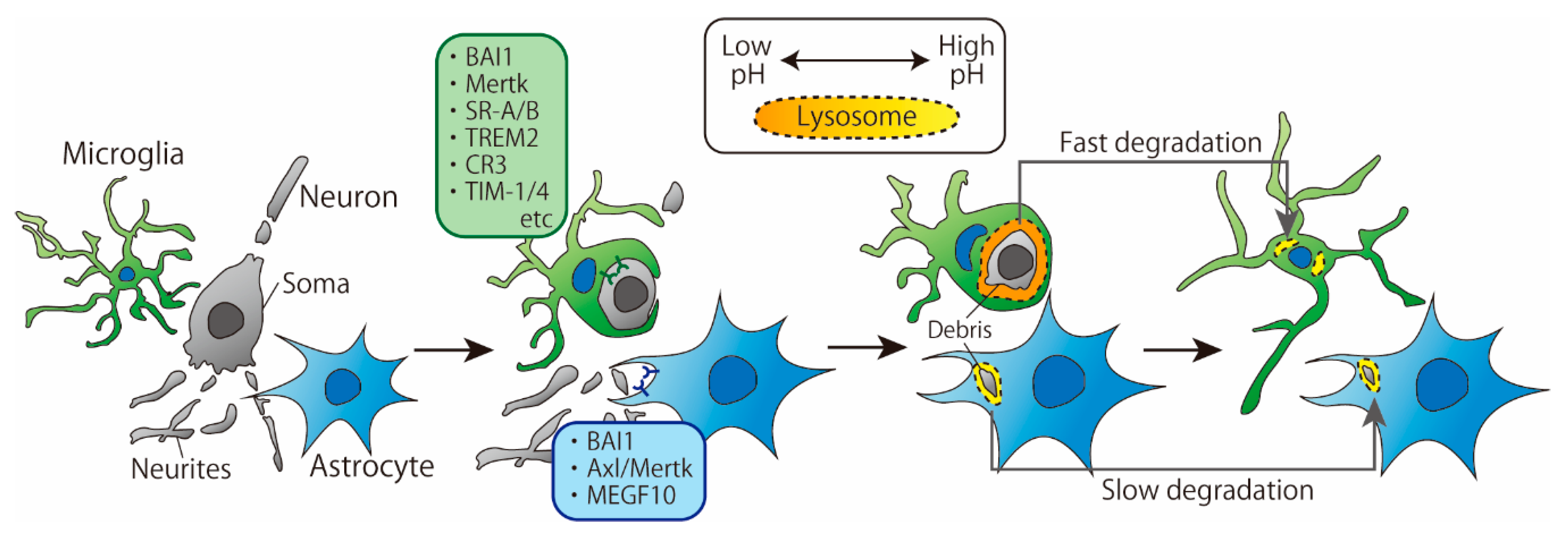
2. Phagocytosis of Cell Debris
2.1. Recognition by Receptors
2.2. Degradation
2.3. The Fate of Both Phagocytes and Phagocytosed Targets after Ingestion
3. Phagocytosis of Amyloid-Beta (Aβ)
3.1. Recognition by Receptors
3.2. Degradation
3.3. The Fate of Both Phagocytes and Phagocytosed Targets after Ingestion
4. Phagocytosis of Synapses
4.1. The dLGN
4.2. The Hippocampus
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Magnus, T.; Chan, A.; Linker, R.A.; Toyka, K.V.; Gold, R. Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: Implications for the role of glial cells in the inflamed central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 760–766. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 1–22. [Google Scholar] [CrossRef]
- Sokolowski, J.D.; Nobles, S.L.; Heffron, D.S.; Park, D.; Ravichandran, K.S.; Mandell, J.W. Brain-specific angiogenesis inhibitor-1 expression in astrocytes and neurons: Implications for its dual function as an apoptotic engulfment receptor. Brain Behav. Immun. 2011. [Google Scholar] [CrossRef]
- Park, D.; Tosello-Trampont, A.C.; Elliott, M.R.; Lu, M.; Haney, L.B.; Ma, Z.; Klibanov, A.L.; Mandell, J.W.; Ravichandran, K.S. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 2007. [Google Scholar] [CrossRef] [PubMed]
- Iram, T.; Ramirez-Ortiz, Z.; Byrne, M.H.; Coleman, U.A.; Kingery, N.D.; Means, T.K.; Frenkel, D.; El Khoury, J. Megf10 Is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J. Neurosci. 2016, 36, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Lööv, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir-Yilmaz, O.E.; Freeman, M.R. Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev. 2014, 28, 20–33. [Google Scholar] [CrossRef]
- Godoy, B.; Murgas, P.; Tichauer, J.; von Bernhardi, R. Scavenger receptor class A ligands induce secretion of IL1β and exert a modulatory effect on the inflammatory activation of astrocytes in culture. J. Neuroimmunol. 2012, 251, 6–13. [Google Scholar] [CrossRef]
- Wakida, N.M.; Cruz, G.M.S.; Pouladian, P.; Berns, M.W.; Preece, D. Fluid Shear Stress Enhances the Phagocytic Response of Astrocytes. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Yap, A.S.; Duszyc, K.; Viasnoff, V. Mechanosensing and mechanotransduction at cell—Cell junctions. Cold Spring Harb. Perspect. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hlavac, N.; VandeVord, P.J. Astrocyte mechano-activation by high-rate overpressure involves alterations in structural and junctional proteins. Front. Neurol. 2019. [Google Scholar] [CrossRef]
- Lööv, C.; Mitchell, C.H.; Simonsson, M.; Erlandsson, A. Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. Glia 2015, 63, 1997–2009. [Google Scholar] [CrossRef]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541. [Google Scholar] [CrossRef]
- DiCiccio, J.E.; Steinberg, B.E. Lysosomal pH and analysis of the counter ion pathways that support acidification. J. Gen. Physiol. 2011, 137, 385–390. [Google Scholar] [CrossRef]
- Majumdar, A.; Cruz, D.; Asamoah, N.; Buxbaum, A.; Sohar, I.; Lobel, P.; Maxfield, F.R. Activation of Microglia Acidifies Lysosomes and Leads to Degradation of Alzheimer Amyloid Fibrils. Mol. Biol. Cell 2007, 18, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. USA 2018, 115, 6640–6649. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Ghassemifar, R.; Brunk, U.T. Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem. J. 1997. [Google Scholar] [CrossRef]
- Magnus, T.; Chan, A.; Grauer, O.; Toyka, K.V.; Gold, R. Microglial Phagocytosis of Apoptotic Inflammatory T Cells Leads to Down-Regulation of Microglial Immune Activation. J. Immunol. 2001, 167, 5004–5010. [Google Scholar] [CrossRef]
- Brown, S.B.; Savill, J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J. Immunol. 1999, 162, 480–485. [Google Scholar]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef]
- Yoon, S.S.; Jo, S.A. Mechanisms of amyloid-β peptide clearance: Potential therapeutic targets for Alzheimer’s disease. Biomol. Ther. 2012. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 2017. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Connor, T.J.; Lynch, M.A. Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 2013, 8, 301–311. [Google Scholar] [CrossRef]
- Zhang, H.; Su, Y.J.; Zhou, W.W.; Wang, S.W.; Xu, P.X.; Yu, X.L.; Liu, R.T. Activated scavenger receptor a promotes glial internalization of Aβ. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef]
- Fujita, Y.; Maeda, T.; Sato, C.; Sato, M.; Hatakeyama, H.; Ota, Y.; Iwabuchi, N.; Tatesawa, K.; Nomura, A.; Zou, K.; et al. Engulfment of Toxic Amyloid β-protein in Neurons and Astrocytes Mediated by MEGF10. Neuroscience 2020, 443, 1–7. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.D.; Nielsen, H.M.; Blankenstein, M.A.; Eikelenboom, P.; Veerhuis, R. Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microglia in vitro. Glia 2014. [Google Scholar] [CrossRef]
- Auderset, L.; Cullen, C.L.; Young, K.M. Low density lipoprotein-receptor related protein 1 is differentially expressed by neuronal and glial populations in the developing and mature mouse central nervous system. PLoS ONE 2016. [Google Scholar] [CrossRef]
- Prakash, P.; Jethava, K.; Korte, N.; Izquierdo, P.; Favuzzi, E.; Rose, I.; Guttenplan, K.A.; Dutta, S.; Rochet, C.; Fishell, G.; et al. Monitoring phagocytic uptake of amyloid β into glial cell lysosomes in real time. bioRxiv 2020, 1, 1–36. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016. [Google Scholar] [CrossRef]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zheng, L.; Zhang, Q.; Li, X.; Zhang, X.; Li, Z.; Bai, X.; Zhang, Z.; Huo, W.; Zhao, X.; et al. Deacetylation of TFEB promotes fibrillar Aβ degradation by upregulating lysosomal biogenesis in microglia. Protein Cell 2016, 7, 417–433. [Google Scholar] [CrossRef]
- Tanaka, Y.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 2013, 250, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Götzl, J.K.; Colombo, A.V.; Fellerer, K.; Reifschneider, A.; Werner, G.; Tahirovic, S.; Haass, C.; Capell, A. Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.W.; McKay, A.; Singh, P.P.; Brunet, A.; Huang, E.J. Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Zheng, L.J.; Shen, J.; Li, X.Y.; Zhang, Q.; Bai, X.; Wang, Q.S.; Ji, J.G. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 2018, 13, 2005–2013. [Google Scholar] [CrossRef]
- Kim, H.N.; Seo, B.R.; Kim, H.; Koh, J.Y. Cilostazol restores autophagy flux in bafilomycin A1-treated, cultured cortical astrocytes through lysosomal reacidification: Roles of PKA, zinc and metallothionein 3. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Guo, X.; Tang, P.; Chen, L.; Liu, P.; Hou, C.; Zhang, X.; Liu, Y.; Chong, L.; Li, X. Amyloid β-induced redistribution of transcriptional factor EB and lysosomal dysfunction in primary microglial cells. Front. Aging Neurosci. 2017. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R.; Xu, S.; Lakshmana, M.K. Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer’s and Amyotrophic Lateral Sclerosis Brains. Neurosci. J. 2016. [Google Scholar] [CrossRef]
- Monasor, L.S.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar aβ triggers microglial proteome alterations and dysfunction in alzheimer mouse models. Elife 2020. [Google Scholar] [CrossRef]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia 2020, 1–15. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 1–19. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015. [Google Scholar] [CrossRef]
- Victoria, G.S.; Zurzolo, C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.F.; Chen, C.; Fu, W.Y.; Qu, J.Y.; Cheung, T.H.; Fu, A.K.Y.; Ip, N.Y. IL-33-PU.1 Transcriptome Reprogramming Drives Functional State Transition and Clearance Activity of Microglia in Alzheimer’s Disease. Cell Rep. 2020, 31, 107530. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Eremenko, E.; Berner, O.; Elyahu, Y.; Strominger, I.; Apelblat, D.; Nemirovsky, A.; Spiegel, I.; Monsonego, A. CD4 T Cells Induce A Subset of MHCII-Expressing Microglia that Attenuates Alzheimer Pathology. Iscience 2019. [Google Scholar] [CrossRef] [PubMed]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflamm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Barres, B. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007. [Google Scholar] [CrossRef]
- Yilmaz, M.; Yalcin, E.; Presumey, J.; Aw, E.; Ma, M.; Whelan, C.W.; Stevens, B.; McCarroll, S.A.; Carroll, M.C. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 2020. [Google Scholar] [CrossRef]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.; et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 2018, 100, 120–134.e6. [Google Scholar] [CrossRef]
- Cong, Q.; Soteros, B.M.; Wollet, M.; Kim, J.H.; Sia, G.M. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci. 2020, 23, 1067–1078. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Chung, W.-S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Schumacher, A.; Snaidero, N.; Gavilanes, E.M.U.; Neziraj, T.; Kocsis-Jutka, V.; Engels, D.; Jürgens, T.; Wagner, I.; Weidinger, J.D.F.; et al. Phagocyte-mediated synapse removal in cortical neuroinflammation is promoted by local calcium accumulation. Nat. Neurosci. 2021, 24, 355–367. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, J.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2020. [Google Scholar] [CrossRef]
- Rice, R.A.; Spangenberg, E.E.; Yamate-Morgan, H.; Lee, R.J.; Arora, R.P.S.; Hernandez, M.X.; Tenner, A.J.; West, B.L.; Green, K.N. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J. Neurosci. 2015. [Google Scholar] [CrossRef]
- Jay, T.R.; von Saucken, V.E.; Muñoz, B.; Codocedo, J.F.; Atwood, B.K.; Lamb, B.T.; Landreth, G.E. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 2019, 67, 1873–1892. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22. [Google Scholar] [CrossRef]
- Bialas, A.R.; Stevens, B. TGF-Beta Signaling Regulates Neuronal C1q Expression and Developmental Syanptic Refinement. Nat. Neurosci. 2013, 16. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Takano, H.; Dong, J.H.; Haydon, P.G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 2007, 27, 6473–6477. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.Y.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kono, R.; Ikegaya, Y.; Koyama, R. Phagocytic Glial Cells in Brain Homeostasis. Cells 2021, 10, 1348. https://doi.org/10.3390/cells10061348
Kono R, Ikegaya Y, Koyama R. Phagocytic Glial Cells in Brain Homeostasis. Cells. 2021; 10(6):1348. https://doi.org/10.3390/cells10061348
Chicago/Turabian StyleKono, Rena, Yuji Ikegaya, and Ryuta Koyama. 2021. "Phagocytic Glial Cells in Brain Homeostasis" Cells 10, no. 6: 1348. https://doi.org/10.3390/cells10061348
APA StyleKono, R., Ikegaya, Y., & Koyama, R. (2021). Phagocytic Glial Cells in Brain Homeostasis. Cells, 10(6), 1348. https://doi.org/10.3390/cells10061348