
The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines

 ,
,  , , ,
, , ,
Abstract
1. Introduction
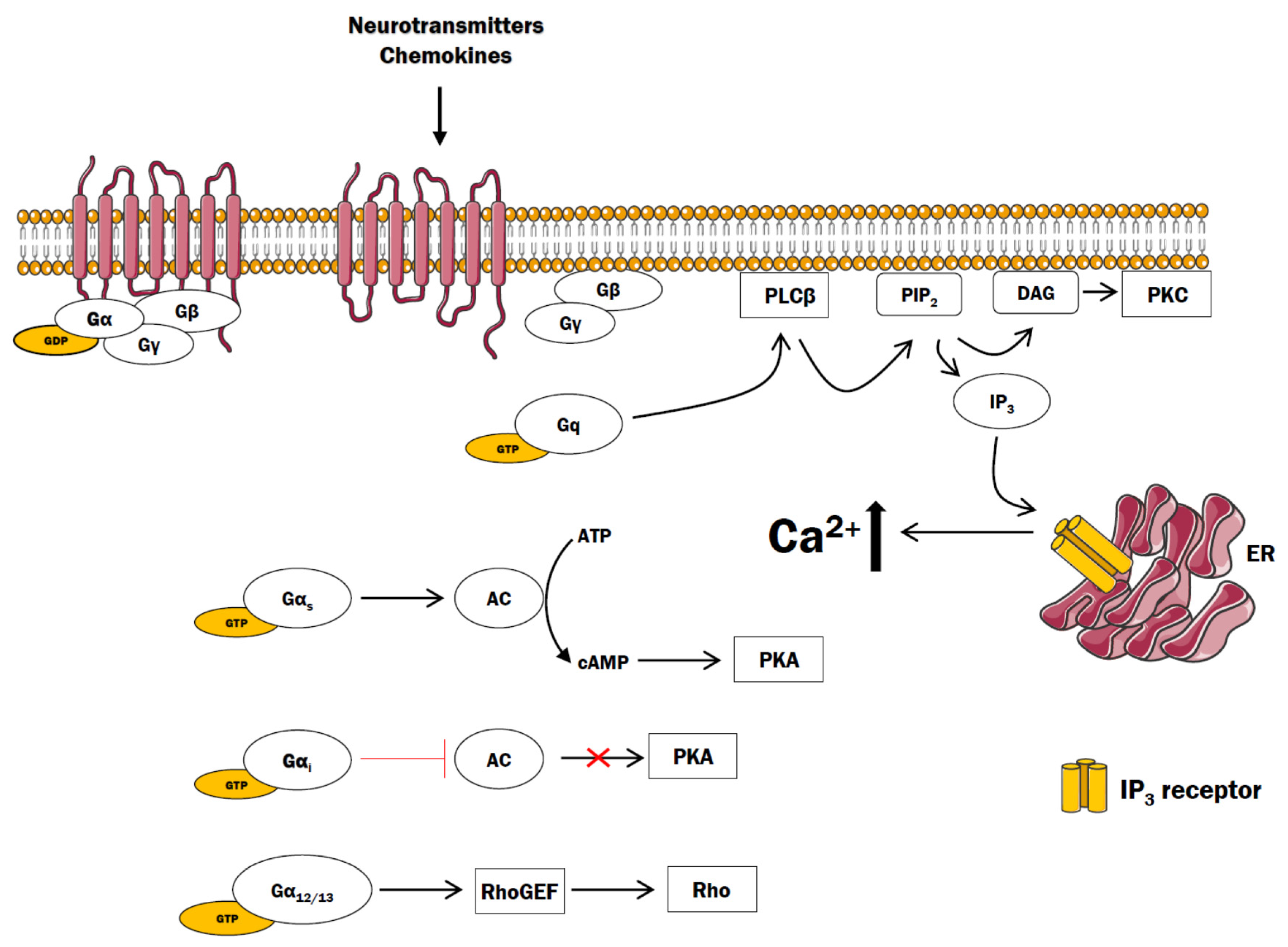
2. GPCR and Ca2+ Signaling
3. Dopaminergic Receptors
4. Adrenergic Receptors
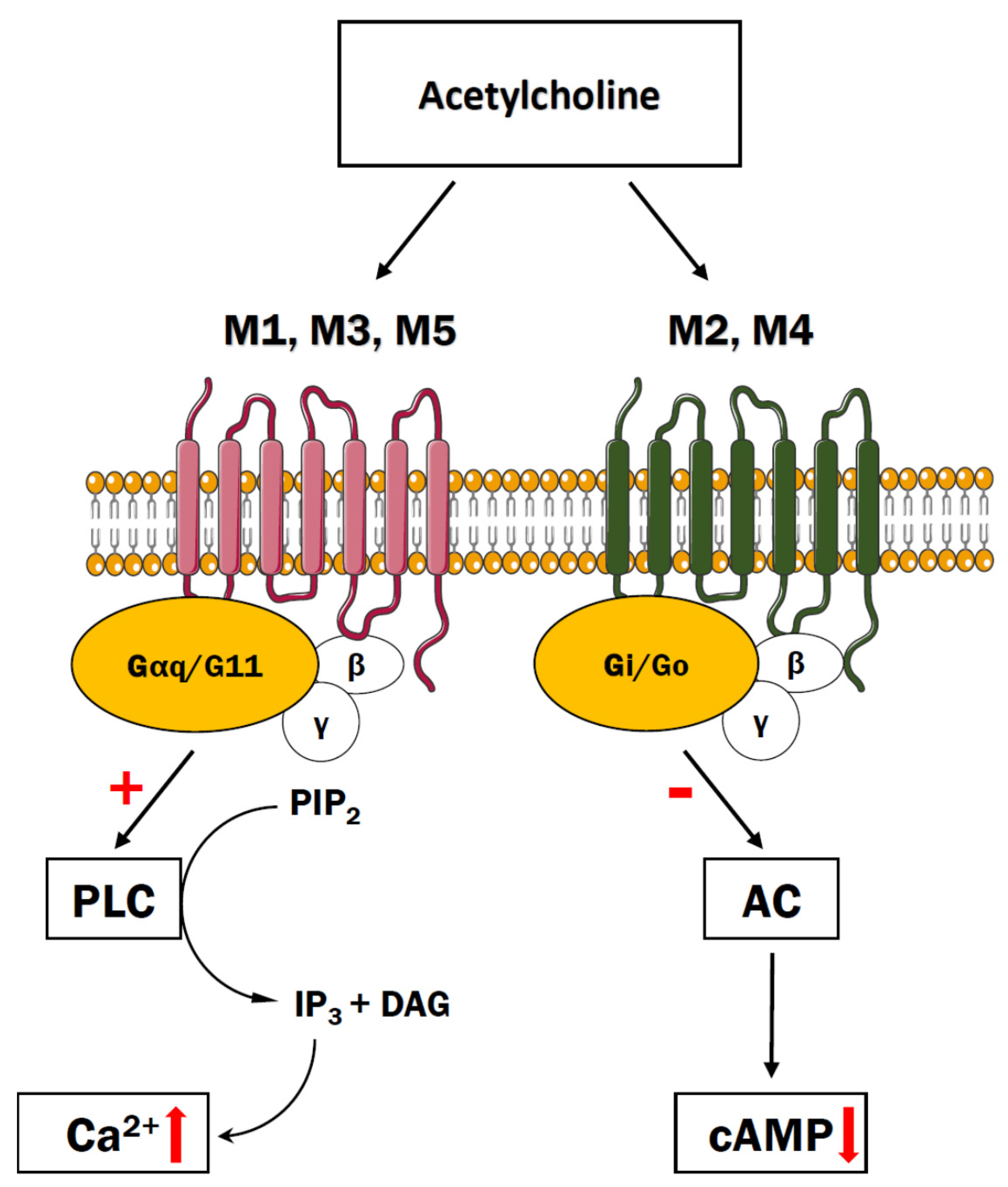
5. Cholinergic Receptors
6. Serotonergic Receptors
7. Glutamate Metabotropic Receptors
8. GABAB Receptors
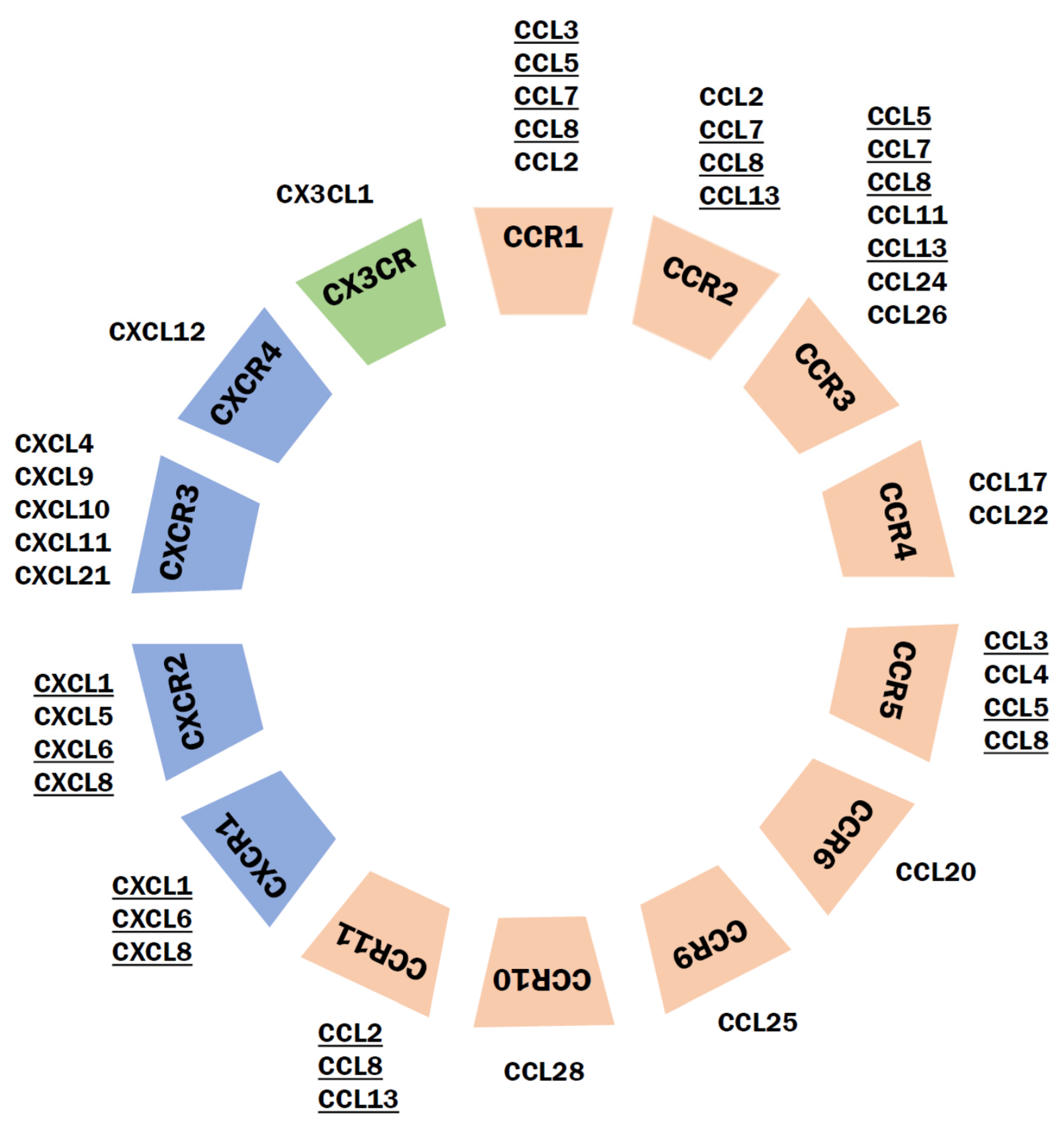
9. Chemokine Receptors
9.1. Classification of Chemokines and Their Receptors
9.2. Chemokines and Their Receptors in Schizophrenia
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global Epidemiology and Burden of Schizophrenia: Findings From the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Bowie, C.R.; Harvey, P.D. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr. Dis. Treat. 2006, 2, 531–536. [Google Scholar] [CrossRef]
- Correll, C.U.; Schooler, N.R. Negative Symptoms in Schizophrenia: A Review and Clinical Guide for Recognition, Assessment, and Treatment. Neuropsychiatr. Dis. Treat. 2020, 16, 519–534. [Google Scholar] [CrossRef]
- Galderisi, S.; Mucci, A.; Buchanan, R.W.; Arango, C. Negative symptoms of schizophrenia: New developments and unanswered research questions. Lancet Psychiatry 2018, 5, 664–677. [Google Scholar] [CrossRef]
- Toda, M.; Abi-Dargham, A. Dopamine hypothesis of schizophrenia: Making sense of it all. Curr. Psychiatry Rep. 2007, 9, 329–336. [Google Scholar] [CrossRef]
- Seeman, P. Schizophrenia and dopamine receptors. Eur. Neuropsychopharmacol. 2013, 23, 999–1009. [Google Scholar] [CrossRef]
- Iasevoli, F.; Tomasetti, C.; Buonaguro, E.F.; de Bartolomeis, A. The glutamatergic aspects of schizophrenia molecular pathophysiology: Role of the postsynaptic density, and implications for treatment. Curr. Neuropharmacol. 2014, 12, 219–238. [Google Scholar] [CrossRef]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef]
- Dickerson, F.; Stallings, C.; Origoni, A.; Schroeder, J.; Katsafanas, E.; Schweinfurth, L.; Savage, C.; Khushalani, S.; Yolken, R. Inflammatory Markers in Recent Onset Psychosis and Chronic Schizophrenia. Schizophr. Bull. 2016, 42, 134–141. [Google Scholar] [CrossRef]
- Stuart, M.J.; Baune, B.T. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: A systematic review of biomarker studies. Neurosci. Biobehav. Rev. 2014, 42, 93–115. [Google Scholar] [CrossRef]
- Hong, S.; Lee, E.E.; Martin, A.S.; Soontornniyomkij, B.; Soontornniyomkij, V.; Achim, C.L.; Reuter, C.; Irwin, M.R.; Eyler, L.T.; Jeste, D.V. Abnormalities in chemokine levels in schizophrenia and their clinical correlates. Schizophr. Res. 2017, 181, 63–69. [Google Scholar] [CrossRef]
- Réaux-Le Goazigo, A.; Van Steenwinckel, J.; Rostène, W.; Mélik Parsadaniantz, S. Current status of chemokines in the adult CNS. Prog. Neurobiol. 2013, 104, 67–92. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic evidence that β-arrestins are dispensable for the initiation of β. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential Utility of Biased GPCR Signaling for Treatment of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef] [PubMed]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.M.; Sung, J.Y.; Hébert, T.E. Gβγ subunits-Different spaces, different faces. Pharmacol. Res. 2016, 111, 434–441. [Google Scholar] [CrossRef]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. (Seoul) 2017, 25, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin) 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Miller, R.J. Activation of the nuclear factor of activated T-cells (NFAT) mediates upregulation of CCR2 chemokine receptors in dorsal root ganglion (DRG) neurons: A possible mechanism for activity-dependent transcription in DRG neurons in association with neuropathic pain. Mol. Cell. Neurosci. 2008, 37, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.U.; Kim, L.K.; Choi, J.M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front. Immunol. 2018, 9, 2747. [Google Scholar] [CrossRef] [PubMed]
- Al Akoum, C.; Akl, I.; Rouas, R.; Fayyad-Kazan, M.; Falha, L.; Renno, T.; Burny, A.; Lewalle, P.; Fayyad-Kazan, H.; Badran, B. NFAT-1, Sp-1, Sp-3, and miR-21: New regulators of chemokine C receptor 7 expression in mature human dendritic cells. Hum. Immunol. 2015, 76, 307–317. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, S.B.; Luo, Y.X.; Yang, Y.L.; Zhang, X.Z.; Li, B.; Meng, Y.; Chen, Y.J.; Guo, R.X.; Xiong, Y.C.; et al. NFATc2-dependent epigenetic upregulation of CXCL14 is involved in the development of neuropathic pain induced by paclitaxel. J. Neuroinflamm. 2020, 17, 310. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Fernandez, S.; Carrero, P.; Garcia-Garcia, M.; Torres-Aleman, I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci. 2007, 27, 8745–8756. [Google Scholar] [CrossRef] [PubMed]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Liencres, C.; Tas, C.; Brown, E.C.; Erdin, S.; Onur, E.; Cubukcoglu, Z.; Aydemir, O.; Esen-Danaci, A.; Brüne, M. Oxidative stress in schizophrenia: A case-control study on the effects on social cognition and neurocognition. BMC Psychiatry 2014, 14, 268. [Google Scholar] [CrossRef]
- Madireddy, S. Regulation of Reactive Oxygen Species-Mediated Damage in the Pathogenesis of Schizophrenia. Brain Sci. 2020, 10, 742. [Google Scholar] [CrossRef]
- Upthegrove, R.; Khandaker, G.M. Cytokines, Oxidative Stress and Cellular Markers of Inflammation in Schizophrenia. Curr. Top. Behav. Neurosci. 2020, 44, 49–66. [Google Scholar] [CrossRef]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxid. Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Keilhoff, G.; Steiner, J.; Dobrowolny, H.; Bogerts, B. Nitric oxide and schizophrenia: Present knowledge and emerging concepts of therapy. CNS Neurol. Disord. Drug Targets 2011, 10, 792–807. [Google Scholar] [CrossRef]
- Nasyrova, R.F.; Ivashchenko, D.V.; Ivanov, M.V.; Neznanov, N.G. Role of nitric oxide and related molecules in schizophrenia pathogenesis: Biochemical, genetic and clinical aspects. Front. Physiol. 2015, 6, 139. [Google Scholar] [CrossRef]
- Bitanihirwe, B.K.; Woo, T.U. Oxidative stress in schizophrenia: An integrated approach. Neurosci. Biobehav. Rev. 2011, 35, 878–893. [Google Scholar] [CrossRef]
- Morris, G.; Walker, A.J.; Walder, K.; Berk, M.; Marx, W.; Carvalho, A.F.; Maes, M.; Puri, B.K. Increasing Nrf2 Activity as a Treatment Approach in Neuropsychiatry. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Boll, K.M.; Noto, C.; Bonifácio, K.L.; Bortolasci, C.C.; Gadelha, A.; Bressan, R.A.; Barbosa, D.S.; Maes, M.; Moreira, E.G. Oxidative and nitrosative stress biomarkers in chronic schizophrenia. Psychiatry Res. 2017, 253, 43–48. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 2017, 187, 26–32. [Google Scholar] [CrossRef]
- Baumeister, A.A.; Francis, J.L. Historical development of the dopamine hypothesis of schizophrenia. J. Hist. Neurosci. 2002, 11, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, A.; Niznik, H.B. Coupling of dopamine receptor subtypes to multiple and diverse G proteins. Int. J. Dev. Neurosci. 2000, 18, 669–677. [Google Scholar] [CrossRef]
- Neve, K.A.; Seamans, J.K.; Trantham-Davidson, H. Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 2004, 24, 165–205. [Google Scholar] [CrossRef]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.A.; Nairn, A.C.; Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 269–296. [Google Scholar] [CrossRef]
- Albert, K.A.; Hemmings, H.C.; Adamo, A.I.; Potkin, S.G.; Akbarian, S.; Sandman, C.A.; Cotman, C.W.; Bunney, W.E.; Greengard, P. Evidence for decreased DARPP-32 in the prefrontal cortex of patients with schizophrenia. Arch. Gen. Psychiatry 2002, 59, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, J.; Takahashi, H.; Ito, H.; Takano, A.; Fujimura, Y.; Matsumoto, R.; Nozaki, S.; Yasuno, F.; Okubo, Y.; Kishimoto, T.; et al. Decreased binding of [11C]NNC112 and [11C]SCH23390 in patients with chronic schizophrenia. Life Sci. 2010, 86, 814–818. [Google Scholar] [CrossRef]
- Takahashi, H.; Kato, M.; Takano, H.; Arakawa, R.; Okumura, M.; Otsuka, T.; Kodaka, F.; Hayashi, M.; Okubo, Y.; Ito, H.; et al. Differential contributions of prefrontal and hippocampal dopamine D(1) and D(2) receptors in human cognitive functions. J. Neurosci. 2008, 28, 12032–12038. [Google Scholar] [CrossRef]
- Domyo, T.; Kurumaji, A.; Toru, M. An increase in [3H]SCH23390 binding in the cerebral cortex of postmortem brains of chronic schizophrenics. J. Neural Transm. (Vienna) 2001, 108, 1475–1484. [Google Scholar] [CrossRef]
- Felsing, D.E.; Jain, M.K.; Allen, J.A. Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr. Top. Med. Chem. 2019, 19, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Saucedo Uribe, E.; Carranza Navarro, F.; Guerrero Medrano, A.F.; García Cervantes, K.I.; Álvarez Villalobos, N.A.; Acuña Rocha, V.D.; Méndez Hernández, M.; Millán Alanís, J.M.; Hinojosa Cavada, C.M.; Zúñiga Hernández, J.A.; et al. Preliminary efficacy and tolerability profiles of first versus second-generation Long-Acting Injectable Antipsychotics in schizophrenia: A systematic review and meta-analysis. J. Psychiatr. Res. 2020, 129, 222–233. [Google Scholar] [CrossRef]
- Lee, E.S.; Kronsberg, H.; Findling, R.L. Psychopharmacologic Treatment of Schizophrenia in Adolescents and Children. Child Adolesc. Psychiatr. Clin. N. Am. 2020, 29, 183–210. [Google Scholar] [CrossRef] [PubMed]
- Muench, J.; Hamer, A.M. Adverse effects of antipsychotic medications. Am. Fam. Physician 2010, 81, 617–622. [Google Scholar]
- Ishigooka, J.; Iwashita, S.; Tadori, Y. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia in Japan: A 6-week, randomized, double-blind, placebo-controlled study. Psychiatry Clin. Neurosci. 2018, 72, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Amada, N.; Akazawa, H.; Ohgi, Y.; Maeda, K.; Sugino, H.; Kurahashi, N.; Kikuchi, T.; Futamura, T. Brexpiprazole has a low risk of dopamine D. Neuropsychopharmacol Rep. 2019, 39, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Vyas, P.; Hwang, B.J.; Brašić, J.R. An evaluation of lumateperone tosylate for the treatment of schizophrenia. Expert Opin. Pharmacother. 2020, 21, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kellendonk, C.; Simpson, E.H.; Kandel, E.R.; Gao, W.J. D2 receptor overexpression in the striatum leads to a deficit in inhibitory transmission and dopamine sensitivity in mouse prefrontal cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 12107–12112. [Google Scholar] [CrossRef]
- Petty, A.; Cui, X.; Tesiram, Y.; Kirik, D.; Howes, O.; Eyles, D. Enhanced Dopamine in Prodromal Schizophrenia (EDiPS): A new animal model of relevance to schizophrenia. NPJ Schizophr. 2019, 5, 6. [Google Scholar] [CrossRef]
- Abi-Dargham, A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. 1), S1–S5. [Google Scholar] [CrossRef]
- Gross, G.; Drescher, K. The role of dopamine D(3) receptors in antipsychotic activity and cognitive functions. Handb. Exp. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Watson, D.J.; Loiseau, F.; Ingallinesi, M.; Millan, M.J.; Marsden, C.A.; Fone, K.C. Selective blockade of dopamine D3 receptors enhances while D2 receptor antagonism impairs social novelty discrimination and novel object recognition in rats: A key role for the prefrontal cortex. Neuropsychopharmacology 2012, 37, 770–786. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Lee, C.C.; Chang, Y.C.; Lu, C.T.; Huang, C.H.; Chang, W.H. Effects of dopamine D2 receptor Ser311Cys polymorphism and clinical factors on risperidone efficacy for positive and negative symptoms and social function. Int. J. Neuropsychopharmacol. 2004, 7, 461–470. [Google Scholar] [CrossRef]
- Staddon, S.; Arranz, M.J.; Mancama, D.; Perez-Nievas, F.; Arrizabalaga, I.; Anney, R.; Buckland, P.; Elkin, A.; Osborne, S.; Munro, J.; et al. Association between dopamine D3 receptor gene polymorphisms and schizophrenia in an isolate population. Schizophr. Res. 2005, 73, 49–54. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Goldman, D.; Buchanan, R.W.; Rooney, W.; Clifton, A.; Kosmidis, M.H.; Breier, A.; Pickar, D. The dopamine D3 receptor (DRD3) Ser9Gly polymorphism and schizophrenia: A haplotype relative risk study and association with clozapine response. Mol. Psychiatry 1998, 3, 72–75. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kapur, S.; Seeman, P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol. Psychiatry 2002, 7, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Lindefors, N.; Barati, S.; O’Connor, W.T. Differential effects of single and repeated ketamine administration on dopamine, serotonin and GABA transmission in rat medial prefrontal cortex. Brain Res. 1997, 759, 205–212. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Can, A.; Zanos, P.; Moaddel, R.; Kang, H.J.; Dossou, K.S.; Wainer, I.W.; Cheer, J.F.; Frost, D.O.; Huang, X.P.; Gould, T.D. Effects of Ketamine and Ketamine Metabolites on Evoked Striatal Dopamine Release, Dopamine Receptors, and Monoamine Transporters. J. Pharmacol. Exp. Ther. 2016, 359, 159–170. [Google Scholar] [CrossRef]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef]
- Bergson, C.; Levenson, R.; Goldman-Rakic, P.S.; Lidow, M.S. Dopamine receptor-interacting proteins: The Ca(2+) connection in dopamine signaling. Trends Pharmacol. Sci. 2003, 24, 486–492. [Google Scholar] [CrossRef]
- Kabbani, N.; Woll, M.P.; Nordman, J.C.; Levenson, R. Dopamine receptor interacting proteins: Targeting neuronal calcium sensor-1/D2 dopamine receptor interaction for antipsychotic drug development. Curr. Drug Targets 2012, 13, 72–79. [Google Scholar] [CrossRef]
- O’Donnell, J.; Zeppenfeld, D.; McConnell, E.; Pena, S.; Nedergaard, M. Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 2012, 37, 2496–2512. [Google Scholar] [CrossRef]
- Saboory, E.; Ghasemi, M.; Mehranfard, N. Norepinephrine, neurodevelopment and behavior. Neurochem. Int. 2020, 135, 104706. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.T.; Mohan, M.L.; Goswami, S.K.; Naga Prasad, S.V. Regulation of β-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 2011, 10, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, F.; Piscitelli, C.L.; Tsai, C.J.; Standfuss, J.; Deupi, X.; Schertler, G.F. Structure of β-adrenergic receptors. Methods Enzymol. 2013, 520, 117–151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shen, H.; Xiong, Y.; Yang, X.; He, J. The beta1-adrenergic receptor mediates extracellular signal-regulated kinase activation via Galphas. Amino Acids 2010, 38, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Knaus, A.E.; Muthig, V.; Schickinger, S.; Moura, E.; Beetz, N.; Gilsbach, R.; Hein, L. Alpha2-adrenoceptor subtypes--unexpected functions for receptors and ligands derived from gene-targeted mouse models. Neurochem. Int. 2007, 51, 277–281. [Google Scholar] [CrossRef]
- Ramos, B.P.; Arnsten, A.F. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef]
- Zhang, H.T.; Whisler, L.R.; Huang, Y.; Xiang, Y.; O’Donnell, J.M. Postsynaptic alpha-2 adrenergic receptors are critical for the antidepressant-like effects of desipramine on behavior. Neuropsychopharmacology 2009, 34, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Hein, L. Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 2006, 326, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Hornykiewicz, O. Proposal for a noradrenaline hypothesis of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 913–922. [Google Scholar] [CrossRef]
- Arnsten, A.F. Adrenergic targets for the treatment of cognitive deficits in schizophrenia. Psychopharmacology 2004, 174, 25–31. [Google Scholar] [CrossRef]
- Atzori, M.; Cuevas-Olguin, R.; Esquivel-Rendon, E.; Garcia-Oscos, F.; Salgado-Delgado, R.C.; Saderi, N.; Miranda-Morales, M.; Treviño, M.; Pineda, J.C.; Salgado, H. Locus Ceruleus Norepinephrine Release: A Central Regulator of CNS Spatio-Temporal Activation? Front. Synaptic. Neurosci. 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.G.; Yuan, P.X.; Wang, M.; Vijayraghavan, S.; Bloom, A.K.; Davis, D.J.; Gobeske, K.T.; Sweatt, J.D.; Manji, H.K.; Arnsten, A.F. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science 2004, 306, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Oranje, B.; Glenthøj, B.Y. Clonidine normalizes sensorimotor gating deficits in patients with schizophrenia on stable medication. Schizophr. Bull. 2013, 39, 684–691. [Google Scholar] [CrossRef]
- Oranje, B.; Glenthøj, B.Y. Clonidine normalizes levels of P50 gating in patients with schizophrenia on stable medication. Schizophr. Bull. 2014, 40, 1022–1029. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baisley, S.K.; Fallace, K.L.; Rajbhandari, A.K.; Bakshi, V.P. Mutual independence of 5-HT(2) and α1 noradrenergic receptors in mediating deficits in sensorimotor gating. Psychopharmacology 2012, 220, 465–479. [Google Scholar] [CrossRef]
- Friedman, J.I.; Adler, D.N.; Temporini, H.D.; Kemether, E.; Harvey, P.D.; White, L.; Parrella, M.; Davis, K.L. Guanfacine treatment of cognitive impairment in schizophrenia. Neuropsychopharmacology 2001, 25, 402–409. [Google Scholar] [CrossRef][Green Version]
- Arnsten, A.F. The Emerging Neurobiology of Attention Deficit Hyperactivity Disorder: The Key Role of the Prefrontal Association Cortex. J. Pediatr. 2009, 154, I-S43. [Google Scholar] [CrossRef]
- Gamo, N.J.; Lur, G.; Higley, M.J.; Wang, M.; Paspalas, C.D.; Vijayraghavan, S.; Yang, Y.; Ramos, B.P.; Peng, K.; Kata, A.; et al. Stress Impairs Prefrontal Cortical Function via D1 Dopamine Receptor Interactions With Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. Biol. Psychiatry 2015, 78, 860–870. [Google Scholar] [CrossRef]
- Phillips, W.A.; Larkum, M.E.; Harley, C.W.; Silverstein, S.M. The effects of arousal on apical amplification and conscious state. Neurosci. Conscious. 2016, 2016, niw015. [Google Scholar] [CrossRef]
- Wang, M.; Gamo, N.J.; Yang, Y.; Jin, L.E.; Wang, X.J.; Laubach, M.; Mazer, J.A.; Lee, D.; Arnsten, A.F. Neuronal basis of age-related working memory decline. Nature 2011, 476, 210–213. [Google Scholar] [CrossRef]
- Valero-Aracama, M.J.; Reboreda, A.; Arboit, A.; Sauvage, M.; Yoshida, M. Noradrenergic suppression of persistent firing in hippocampal CA1 pyramidal cells through cAMP-PKA pathway. eNeuro 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ramos, B.P.; Paspalas, C.D.; Shu, Y.; Simen, A.; Duque, A.; Vijayraghavan, S.; Brennan, A.; Dudley, A.; Nou, E.; et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell 2007, 129, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Jin, L.E. Guanfacine for the treatment of cognitive disorders: A century of discoveries at Yale. Yale J. Biol. Med. 2012, 85, 45–58. [Google Scholar] [PubMed]
- O’Dell, T.J.; Connor, S.A.; Guglietta, R.; Nguyen, P.V. β-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learn. Mem. 2015, 22, 461–471. [Google Scholar] [CrossRef]
- Zhou, H.C.; Sun, Y.Y.; Cai, W.; He, X.T.; Yi, F.; Li, B.M.; Zhang, X.H. Activation of β2-adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP-PKA signaling in the medial prefrontal cortex of rats. Learn. Mem. 2013, 20, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Shek, E.; Bardhan, S.; Cheine, M.V.; Ahonen, J.; Wahlbeck, K. Beta-blocker supplementation of standard drug treatment for schizophrenia. Schizophr. Bull. 2010, 36, 1079–1080. [Google Scholar] [CrossRef]
- Mico’, U.; Bruno, A.; Pandolfo, G.; Maria Romeo, V.; Mallamace, D.; D’Arrigo, C.; Spina, E.; Zoccali, R.A.; Muscatello, M.R. Duloxetine as adjunctive treatment to clozapine in patients with schizophrenia: A randomized, placebo-controlled trial. Int. Clin. Psychopharmacol. 2011, 26, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Terevnikov, V.; Stenberg, J.H.; Joffe, M.; Tiihonen, J.; Burkin, M.; Tchoukhine, E.; Joffe, G. More evidence on additive antipsychotic effect of adjunctive mirtazapine in schizophrenia: An extension phase of a randomized controlled trial. Hum. Psychopharmacol. 2010, 25, 431–438. [Google Scholar] [CrossRef]
- Abbasi, S.H.; Behpournia, H.; Ghoreshi, A.; Salehi, B.; Raznahan, M.; Rezazadeh, S.A.; Rezaei, F.; Akhondzadeh, S. The effect of mirtazapine add on therapy to risperidone in the treatment of schizophrenia: A double-blind randomized placebo-controlled trial. Schizophr. Res. 2010, 116, 101–106. [Google Scholar] [CrossRef]
- Boyda, H.N.; Ho, A.A.; Tse, L.; Procyshyn, R.M.; Yuen, J.W.Y.; Kim, D.D.; Honer, W.G.; Barr, A.M. Differential Effects of Acute Treatment With Antipsychotic Drugs on Peripheral Catecholamines. Front. Psychiatry 2020, 11, 617428. [Google Scholar] [CrossRef]
- Clark, D.A.; Arranz, M.J.; Mata, I.; Lopéz-Ilundain, J.; Pérez-Nievas, F.; Kerwin, R.W. Polymorphisms in the promoter region of the alpha1A-adrenoceptor gene are associated with schizophrenia/schizoaffective disorder in a Spanish isolate population. Biol. Psychiatry 2005, 58, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Lochman, J.; Plesník, J.; Janout, V.; Povová, J.; Míšek, I.; Dvořáková, D.; Šerý, O. Interactive effect of MTHFR and ADRA2A gene polymorphisms on pathogenesis of schizophrenia. Neuro Endocrinol. Lett. 2013, 34, 792–797. [Google Scholar]
- Wang, L.J.; Lee, S.Y.; Chen, S.L.; Chang, Y.H.; Chen, P.S.; Huang, S.Y.; Tzeng, N.S.; Chen, K.C.; Lee, I.H.; Wang, T.Y.; et al. A potential interaction between COMT and MTHFR genetic variants in Han Chinese patients with bipolar II disorder. Sci. Rep. 2015, 5, 8813. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Eglen, R.M. Muscarinic receptor subtype pharmacology and physiology. Prog. Med. Chem. 2005, 43, 105–136. [Google Scholar] [CrossRef]
- Resende, R.R.; Adhikari, A. Cholinergic receptor pathways involved in apoptosis, cell proliferation and neuronal differentiation. Cell Commun. Signal. 2009, 7, 20. [Google Scholar] [CrossRef]
- Espada, S.; Rojo, A.I.; Salinas, M.; Cuadrado, A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: Connecting neurotransmission with neuroprotection. J. Neurochem. 2009, 110, 1107–1119. [Google Scholar] [CrossRef]
- Rouse, S.T.; Hamilton, S.E.; Potter, L.T.; Nathanson, N.M.; Conn, P.J. Muscarinic-induced modulation of potassium conductances is unchanged in mouse hippocampal pyramidal cells that lack functional M1 receptors. Neurosci. Lett. 2000, 278, 61–64. [Google Scholar] [CrossRef]
- Volpicelli, L.A.; Levey, A.I. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004, 145, 59–66. [Google Scholar] [CrossRef]
- Hersch, S.M.; Levey, A.I. Diverse pre- and post-synaptic expression of m1-m4 muscarinic receptor proteins in neurons and afferents in the rat neostriatum. Life Sci. 1995, 56, 931–938. [Google Scholar] [CrossRef]
- Nair, A.G.; Castro, L.R.V.; El Khoury, M.; Gorgievski, V.; Giros, B.; Tzavara, E.T.; Hellgren-Kotaleski, J.; Vincent, P. The high efficacy of muscarinic M4 receptor in D1 medium spiny neurons reverses striatal hyperdopaminergia. Neuropharmacology 2019, 146, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Erskine, D.; Taylor, J.P.; Bakker, G.; Brown, A.J.H.; Tasker, T.; Nathan, P.J. Cholinergic muscarinic M. Drug Discov. Today 2019, 24, 2307–2314. [Google Scholar] [CrossRef]
- Scarr, E.; Sundram, S.; Keriakous, D.; Dean, B. Altered hippocampal muscarinic M4, but not M1, receptor expression from subjects with schizophrenia. Biol. Psychiatry 2007, 61, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A.S.; Scarr, E.; Boer, S.; Money, T.; Jeon, W.J.; Felder, C.; Dean, B. Widespread decreases in cortical muscarinic receptors in a subset of people with schizophrenia. Int. J. Neuropsychopharmacol. 2013, 16, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bakker, G.; Vingerhoets, C.; Boucherie, D.; Caan, M.; Bloemen, O.; Eersels, J.; Booij, J.; van Amelsvoort, T. Relationship between muscarinic M. Neuroimage Clin. 2018, 18, 713–719. [Google Scholar] [CrossRef]
- Odagaki, Y.; Kinoshita, M.; Meana, J.J.; Callado, L.F.; García-Sevilla, J.A. Functional coupling of M. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Newell, K.A.; Zavitsanou, K.; Jew, S.K.; Huang, X.F. Alterations of muscarinic and GABA receptor binding in the posterior cingulate cortex in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 225–233. [Google Scholar] [CrossRef]
- Zavitsanou, K.; Katerina, Z.; Katsifis, A.; Andrew, K.; Mattner, F.; Filomena, M.; Huang, X.F.; Xu-Feng, H. Investigation of m1/m4 muscarinic receptors in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression disorder. Neuropsychopharmacology 2004, 29, 619–625. [Google Scholar] [CrossRef]
- Ghoshal, A.; Rook, J.M.; Dickerson, J.W.; Roop, G.N.; Morrison, R.D.; Jalan-Sakrikar, N.; Lamsal, A.; Noetzel, M.J.; Poslusney, M.S.; Wood, M.R.; et al. Potentiation of M1 Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology 2016, 41, 598–610. [Google Scholar] [CrossRef]
- Scarr, E.; Keriakous, D.; Crossland, N.; Dean, B. No change in cortical muscarinic M2, M3 receptors or [35S]GTPgammaS binding in schizophrenia. Life Sci. 2006, 78, 1231–1237. [Google Scholar] [CrossRef]
- Abad, N.H.; Doulatabad, N.S.; Mohammadi, A.; Srazi, H.R. Treatment of Visual Hallucinations in Schizophrenia by Acetylcholinesterase Inhibitors: A case report. Iran. J. Psychiatry 2011, 6, 161–163. [Google Scholar] [PubMed]
- Patel, S.S.; Attard, A.; Jacobsen, P.; Shergill, S. Acetylcholinesterase Inhibitors (AChEI’s) for the treatment of visual hallucinations in schizophrenia: A case report. BMC Psychiatry 2010, 10, 68. [Google Scholar] [CrossRef]
- Buchanan, R.W.; Conley, R.R.; Dickinson, D.; Ball, M.P.; Feldman, S.; Gold, J.M.; McMahon, R.P. Galantamine for the treatment of cognitive impairments in people with schizophrenia. Am. J. Psychiatry 2008, 165, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Freudenreich, O.; Culhane, M.A.; Pachas, G.N.; Deckersbach, T.; Murphy, E.; Goff, D.C.; Evins, A.E. High-dose galantamine augmentation inferior to placebo on attention, inhibitory control and working memory performance in nonsmokers with schizophrenia. Schizophr. Res. 2008, 102, 88–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keefe, R.S.; Malhotra, A.K.; Meltzer, H.Y.; Kane, J.M.; Buchanan, R.W.; Murthy, A.; Sovel, M.; Li, C.; Goldman, R. Efficacy and safety of donepezil in patients with schizophrenia or schizoaffective disorder: Significant placebo/practice effects in a 12-week, randomized, double-blind, placebo-controlled trial. Neuropsychopharmacology 2008, 33, 1217–1228. [Google Scholar] [CrossRef]
- Scarr, E.; Gibbons, A.S.; Neo, J.; Udawela, M.; Dean, B. Cholinergic connectivity: It’s implications for psychiatric disorders. Front. Cell. Neurosci. 2013, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.R.; Ellis, K.A.; Bartholomeusz, C.F.; Harrison, B.J.; Wesnes, K.A.; Erskine, F.F.; Vitetta, L.; Nathan, P.J. Muscarinic and nicotinic receptors synergistically modulate working memory and attention in humans. Int. J. Neuropsychopharmacol. 2006, 9, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Klinkenberg, I.; Blokland, A. The validity of scopolamine as a pharmacological model for cognitive impairment: A review of animal behavioral studies. Neurosci. Biobehav. Rev. 2010, 34, 1307–1350. [Google Scholar] [CrossRef]
- Sambeth, A.; Riedel, W.J.; Klinkenberg, I.; Kähkönen, S.; Blokland, A. Biperiden selectively induces memory impairment in healthy volunteers: No interaction with citalopram. Psychopharmacology 2015, 232, 1887–1897. [Google Scholar] [CrossRef]
- Bradley, S.R.; Lameh, J.; Ohrmund, L.; Son, T.; Bajpai, A.; Nguyen, D.; Friberg, M.; Burstein, E.S.; Spalding, T.A.; Ott, T.R.; et al. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology 2010, 58, 365–373. [Google Scholar] [CrossRef]
- Fernández de Sevilla, D.; Núñez, A.; Borde, M.; Malinow, R.; Buño, W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J. Neurosci. 2008, 28, 1469–1478. [Google Scholar] [CrossRef]
- Brown, D.A. Regulation of neural ion channels by muscarinic receptors. Neuropharmacology 2018, 136, 383–400. [Google Scholar] [CrossRef]
- Giessel, A.J.; Sabatini, B.L. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron 2010, 68, 936–947. [Google Scholar] [CrossRef]
- Buchanan, K.A.; Petrovic, M.M.; Chamberlain, S.E.; Marrion, N.V.; Mellor, J.R. Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron 2010, 68, 948–963. [Google Scholar] [CrossRef]
- Zhao, L.X.; Ge, Y.H.; Li, J.B.; Xiong, C.H.; Law, P.Y.; Xu, J.R.; Qiu, Y.; Chen, H.Z. M1 muscarinic receptors regulate the phosphorylation of AMPA receptor subunit GluA1. FASEB J. 2019, 33, 6622–6631. [Google Scholar] [CrossRef]
- Zhao, L.X.; Ge, Y.H.; Xiong, C.H.; Tang, L.; Yan, Y.H.; Law, P.Y.; Qiu, Y.; Chen, H.Z. M1 muscarinic receptor facilitates cognitive function by interplay with AMPA receptor GluA1 subunit. FASEB J. 2018, 32, 4247–4257. [Google Scholar] [CrossRef]
- Zeppillo, T.; Schulmann, A.; Macciardi, F.; Hjelm, B.E.; Föcking, M.; Sequeira, P.A.; Guella, I.; Cotter, D.; Bunney, W.E.; Limon, A.; et al. Functional impairment of cortical AMPA receptors in schizophrenia. Schizophr Res. 2020. [Google Scholar] [CrossRef]
- Gururajan, A.; van den Buuse, M. Is the mTOR-signalling cascade disrupted in Schizophrenia? J. Neurochem. 2014, 129, 377–387. [Google Scholar] [CrossRef]
- Jeon, J.; Dencker, D.; Wörtwein, G.; Woldbye, D.P.; Cui, Y.; Davis, A.A.; Levey, A.I.; Schütz, G.; Sager, T.N.; Mørk, A.; et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J. Neurosci. 2010, 30, 2396–2405. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef]
- Palacios, J.M. Serotonin receptors in brain revisited. Brain Res. 2016, 1645, 46–49. [Google Scholar] [CrossRef]
- Hoyer, D.; Hannon, J.P.; Martin, G.R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 2002, 71, 533–554. [Google Scholar] [CrossRef]
- Geyer, M.A.; Vollenweider, F.X. Serotonin research: Contributions to understanding psychoses. Trends Pharmacol. Sci. 2008, 29, 445–453. [Google Scholar] [CrossRef]
- Green, A.R. Neuropharmacology of 5-hydroxytryptamine. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S145–S152. [Google Scholar] [CrossRef]
- González-Maeso, J.; Weisstaub, N.V.; Zhou, M.; Chan, P.; Ivic, L.; Ang, R.; Lira, A.; Bradley-Moore, M.; Ge, Y.; Zhou, Q.; et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron 2007, 53, 439–452. [Google Scholar] [CrossRef]
- González-Maeso, J.; Yuen, T.; Ebersole, B.J.; Wurmbach, E.; Lira, A.; Zhou, M.; Weisstaub, N.; Hen, R.; Gingrich, J.A.; Sealfon, S.C. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J. Neurosci. 2003, 23, 8836–8843. [Google Scholar] [CrossRef]
- Mahesh, G.; Jaiswal, P.; Dey, S.; Sengupta, J.; Mukherjee, S. Cloning, Expression, Purification and Characterization of Oligomeric States of the Native 5HT2A G-Protein-Coupled Receptor. Protein Pept. Lett. 2018, 25, 390–397. [Google Scholar] [CrossRef]
- Rasmussen, H.; Frokjaer, V.G.; Hilker, R.W.; Madsen, J.; Anhøj, S.; Oranje, B.; Pinborg, L.H.; Glenthøj, B.; Knudsen, G.M. Low frontal serotonin 2A receptor binding is a state marker for schizophrenia? Eur. Neuropsychopharmacol. 2016, 26, 1248–1250. [Google Scholar] [CrossRef]
- Liégeois, J.F.; Ichikawa, J.; Meltzer, H.Y. 5-HT(2A) receptor antagonism potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and inhibits that in the nucleus accumbens in a dose-dependent manner. Brain Res. 2002, 947, 157–165. [Google Scholar] [CrossRef]
- Meltzer, H.Y. What’s atypical about atypical antipsychotic drugs? Curr. Opin. Pharmacol. 2004, 4, 53–57. [Google Scholar] [CrossRef]
- Kapur, S.; Remington, G. Dopamine D(2) receptors and their role in atypical antipsychotic action: Still necessary and may even be sufficient. Biol. Psychiatry 2001, 50, 873–883. [Google Scholar] [CrossRef]
- Kapur, S.; Wadenberg, M.L.; Remington, G. Are animal studies of antipsychotics appropriately dosed? Lessons from the bedside to the bench. Can. J. Psychiatry 2000, 45, 241–246. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Q.; Meng, L.; Ren, Y. Design of novel dopamine D. J. Biomol. Struct. Dyn. 2020, 38, 860–885. [Google Scholar] [CrossRef]
- Krause, M.; Zhu, Y.; Huhn, M.; Schneider-Thoma, J.; Bighelli, I.; Nikolakopoulou, A.; Leucht, S. Antipsychotic drugs for patients with schizophrenia and predominant or prominent negative symptoms: A systematic review and meta-analysis. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 268, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Tarsy, D.; Baldessarini, R.J.; Tarazi, F.I. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs 2002, 16, 23–45. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar] [CrossRef]
- McOmish, C.E.; Lira, A.; Hanks, J.B.; Gingrich, J.A. Clozapine-induced locomotor suppression is mediated by 5-HT2A receptors in the forebrain. Neuropsychopharmacology 2012, 37, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Creed-Carson, M.; Oraha, A.; Nobrega, J.N. Effects of 5-HT(2A) and 5-HT(2C) receptor antagonists on acute and chronic dyskinetic effects induced by haloperidol in rats. Behav. Brain Res. 2011, 219, 273–279. [Google Scholar] [CrossRef]
- Tsartsalis, S.; Tournier, B.B.; Gloria, Y.; Millet, P.; Ginovart, N. Effect of 5-HT2A receptor antagonism on levels of D2/3 receptor occupancy and adverse behavioral side-effects induced by haloperidol: A SPECT imaging study in the rat. Transl. Psychiatry 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef]
- Moreno, J.L.; Miranda-Azpiazu, P.; García-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.H.; González-Maeso, J. Serotonin and Glutamate Interactions in Preclinical Schizophrenia Models. ACS Chem. Neurosci. 2019, 10, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Holloway, T.; Albizu, L.; Sealfon, S.C.; González-Maeso, J. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci. Lett. 2011, 493, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Arnone, D.; Cappai, A.; Howes, O. Alterations in the serotonin system in schizophrenia: A systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci. Biobehav. Rev. 2014, 45, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Suhara, T.; Ichimiya, T.; Takano, A.; Ando, T.; Okubo, Y. Decreased 5-HT1A receptor binding in amygdala of schizophrenia. Biol. Psychiatry 2004, 55, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Mataix, L.; Scorza, M.C.; Bortolozzi, A.; Toth, M.; Celada, P.; Artigas, F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: Role in atypical antipsychotic action. J. Neurosci. 2005, 25, 10831–10843. [Google Scholar] [CrossRef]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Reavill, C.; Rogers, D.C. The therapeutic potential of 5-HT6 receptor antagonists. Curr. Opin. Investig. Drugs 2001, 2, 104–109. [Google Scholar] [PubMed]
- Nikiforuk, A. Serotonergic and Cholinergic Strategies as Potential Targets for the Treatment of Schizophrenia. Curr. Pharm. Des. 2016, 22, 2093–2116. [Google Scholar] [CrossRef]
- Murray, R.M.; Bhavsar, V.; Tripoli, G.; Howes, O. 30 Years on: How the Neurodevelopmental Hypothesis of Schizophrenia Morphed Into the Developmental Risk Factor Model of Psychosis. Schizophr. Bull. 2017, 43, 1190–1196. [Google Scholar] [CrossRef]
- Owen, M.J.; O’Donovan, M.C.; Thapar, A.; Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 2011, 198, 173–175. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Yao, Z.; Zhang, X.; Sun, J.; Zhang, Z. Pharmacogenetics of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor polymorphisms separately associate with positive and negative symptom response. Eur. Neuropsychopharmacol. 2005, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Arranz, M.; Collier, D.; Sodhi, M.; Ball, D.; Roberts, G.; Price, J.; Sham, P.; Kerwin, R. Association between clozapine response and allelic variation in 5-HT2A receptor gene. Lancet 1995, 346, 281–282. [Google Scholar] [CrossRef]
- Kim, J.H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25, 4749. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Hermans, E.; Challiss, R.A. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: Prototypic family C G-protein-coupled receptors. Biochem. J. 2001, 359, 465–484. [Google Scholar] [CrossRef]
- Page, G.; Khidir, F.A.; Pain, S.; Barrier, L.; Fauconneau, B.; Guillard, O.; Piriou, A.; Hugon, J. Group I metabotropic glutamate receptors activate the p70S6 kinase via both mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK 1/2) signaling pathways in rat striatal and hippocampal synaptoneurosomes. Neurochem. Int. 2006, 49, 413–421. [Google Scholar] [CrossRef]
- Correa, A.M.B.; Guimarães, J.D.S.; Dos Santos E Alhadas, E.; Kushmerick, C. Control of neuronal excitability by Group I metabotropic glutamate receptors. Biophys. Rev. 2017, 9, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A.; Angelicheva, D.; Vile, D.; Chandler, D.; Morar, B.; Cavanaugh, J.A.; Visscher, P.M.; Jablensky, A.; Pfleger, K.D.; Kalaydjieva, L. Deleterious GRM1 mutations in schizophrenia. PLoS ONE 2012, 7, e32849. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Eggan, S.M.; Lewis, D.A. Alterations in metabotropic glutamate receptor 1α and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. Am. J. Psychiatry 2010, 167, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Li, Y.; Takagi, S.; Presti, K.T.; Ramikie, T.S.; Rook, J.M.; Jones, C.K.; Lindsley, C.W.; Conn, P.J.; Bolshakov, V.Y.; et al. An mGlu5-Positive Allosteric Modulator Rescues the Neuroplasticity Deficits in a Genetic Model of NMDA Receptor Hypofunction in Schizophrenia. Neuropsychopharmacology 2016, 41, 2052–2061. [Google Scholar] [CrossRef]
- Ohishi, H.; Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: An in situ hybridization study. J. Comp. Neurol. 1993, 335, 252–266. [Google Scholar] [CrossRef]
- Mazzitelli, M.; Palazzo, E.; Maione, S.; Neugebauer, V. Group II Metabotropic Glutamate Receptors: Role in Pain Mechanisms and Pain Modulation. Front. Mol. Neurosci 2018, 11, 383. [Google Scholar] [CrossRef] [PubMed]
- Uslaner, J.M.; Smith, S.M.; Huszar, S.L.; Pachmerhiwala, R.; Hinchliffe, R.M.; Vardigan, J.D.; Hutson, P.H. Combined administration of an mGlu2/3 receptor agonist and a 5-HT 2A receptor antagonist markedly attenuate the psychomotor-activating and neurochemical effects of psychostimulants. Psychopharmacology 2009, 206, 641–651. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.W. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef]
- Cartmell, J.; Monn, J.A.; Schoepp, D.D. Attenuation of specific PCP-evoked behaviors by the potent mGlu2/3 receptor agonist, LY379268 and comparison with the atypical antipsychotic, clozapine. Psychopharmacology 2000, 148, 423–429. [Google Scholar] [CrossRef]
- Krystal, J.H.; Abi-Saab, W.; Perry, E.; D’Souza, D.C.; Liu, N.; Gueorguieva, R.; McDougall, L.; Hunsberger, T.; Belger, A.; Levine, L.; et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology 2005, 179, 303–309. [Google Scholar] [CrossRef]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Sealfon, S.C.; González-Maeso, J. Group II metabotropic glutamate receptors and schizophrenia. Cell. Mol. Life Sci. 2009, 66, 3777–3785. [Google Scholar] [CrossRef] [PubMed]
- Galici, R.; Echemendia, N.G.; Rodriguez, A.L.; Conn, P.J. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J. Pharmacol. Exp. Ther. 2005, 315, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Benneyworth, M.A.; Xiang, Z.; Smith, R.L.; Garcia, E.E.; Conn, P.J.; Sanders-Bush, E. A selective positive allosteric modulator of metabotropic glutamate receptor subtype 2 blocks a hallucinogenic drug model of psychosis. Mol. Pharmacol. 2007, 72, 477–484. [Google Scholar] [CrossRef]
- Kinon, B.J.; Gómez, J.C. Clinical development of pomaglumetad methionil: A non-dopaminergic treatment for schizophrenia. Neuropharmacology 2013, 66, 82–86. [Google Scholar] [CrossRef]
- Adams, D.H.; Kinon, B.J.; Baygani, S.; Millen, B.A.; Velona, I.; Kollack-Walker, S.; Walling, D.P. A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry 2013, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.R. Is there a path forward for mGlu(2) positive allosteric modulators for the treatment of schizophrenia? ACS Chem. Neurosci. 2013, 4, 211–213. [Google Scholar] [CrossRef][Green Version]
- Salih, H.; Anghelescu, I.; Kezic, I.; Sinha, V.; Hoeben, E.; Van Nueten, L.; De Smedt, H.; De Boer, P. Pharmacokinetic and pharmacodynamic characterisation of JNJ-40411813, a positive allosteric modulator of mGluR2, in two randomised, double-blind phase-I studies. J. Psychopharmacol. 2015, 29, 414–425. [Google Scholar] [CrossRef]
- Litman, R.E.; Smith, M.A.; Doherty, J.J.; Cross, A.; Raines, S.; Gertsik, L.; Zukin, S.R. AZD8529, a positive allosteric modulator at the mGluR2 receptor, does not improve symptoms in schizophrenia: A proof of principle study. Schizophr. Res. 2016, 172, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, L.; Bruno, V.; Salvatore, L.; Melchiorri, D.; Gradini, R.; Caricasole, A.; Barletta, E.; De Blasi, A.; Nicoletti, F. Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J. Neurochem. 2002, 82, 216–223. [Google Scholar] [CrossRef]
- Senter, R.K.; Ghoshal, A.; Walker, A.G.; Xiang, Z.; Niswender, C.M.; Conn, P.J. The Role of mGlu Receptors in Hippocampal Plasticity Deficits in Neurological and Psychiatric Disorders: Implications for Allosteric Modulators as Novel Therapeutic Strategies. Curr. Neuropharmacol. 2016, 14, 455–473. [Google Scholar] [CrossRef]
- Mena, A.; Ruiz-Salas, J.C.; Puentes, A.; Dorado, I.; Ruiz-Veguilla, M.; De la Casa, L.G. Reduced Prepulse Inhibition as a Biomarker of Schizophrenia. Front. Behav. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Wierońska, J.M.; Acher, F.C.; Sławińska, A.; Gruca, P.; Lasoń-Tyburkiewicz, M.; Papp, M.; Pilc, A. The antipsychotic-like effects of the mGlu group III orthosteric agonist, LSP1-2111, involves 5-HT1A signalling. Psychopharmacology 2013, 227, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.; Gołembiowska, K.; Noworyta-Sokołowska, K.; Acher, F.; Cieślik, P.; Kusek, M.; Tokarski, K.; Pilc, A.; Wierońska, J.M. Neurochemical and behavioral studies on the 5-HT. Neuropharmacology 2017, 115, 149–165. [Google Scholar] [CrossRef]
- Sławińska, A.; Wierońska, J.M.; Stachowicz, K.; Marciniak, M.; Lasoń-Tyburkiewicz, M.; Gruca, P.; Papp, M.; Kusek, M.; Tokarski, K.; Doller, D.; et al. The antipsychotic-like effects of positive allosteric modulators of metabotropic glutamate mGlu4 receptors in rodents. Br. J. Pharmacol. 2013, 169, 1824–1839. [Google Scholar] [CrossRef] [PubMed]
- Kalinichev, M.; Le Poul, E.; Boléa, C.; Girard, F.; Campo, B.; Fonsi, M.; Royer-Urios, I.; Browne, S.E.; Uslaner, J.M.; Davis, M.J.; et al. Characterization of the novel positive allosteric modulator of the metabotropic glutamate receptor 4 ADX88178 in rodent models of neuropsychiatric disorders. J. Pharmacol. Exp. Ther. 2014, 350, 495–505. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.M.; Finger, B.C.; Flor, P.J.; Cryan, J.F. Metabotropic glutamate receptor 7: At the interface of cognition and emotion. Eur. J. Pharmacol. 2010, 639, 123–131. [Google Scholar] [CrossRef]
- Suzuki, G.; Tsukamoto, N.; Fushiki, H.; Kawagishi, A.; Nakamura, M.; Kurihara, H.; Mitsuya, M.; Ohkubo, M.; Ohta, H. In vitro pharmacological characterization of novel isoxazolopyridone derivatives as allosteric metabotropic glutamate receptor 7 antagonists. J. Pharmacol. Exp. Ther. 2007, 323, 147–156. [Google Scholar] [CrossRef]
- Kalinichev, M.; Rouillier, M.; Girard, F.; Royer-Urios, I.; Bournique, B.; Finn, T.; Charvin, D.; Campo, B.; Le Poul, E.; Mutel, V.; et al. ADX71743, a potent and selective negative allosteric modulator of metabotropic glutamate receptor 7: In vitro and in vivo characterization. J. Pharmacol. Exp. Ther. 2013, 344, 624–636. [Google Scholar] [CrossRef]
- Cieślik, P.; Woźniak, M.; Kaczorowska, K.; Brański, P.; Burnat, G.; Chocyk, A.; Bobula, B.; Gruca, P.; Litwa, E.; Pałucha-Poniewiera, A.; et al. Negative Allosteric Modulators of mGlu. Front. Mol. Neurosci. 2018, 11, 316. [Google Scholar] [CrossRef]
- Gerlai, R.; Adams, B.; Fitch, T.; Chaney, S.; Baez, M. Performance deficits of mGluR8 knockout mice in learning tasks: The effects of null mutation and the background genotype. Neuropharmacology 2002, 43, 235–249. [Google Scholar] [CrossRef]
- Duvoisin, R.M.; Zhang, C.; Pfankuch, T.F.; O’Connor, H.; Gayet-Primo, J.; Quraishi, S.; Raber, J. Increased measures of anxiety and weight gain in mice lacking the group III metabotropic glutamate receptor mGluR8. Eur. J. Neurosci. 2005, 22, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Duvoisin, R.M.; Raber, J. Related functions of mGlu4 and mGlu8. Pharmacol. Biochem. Behav. 2013, 111, 11–16. [Google Scholar] [CrossRef][Green Version]
- Fendt, M.; Bürki, H.; Imobersteg, S.; van der Putten, H.; McAllister, K.; Leslie, J.C.; Shaw, D.; Hölscher, C. The effect of mGlu8 deficiency in animal models of psychiatric diseases. Genes Brain Behav. 2010, 9, 33–44. [Google Scholar] [CrossRef]
- Ossowska, K.; Pietraszek, M.; Wardas, J.; Wolfarth, S. Potential antipsychotic and extrapyramidal effects of (R,S)-3,4-dicarboxyphenylglycine [(R,S)-3,4-DCPG], a mixed AMPA antagonist/mGluR8 agonist. Pol. J. Pharmacol. 2004, 56, 295–304. [Google Scholar]
- Robbins, M.J.; Starr, K.R.; Honey, A.; Soffin, E.M.; Rourke, C.; Jones, G.A.; Kelly, F.M.; Strum, J.; Melarange, R.A.; Harris, A.J.; et al. Evaluation of the mGlu8 receptor as a putative therapeutic target in schizophrenia. Brain Res. 2007, 1152, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Mizukami, K.; Iwakiri, M.; Asada, T. Immunohistochemical and immunoblot analysis of gamma-aminobutyric acid B receptor in the prefrontal cortex of subjects with schizophrenia and bipolar disorder. Neurosci. Lett. 2005, 383, 272–277. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D.; Thuras, P.D. Deficits in GABA(B) receptor system in schizophrenia and mood disorders: A postmortem study. Schizophr. Res. 2011, 128, 37–43. [Google Scholar] [CrossRef]
- Chalifoux, J.R.; Carter, A.G. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron 2010, 66, 101–113. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D.; Thuras, P.D. GABAA and GABAB receptor dysregulation in superior frontal cortex of subjects with schizophrenia and bipolar disorder. Synapse 2017, 71. [Google Scholar] [CrossRef]
- Li, P.; Stewart, R.; Butler, A.; Gonzalez-Cota, A.L.; Harmon, S.; Salkoff, L. GABA-B Controls Persistent Na. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Mizukami, K.; Ishikawa, M.; Hidaka, S.; Iwakiri, M.; Sasaki, M.; Iritani, S. Immunohistochemical localization of GABAB receptor in the entorhinal cortex and inferior temporal cortex of schizophrenic brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2002, 26, 393–396. [Google Scholar] [CrossRef]
- Zai, G.; King, N.; Wong, G.W.; Barr, C.L.; Kennedy, J.L. Possible association between the gamma-aminobutyric acid type B receptor 1 (GABBR1) gene and schizophrenia. Eur. Neuropsychopharmacol. 2005, 15, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Harada, S.; Kawanishi, Y.; Tachikawa, H.; Okubo, T.; Asada, T. Association analysis of an (AC)n repeat polymorphism in the GABA(B) receptor gene and schizophrenia. Am. J. Med. Genet. 2002, 114, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qin, S.; Shi, Y.; Zhang, A.; Zhang, J.; Bian, L.; Wan, C.; Feng, G.; Gu, N.; Zhang, G.; et al. Systematic study of association of four GABAergic genes: Glutamic acid decarboxylase 1 gene, glutamic acid decarboxylase 2 gene, GABA(B) receptor 1 gene and GABA(A) receptor subunit beta2 gene, with schizophrenia using a universal DNA microarray. Schizophr. Res. 2007, 93, 374–384. [Google Scholar] [CrossRef]
- Klempan, T.A.; Sequeira, A.; Canetti, L.; Lalovic, A.; Ernst, C.; ffrench-Mullen, J.; Turecki, G. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 2009, 14, 175–189. [Google Scholar] [CrossRef]
- Kantrowitz, J.; Citrome, L.; Javitt, D. GABA(B) receptors, schizophrenia and sleep dysfunction: A review of the relationship and its potential clinical and therapeutic implications. CNS Drugs 2009, 23, 681–691. [Google Scholar] [CrossRef]
- Arai, S.; Takuma, K.; Mizoguchi, H.; Ibi, D.; Nagai, T.; Takahashi, K.; Kamei, H.; Nabeshima, T.; Yamada, K. Involvement of pallidotegmental neurons in methamphetamine- and MK-801-induced impairment of prepulse inhibition of the acoustic startle reflex in mice: Reversal by GABAB receptor agonist baclofen. Neuropsychopharmacology 2008, 33, 3164–3175. [Google Scholar] [CrossRef]
- Bortolato, M.; Frau, R.; Aru, G.N.; Orrù, M.; Gessa, G.L. Baclofen reverses the reduction in prepulse inhibition of the acoustic startle response induced by dizocilpine, but not by apomorphine. Psychopharmacology 2004, 171, 322–330. [Google Scholar] [CrossRef]
- Fejgin, K.; Pålsson, E.; Wass, C.; Finnerty, N.; Lowry, J.; Klamer, D. Prefrontal GABA(B) receptor activation attenuates phencyclidine-induced impairments of prepulse inhibition: Involvement of nitric oxide. Neuropsychopharmacology 2009, 34, 1673–1684. [Google Scholar] [CrossRef][Green Version]
- Kaupmann, K.; Cryan, J.F.; Wellendorph, P.; Mombereau, C.; Sansig, G.; Klebs, K.; Schmutz, M.; Froestl, W.; van der Putten, H.; Mosbacher, J.; et al. Specific gamma-hydroxybutyrate-binding sites but loss of pharmacological effects of gamma-hydroxybutyrate in GABA(B)(1)-deficient mice. Eur. J. Neurosci. 2003, 18, 2722–2730. [Google Scholar] [CrossRef]
- Ma, J.; Stan Leung, L. Effects of GABA-B receptor positive modulator on ketamine-induced psychosis-relevant behaviors and hippocampal electrical activity in freely moving rats. Psychopharmacology 2017, 234, 3129–3142. [Google Scholar] [CrossRef] [PubMed]
- Helm, K.A.; Haberman, R.P.; Dean, S.L.; Hoyt, E.C.; Melcher, T.; Lund, P.K.; Gallagher, M. GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharmacology 2005, 48, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Leung, L.S. GABA(B) receptor blockade in the hippocampus affects sensory and sensorimotor gating in Long-Evans rats. Psychopharmacology 2011, 217, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Selten, M.M.; Meyer, F.; Ba, W.; Vallès, A.; Maas, D.A.; Negwer, M.; Eijsink, V.D.; van Vugt, R.W.M.; van Hulten, J.A.; van Bakel, N.H.M.; et al. Increased GABAB receptor signaling in a rat model for schizophrenia. Sci. Rep. 2016, 6, 34240. [Google Scholar] [CrossRef]
- Wierońska, J.M.; Kusek, M.; Tokarski, K.; Wabno, J.; Froestl, W.; Pilc, A. The GABA B receptor agonist CGP44532 and the positive modulator GS39783 reverse some behavioural changes related to positive syndromes of psychosis in mice. Br. J. Pharmacol. 2011, 163, 1034–1047. [Google Scholar] [CrossRef]
- Cedillo, L.N.; Miranda, F. Effects of co-administration of the GABAB receptor agonist baclofen and a positive allosteric modulator of the GABAB receptor, CGP7930, on the development and expression of amphetamine-induced locomotor sensitization in rats. Pharmacol. Rep. 2013, 65, 1132–1143. [Google Scholar] [CrossRef]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O.; Bastiampillai, T. Binding of clozapine to the GABA. Mol. Psychiatry 2020, 25, 1910–1919. [Google Scholar] [CrossRef]
- Otmakhova, N.A.; Lisman, J.E. Contribution of Ih and GABAB to synaptically induced afterhyperpolarizations in CA1: A brake on the NMDA response. J. Neurophysiol. 2004, 92, 2027–2039. [Google Scholar] [CrossRef]
- Pérez-Garci, E.; Gassmann, M.; Bettler, B.; Larkum, M.E. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron 2006, 50, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Vida, I.; Luján, R.; Haas, C.A.; López-Bendito, G.; Shigemoto, R.; Frotscher, M. Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J. Neurosci. 2003, 23, 11026–11035. [Google Scholar] [CrossRef]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef]
- Eiger, D.S.; Boldizsar, N.; Honeycutt, C.C.; Gardner, J.; Rajagopal, S. Biased agonism at chemokine receptors. Cell. Signal. 2021, 78, 109862. [Google Scholar] [CrossRef]
- Bachelerie, F.; Graham, G.J.; Locati, M.; Mantovani, A.; Murphy, P.M.; Nibbs, R.; Rot, A.; Sozzani, S.; Thelen, M. New nomenclature for atypical chemokine receptors. Nat. Immunol. 2014, 15, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Graham, G.J. Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Kang, D.S.; Benovic, J.L. β-arrestins and G protein-coupled receptor trafficking. Handb. Exp. Pharmacol. 2014, 219, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef]
- Bennett, L.D.; Fox, J.M.; Signoret, N. Mechanisms regulating chemokine receptor activity. Immunology 2011, 134, 246–256. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Stone, M.J.; Hayward, J.A.; Huang, C.; E Huma, Z.; Sanchez, J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef]
- Stephens, B.; Handel, T.M. Chemokine receptor oligomerization and allostery. Prog. Mol. Biol. Transl. Sci. 2013, 115, 375–420. [Google Scholar] [CrossRef]
- Yang, L.K.; Hou, Z.S.; Tao, Y.X. Biased signaling in naturally occurring mutations of G protein-coupled receptors associated with diverse human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165973. [Google Scholar] [CrossRef] [PubMed]
- Rostène, W.; Dansereau, M.A.; Godefroy, D.; Van Steenwinckel, J.; Reaux-Le Goazigo, A.; Mélik-Parsadaniantz, S.; Apartis, E.; Hunot, S.; Beaudet, N.; Sarret, P. Neurochemokines: A menage a trois providing new insights on the functions of chemokines in the central nervous system. J. Neurochem. 2011, 118, 680–694. [Google Scholar] [CrossRef]
- Rostène, W.; Guyon, A.; Kular, L.; Godefroy, D.; Barbieri, F.; Bajetto, A.; Banisadr, G.; Callewaere, C.; Conductier, G.; Rovère, C.; et al. Chemokines and chemokine receptors: New actors in neuroendocrine regulations. Front. Neuroendocrinol. 2011, 32, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Najjar, S.; Pahlajani, S.; De Sanctis, V.; Stern, J.N.H.; Najjar, A.; Chong, D. Neurovascular Unit Dysfunction and Blood-Brain Barrier Hyperpermeability Contribute to Schizophrenia Neurobiology: A Theoretical Integration of Clinical and Experimental Evidence. Front. Psychiatry 2017, 8, 83. [Google Scholar] [CrossRef]
- Ochoa, S.; Usall, J.; Cobo, J.; Labad, X.; Kulkarni, J. Gender differences in schizophrenia and first-episode psychosis: A comprehensive literature review. Schizophr. Res. Treat. 2012, 2012, 916198. [Google Scholar] [CrossRef]
- Mendrek, A.; Mancini-Marïe, A. Sex/gender differences in the brain and cognition in schizophrenia. Neurosci. Biobehav. Rev. 2016, 67, 57–78. [Google Scholar] [CrossRef]
- Cartier, L.; Hartley, O.; Dubois-Dauphin, M.; Krause, K.H. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res. Brain Res. Rev. 2005, 48, 16–42. [Google Scholar] [CrossRef]
- Pedemonte, E.; Mancardi, G.; Giunti, D.; Corcione, A.; Benvenuto, F.; Pistoia, V.; Uccelli, A. Mechanisms of the adaptive immune response inside the central nervous system during inflammatory and autoimmune diseases. Pharmacol. Ther. 2006, 111, 555–566. [Google Scholar] [CrossRef]
- Ivanovska, M.; Abdi, Z.; Murdjeva, M.; Macedo, D.; Maes, A.; Maes, M. CCL-11 or Eotaxin-1: An Immune Marker for Ageing and Accelerated Ageing in Neuro-Psychiatric Disorders. Pharmaceuticals 2020, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Sirivichayakul, S.; Kanchanatawan, B.; Thika, S.; Carvalho, A.F.; Maes, M. Eotaxin, an Endogenous Cognitive Deteriorating Chemokine (ECDC), Is a Major Contributor to Cognitive Decline in Normal People and to Executive, Memory, and Sustained Attention Deficits, Formal Thought Disorders, and Psychopathology in Schizophrenia Patients. Neurotox. Res. 2019, 35, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.; Massuda, R.; de Lucena, D.; Macêdo, D.; Paz, A.V.; Lobato, M.I.; Belmonte-de-Abreu, P.S.; Ceresér, K.M.; Rocha, N.P.; Curra, M.D.; et al. Differences in eotaxin serum levels patients with recent onset and in chronic stable schizophrenia: A clue for understanding accelerating aging profile. Schizophr. Res. 2014, 152, 528–529. [Google Scholar] [CrossRef]
- Frydecka, D.; Krzystek-Korpacka, M.; Lubeiro, A.; Stramecki, F.; Stańczykiewicz, B.; Beszłej, J.A.; Piotrowski, P.; Kotowicz, K.; Szewczuk-Bogusławska, M.; Pawlak-Adamska, E.; et al. Profiling inflammatory signatures of schizophrenia: A cross-sectional and meta-analysis study. Brain Behav. Immun. 2018, 71, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Al-Hakeim, H.K.; Almulla, A.F.; Maes, M. The Neuroimmune and Neurotoxic Fingerprint of Major Neurocognitive Psychosis or Deficit Schizophrenia: A Supervised Machine Learning Study. Neurotox. Res. 2020, 37, 753–771. [Google Scholar] [CrossRef]
- Al-Dujaili, A.H.; Mousa, R.F.; Al-Hakeim, H.K.; Maes, M. High Mobility Group Protein 1 and Dickkopf-Related Protein 1 in Schizophrenia and Treatment-Resistant Schizophrenia: Associations With Interleukin-6, Symptom Domains, and Neurocognitive Impairments. Schizophr. Bull. 2021, 47, 530–541. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Reis, H.J.; Nicolato, R.; Brito-Melo, G.; Correa, H.; Teixeira, M.M.; Romano-Silva, M.A. Increased serum levels of CCL11/eotaxin in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 710–714. [Google Scholar] [CrossRef]
- Cronshaw, D.G.; Kouroumalis, A.; Parry, R.; Webb, A.; Brown, Z.; Ward, S.G. Evidence that phospholipase-C-dependent, calcium-independent mechanisms are required for directional migration of T-lymphocytes in response to the CCR4 ligands CCL17 and CCL22. J. Leukoc. Biol. 2006, 79, 1369–1380. [Google Scholar] [CrossRef]
- Smit, M.J.; Verdijk, P.; van der Raaij-Helmer, E.M.; Navis, M.; Hensbergen, P.J.; Leurs, R.; Tensen, C.P. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 2003, 102, 1959–1965. [Google Scholar] [CrossRef]
- Soriano, S.F.; Serrano, A.; Hernanz-Falcón, P.; Martín de Ana, A.; Monterrubio, M.; Martínez, C.; Rodríguez-Frade, J.M.; Mellado, M. Chemokines integrate JAK/STAT and G-protein pathways during chemotaxis and calcium flux responses. Eur. J. Immunol. 2003, 33, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Fillman, S.G.; Cloonan, N.; Miller, L.C.; Weickert, C.S. Markers of inflammation in the prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2013, 18, 133. [Google Scholar] [CrossRef][Green Version]
- Brown, A.S. Prenatal infection as a risk factor for schizophrenia. Schizophr. Bull. 2006, 32, 200–202. [Google Scholar] [CrossRef]
- Ellman, L.M.; Deicken, R.F.; Vinogradov, S.; Kremen, W.S.; Poole, J.H.; Kern, D.M.; Tsai, W.Y.; Schaefer, C.A.; Brown, A.S. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 2010, 121, 46–54. [Google Scholar] [CrossRef]
- Martinelli, R.; Sabroe, I.; LaRosa, G.; Williams, T.J.; Pease, J.E. The CC chemokine eotaxin (CCL11) is a partial agonist of CC chemokine receptor 2b. J. Biol. Chem. 2001, 276, 42957–42964. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Bechter, K.; Fuxe, K. IL1R2, CCR2, and CXCR4 May Form Heteroreceptor Complexes with NMDAR and D2R: Relevance for Schizophrenia. Front. Psychiatry 2017, 8, 24. [Google Scholar] [CrossRef]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef]
- Cardona, A.E.; Sasse, M.E.; Liu, L.; Cardona, S.M.; Mizutani, M.; Savarin, C.; Hu, T.; Ransohoff, R.M. Scavenging roles of chemokine receptors: Chemokine receptor deficiency is associated with increased levels of ligand in circulation and tissues. Blood 2008, 112, 256–263. [Google Scholar] [CrossRef]
- Reshef, R.; Kudryavitskaya, E.; Shani-Narkiss, H.; Isaacson, B.; Rimmerman, N.; Mizrahi, A.; Yirmiya, R. The role of microglia and their CX3CR1 signaling in adult neurogenesis in the olfactory bulb. Elife 2017, 6. [Google Scholar] [CrossRef]
- Chamera, K.; Trojan, E.; Szuster-Głuszczak, M.; Basta-Kaim, A. The Potential Role of Dysfunctions in Neuron-Microglia Communication in the Pathogenesis of Brain Disorders. Curr. Neuropharmacol. 2020, 18, 408–430. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Stuart, M.J.; Singhal, G.; Baune, B.T. Systematic Review of the Neurobiological Relevance of Chemokines to Psychiatric Disorders. Front. Cell. Neurosci. 2015, 9, 357. [Google Scholar] [CrossRef]
- Asevedo, E.; Gadelha, A.; Noto, C.; Mansur, R.B.; Zugman, A.; Belangero, S.I.; Berberian, A.A.; Scarpato, B.S.; Leclerc, E.; Teixeira, A.L.; et al. Impact of peripheral levels of chemokines, BDNF and oxidative markers on cognition in individuals with schizophrenia. J. Psychiatr. Res. 2013, 47, 1376–1382. [Google Scholar] [CrossRef]
- Ishizuka, K.; Fujita, Y.; Kawabata, T.; Kimura, H.; Iwayama, Y.; Inada, T.; Okahisa, Y.; Egawa, J.; Usami, M.; Kushima, I.; et al. Rare genetic variants in CX3CR1 and their contribution to the increased risk of schizophrenia and autism spectrum disorders. Transl Psychiatry 2017, 7, e1184. [Google Scholar] [CrossRef]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Jiang, P.; Zhang, R.; Xu, L.; Gou, X. Glioma in Schizophrenia: Is the Risk Higher or Lower? Front. Cell. Neurosci 2018, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- García-Cuesta, E.M.; Santiago, C.A.; Vallejo-Díaz, J.; Juarranz, Y.; Rodríguez-Frade, J.M.; Mellado, M. The Role of the CXCL12/CXCR4/ACKR3 Axis in Autoimmune Diseases. Front. Endocrinol. (Lausanne) 2019, 10, 585. [Google Scholar] [CrossRef]
- Malmqvist, A.; Schwieler, L.; Orhan, F.; Fatouros-Bergman, H.; Bauer, M.; Flyckt, L.; Cervenka, S.; Engberg, G.; Piehl, F.; Erhardt, S.; et al. Increased peripheral levels of TARC/CCL17 in first episode psychosis patients. Schizophr. Res. 2019, 210, 221–227. [Google Scholar] [CrossRef]
- Laurikainen, H.; Vuorela, A.; Toivonen, A.; Reinert-Hartwall, L.; Trontti, K.; Lindgren, M.; Keinänen, J.; Mäntylä, T.; Paju, J.; Ilonen, T.; et al. Elevated serum chemokine CCL22 levels in first-episode psychosis: Associations with symptoms, peripheral immune state and in vivo brain glial cell function. Transl. Psychiatry 2020, 10, 94. [Google Scholar] [CrossRef]
- Hill, S.L.; Shao, L.; Beasley, C.L. Diminished levels of the chemokine fractalkine in post-mortem prefrontal cortex in schizophrenia but not bipolar disorder. World J. Biol. Psychiatry 2020, 1–10. [Google Scholar] [CrossRef]
- Chamera, K.; Szuster-Głuszczak, M.; Trojan, E.; Basta-Kaim, A. Maternal Immune Activation Sensitizes Male Offspring Rats to Lipopolysaccharide-Induced Microglial Deficits Involving the Dysfunction of CD200-CD200R and CX3CL1-CX3CR1 Systems. Cells 2020, 9, 1676. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, J.; Zhang, Y.; Shao, F.; Wang, W. The Role of Microglial CX3CR1 in Schizophrenia-Related Behaviors Induced by Social Isolation. Front. Integr. Neurosci. 2020, 14, 551676. [Google Scholar] [CrossRef]
- Cathomas, F.; Klaus, F.; Guetter, K.; Chung, H.K.; Raja Beharelle, A.; Spiller, T.R.; Schlegel, R.; Seifritz, E.; Hartmann-Riemer, M.N.; Tobler, P.N.; et al. Increased random exploration in schizophrenia is associated with inflammation. NPJ Schizophr 2021, 7, 6. [Google Scholar] [CrossRef]
- Ranasinghe, R.; Eri, R. Pleiotropic Immune Functions of Chemokine Receptor 6 in Health and Disease. Medicines 2018, 5, 69. [Google Scholar] [CrossRef]
- Na, K.S.; Jung, H.Y.; Kim, Y.K. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 277–286. [Google Scholar] [CrossRef]
- Fillman, S.G.; Sinclair, D.; Fung, S.J.; Webster, M.J.; Shannon Weickert, C. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl. Psychiatry 2014, 4, e365. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, D.; Weickert, T.W.; Lenroot, R.; O’Donnell, M.; Galletly, C.; Liu, D.; Burgess, M.; Cadiz, R.; Jacomb, I.; Catts, V.S.; et al. Using blood cytokine measures to define high inflammatory biotype of schizophrenia and schizoaffective disorder. J. Neuroinflamm. 2017, 14, 188. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Potential | Type | Mechanism of Action | CNS Distribution |
|---|---|---|---|---|
| 5-HT1 (5-HT1A, 1B, 1D–F) | Inhibitory | Gi/G0-protein coupled | Inhibition of AC and decreasing intracellular concentration of cAMP | cerebral and frontal cortex, hippocampus, striatum, olfactory bulb, substantia nigra |
| 5-HT2 (5-HT2A–C) | Excitatory | Gq11- protein coupled | Activation of PLC, increasing intracellular concentration of IP3 and DAG, and increasing intracellular calcium | nucleus accumbens, basal ganglia, cerebellum, hypothalamus |
| 5-HT3 (5-HT3A,3B) | Excitatory | Ligand-gated Na+/K+ channel | Depolarization of cell plasma membrane | hippocampus, amygdala, nucleus accumbens |
| 5-HT4 (5-HT4A–H) | Excitatory | Gs-protein coupled | Activation of AC and increasing intracellular concentration of cAMP | hippocampal membranes |
| 5-HT5 (5-HT5A) | Inhibitory | Gi/G0-protein coupled | Inhibition of AC and decreasing intracellular concentration of cAMP | olfactory bulb, neocortex, hippocampus, caudate putamen |
| 5-HT6 | Excitatory | Gs-protein coupled | Activation of AC and increasing intracellular concentration of cAMP | thalamus, hypothalamus, hippocampus |
| 5-HT7 (5-HT7A–D) | Excitatory | Gs-protein coupled | Activation of AC and increasing intracellular concentration of cAMP | thalamus, hypothalamus, hippocampus |
| CCL2 | CCL3 | CCL4 | CCL5 | CCL7 | CCL8 | CCL11 | CCL13 | CCL17 | CCL20 |
| MCP-1 | MIP-1α | MIP-1β | RANTES | MCP-3 | MCP-2 | Eotaxin-1 | MCP-4 | TARC | MIP-3α |
| CCL22 | CCL23 | CCL24 | CCL25 | CCL26 | CCL28 | ||||
| ABCD-1 | MPIF-1 | MPIF-2 | TECK | MIP-4α | MEC | ||||
| Eotaxin-2 | Eotaxin-3 | ||||||||
| CXCL1 | CXCL4 | CXCL5 | CXCL6 | CXCL8 | CXCL9 | CXCL10 | CXCL11 | CXCL12 | CXCL21 |
| MGSA | PF4 | ENA78 | GCP-2 | IL-8 | MIG | IP-10 | I-TAC | SDF1 | SLC |
| CX3CL1 | |||||||||
| Fractalkine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczek, T.; Mackiewicz, J.; Sobolczyk, M.; Wawrzyniak, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Cells 2021, 10, 1228. https://doi.org/10.3390/cells10051228
Boczek T, Mackiewicz J, Sobolczyk M, Wawrzyniak J, Lisek M, Ferenc B, Guo F, Zylinska L. The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Cells. 2021; 10(5):1228. https://doi.org/10.3390/cells10051228
Chicago/Turabian StyleBoczek, Tomasz, Joanna Mackiewicz, Marta Sobolczyk, Julia Wawrzyniak, Malwina Lisek, Bozena Ferenc, Feng Guo, and Ludmila Zylinska. 2021. "The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines" Cells 10, no. 5: 1228. https://doi.org/10.3390/cells10051228
APA StyleBoczek, T., Mackiewicz, J., Sobolczyk, M., Wawrzyniak, J., Lisek, M., Ferenc, B., Guo, F., & Zylinska, L. (2021). The Role of G Protein-Coupled Receptors (GPCRs) and Calcium Signaling in Schizophrenia. Focus on GPCRs Activated by Neurotransmitters and Chemokines. Cells, 10(5), 1228. https://doi.org/10.3390/cells10051228