
Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition

 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Langendorff Perfusion of Isolated Rat Heart
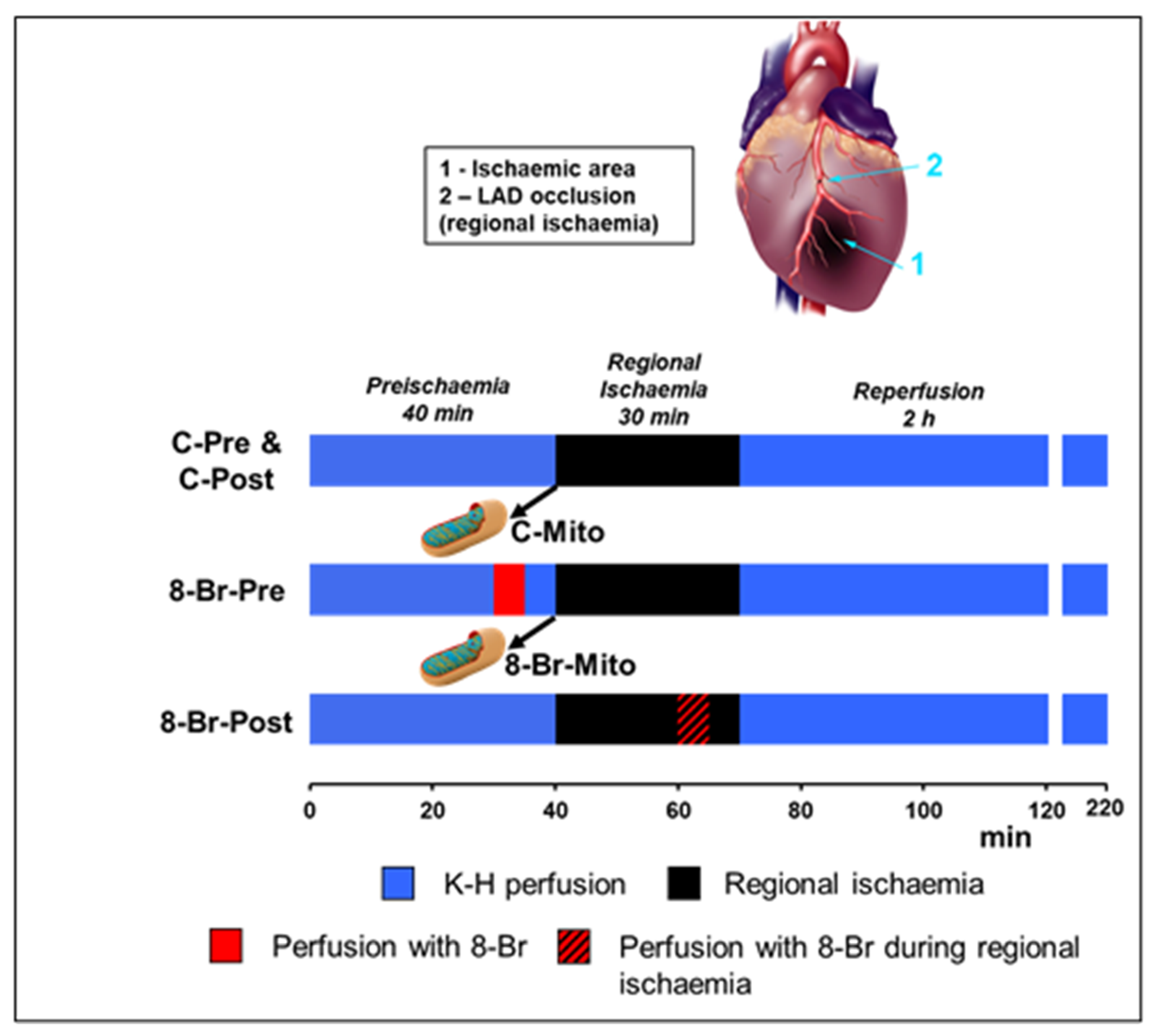
2.4. Heart Perfusion Protocols
2.5. Analysis of the Left Ventricular Arrhythmias
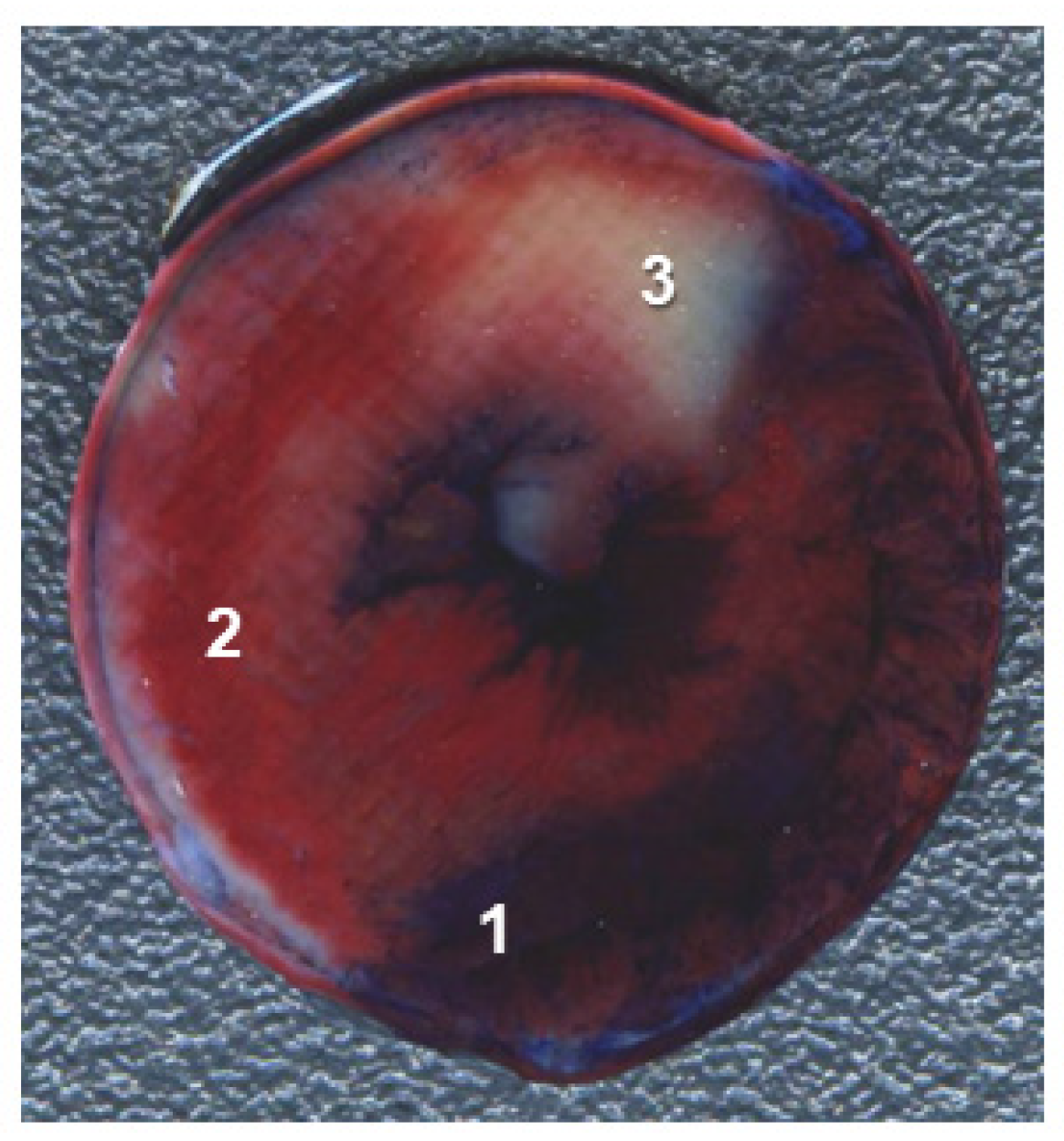
2.6. Infarct Size
2.7. Ca2+-Induced Mitochondria Swelling Assay
2.8. Evaluation of Mitochondria-Bound Hexokinase II (HKII)
2.9. Western Blots (Binding of Hexokinase II to Mitochondria)
2.10. Mitochondrial Permeability Transition Pore (MPTP) Opening
2.11. Statistical Analysis
3. Results
3.1. The Effects of 8-Br Added before Ischemia
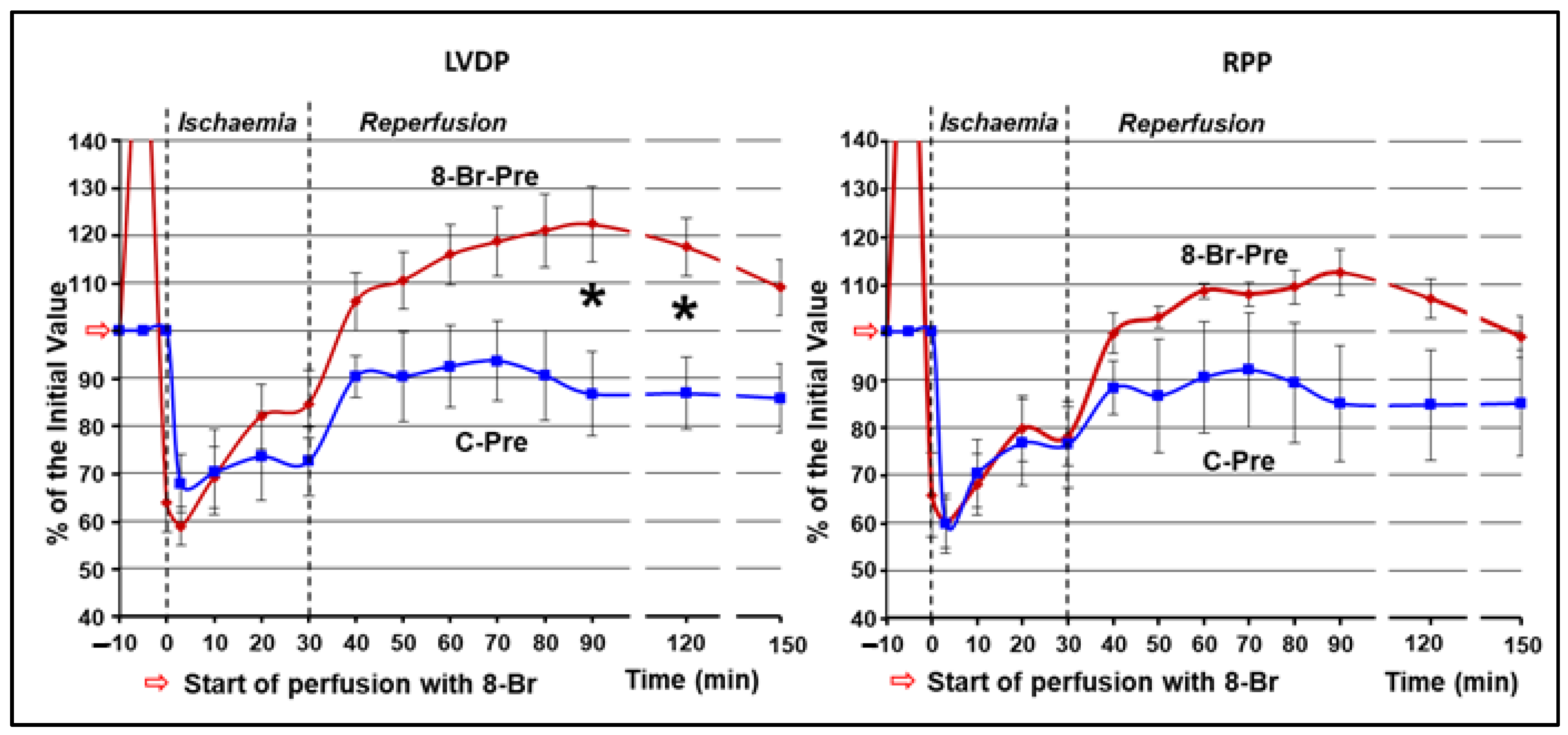
3.1.1. Changes in Haemodynamic Function
3.1.2. Changes in Haemodynamic Function during Ischaemia and Reperfusion
3.2. The Effects of 8-Br Added at the End of Ischemia
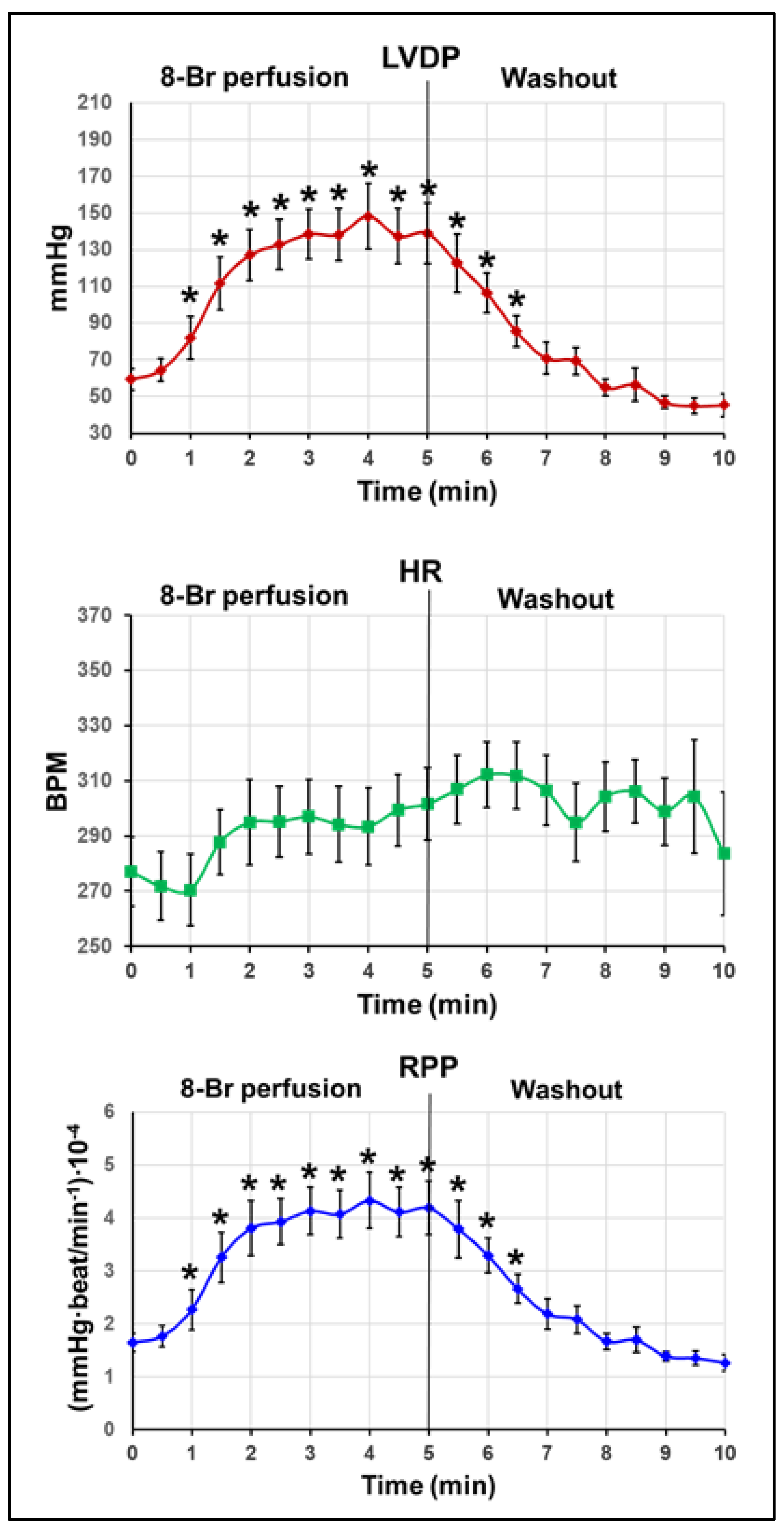
3.2.1. Changes in the Haemodynamic Function during 8-Br Treatment
3.2.2. Changes in Haemodynamic Function during Regional Ischaemia and Reperfusion
3.2.3. Effect of 8-Br on Ventricular Arrhythmias during I/R
3.2.4. Cardiac Injury
3.3. Ca2+-Induced Mitochondria Swelling
3.4. Hexokinase II Binding to Mitochondria
4. Discussion
4.1. 8-Br Protects the Heart against Regional Ischaemia when Applied Either before Ischaemia or by the Onset of Reperfusion
4.2. Perfusion of Hearts with 8-Br Inhibits MPTP
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Serruys, P.W.; Morice, M.-C.; Kappetein, A.P.; Colombo, A.; Holmes, D.R.; Mack, M.J.; Ståhle, E.; Feldman, T.E.; van den Brand, M.V.D.; Bass, E.J.; et al. Percutaneous Coronary Intervention versus Coronary-Artery Bypass Grafting for Severe Coronary Artery Disease. N. Engl. J. Med. 2009, 360, 961–972. [Google Scholar] [CrossRef]
- Joyal, D.; Afilalo, J.; Rinfret, S. Effectiveness of recanalization of chronic total occlusions: A systematic review and meta-analysis. Am. Heart J. 2010, 160, 179–187. [Google Scholar] [CrossRef]
- Ito, H.; Okamura, A.; Iwakura, K.; Masuyama, T.; Hori, M.; Takiuchi, S.; Negoro, S.; Nakatsuchi, Y.; Taniyama, Y.; Higashi-no, Y. Myocardial perfusion patterns related to thrombolysis in myocardial infarction perfusion grades after coronary angio-plasty in patients with acute anterior wall myocardial infarction. Circulation 1996, 93, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.S.; Curtis, J.P.; Chen, J.; Wang, Y.; Nallamothu, B.K.; Epstein, A.J.; Krumholz, H.M.; Registry, F.T.N.C.D. Association of door-to-balloon time and mortality in patients admitted to hospital with ST elevation myocardial infarction: National cohort study. BMJ 2009, 338, b1807. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfu-sion-a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1402–1415. [Google Scholar] [CrossRef]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and Ischemia/Reperfusion Injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Genade, S.; Tromp, E.; Podzuweit, T.; Moolman, J.A. Ischemic Preconditioning and the {beta}-Adrenergic Signal Transduction Pathway. Circulation 1999, 100, 958–966. [Google Scholar] [CrossRef]
- Khaliulin, I.; Halestrap, A.P.; Bryant, S.M.; Dudley, D.J.; James, A.F.; Suleiman, M.-S. Clinically-relevant consecutive treat-ment with isoproterenol and adenosine protects the failing heart against ischaemia and reperfusion. J. Transl. Med. 2014, 12, 1–13. [Google Scholar] [CrossRef]
- Khaliulin, I.; Parker, J.E.; Halestrap, A.P. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc. Res. 2010, 88, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 2013, 4, 264. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Going to cAMP just got more complicated. J. Physiol. 2007, 583, 415. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Khaliulin, I.; Bond, M.; James, A.F.; Dyar, Z.; Amini, R.; Johnson, J.L.; Suleiman, M. Functional and cardioprotective effects of simultaneous and individual activation of protein kinase A and Epac. Br. J. Pharmacol. 2017, 174, 438–453. [Google Scholar] [CrossRef]
- Fishbein, G.; Fishbein, M.; Buja, L. Myocardial ischemia and its complications. In Cardiovascular Pathology; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 239–270. [Google Scholar]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [PubMed]
- Khaliulin, I.; Halestrap, A.P.; Suleiman, M.-S. Temperature preconditioning is optimal at 26 C and confers additional protec-tion to hypothermic cardioplegic ischemic arrest. Exper. Biol. Med. 2011, 236, 736–745. [Google Scholar] [CrossRef]
- Klocke, R.; Tian, W.; Kuhlmann, M.T.; Nikol, S. Surgical animal models of heart failure related to coronary heart disease. Cardiovasc. Res. 2007, 74, 29–38. [Google Scholar] [CrossRef]
- Walker, M.; Curtis, M.; Hearse, D.; Campbell, R.; Janse, M.; Yellon, D.; Cobbe, S.; Coker, S.; Harness, J.; Harron, D. The Lam-beth Conventions: Guidelines for the study of arrhythmias in ischaemia, infarction, and reperfusion. Cardiovasc. Res. 1988, 22, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Walker, M.J. Quantification of arrhythmias using scoring systems: An examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc. Res. 1988, 22, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Van Vuuren, D.; Genis, A.; Genade, S.; Lochner, A. Postconditioning the Isolated Working Rat Heart. Cardiovasc. Drugs Ther. 2008, 22, 391–397. [Google Scholar] [CrossRef]
- Halestrap, A.P. The regulation of the oxidation of fatty acids and other substrates in rat heart mitochondria by changes in the matrix volume induced by osmotic strength, valinomycin and Ca2+. Biochem. J. 1987, 244, 159–164. [Google Scholar] [CrossRef]
- Clarke, S.J.; Khaliulin, I.; Das, M.; Parker, J.E.; Heesom, K.J.; Halestrap, A.P. Inhibition of mitochondrial permeability transi-tion pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondri-al protein phosphorylation. Circ. Res. 2008, 102, 1082–1090. [Google Scholar] [CrossRef]
- Pasdois, P.; Parker, J.E.; Halestrap, A.P. Extent of Mitochondrial Hexokinase II Dissociation During Ischemia Correlates with Mitochondrial Cytochrome c Release, Reactive Oxygen Species Production, and Infarct Size on Reperfusion. J. Am. Hear. Assoc. 2013, 2, e005645. [Google Scholar] [CrossRef]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transi-tion pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.A.; Clarke, S.; Das, M.; Griffiths, E.J.; Lim, K.H.H.; Halestrap, A.P. Ischaemic Preconditioning Inhibits Opening of Mitochondrial Permeability Transition Pores in the Reperfused Rat Heart. J. Physiol. 2003, 549, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef]
- Wong, R.; Steenbergen, C.; Murphy, E. Mitochondrial permeability transition pore and calcium handling. In Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin, Germany, 2012; Volume 810, pp. 235–242. [Google Scholar]
- Sheehan, F.H.; Doerr, R.; Schmidt, W.G.; Bolson, E.L.; Uebis, R.; von Essen, R.; Effert, S.; Dodge, H.T. Early recovery of left ventricular function after thrombolytic therapy for acute myocardial infarction: An important determinant of survival. J. Am. Coll. Cardiol. 1988, 12, 289–300. [Google Scholar] [CrossRef]
- Pereira, L.; Métrich, M.; Fernández-Velasco, M.; Lucas, A.; Leroy, J.; Perrier, R.; Morel, E.; Fischmeister, R.; Richard, S.; Bénitah, J.-P.; et al. The cAMP binding protein Epac modulates Ca2+sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 2007, 583, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Cheng, H.; Lao, D.H.; Na, L.; van Oort, R.J.; Brown, J.H.; Wehrens, X.H.T.; Chen, J.; Bers, D.M. Epac2 Mediates Cardiac β1-Adrenergic–Dependent Sarcoplasmic Reticulum Ca 2+ Leak and Arrhythmia. Circulation 2013, 127, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.H.; Owen, J.; Borgman, K.Y.; Fish, F.A.; Kannankeril, P.J. Relation of Milrinone After Surgery for Congenital Heart Disease to Significant Postoperative Tachyarrhythmias. Am. J. Cardiol. 2011, 108, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P. Phos-phodiesterase 4B in the cardiac L-type Ca 2+ channel complex regulates Ca 2+ current and protects against ventricular ar-rhythmias in mice. J. Clin. Investig. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.-L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D Deficiency in the Ryanodine-Receptor Complex Promotes Heart Failure and Arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Varin, A.; Lefebvre, F.; Fischmeister, R.; Vandecasteele, G.; Leroy, J. Calmodulin kinase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors. Cardiovasc. Res. 2016, 110, 151–161. [Google Scholar] [CrossRef]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional Mitochondrial Epac1 Controls Myocardial Cell Death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Taggart, P.; Yellon, D.M. Preconditioning and Arrhythmias. Circulation 2002, 106, 2999–3001. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-K.; Iivainen, T.; Pehkonen, E.; Laurikka, J.; Tarkka, M.R. Antiarrhythmic Effect of Ischemic Preconditioning in Recent Unstable Angina Patients Undergoing Coronary Artery Bypass Grafting. World J. Surg. 2004, 28, 74–79. [Google Scholar] [CrossRef]
- Babai, L.; Papp, J.G.; Parratt, J.R.; Végh, Á. The antiarrhythmic effects of ischaemic preconditioning in anaesthetized dogs are prevented by atropine; role of changes in baroreceptor reflex sensitivity. Br. J. Pharm. 2002, 135, 55–64. [Google Scholar] [CrossRef]
- Khaliulin, I.; Clarke, S.J.; Lin, H.; Parker, J.E.; Suleiman, M.S.; Halestrap, A.P. Temperature Preconditioning of Isolated Rat Hearts—A Potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. J. Physiol. 2007, 581, 1147–1161. [Google Scholar] [CrossRef]
- Yang, C.; Talukder, M.H.; Varadharaj, S.; Velayutham, M.; Zweier, J.L. Early ischaemic preconditioning requires Akt- and PKA-mediated activation of eNOS via serine1176 phosphorylation. Cardiovasc. Res. 2012, 97, 33–43. [Google Scholar] [CrossRef]
- Marais, E.; Genade, S.; Lochner, A. CREB Activation and Ischaemic Preconditioning. Cardiovasc. Drugs Ther. 2008, 22, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Ronca-Testoni, S.; Yu, G.; Galbani, P.; Ronca, G.; Mariani, M. Postischemic changes in cardiac sarcoplasmic reticu-lum Ca2+ channels: A possible mechanism of ischemic preconditioning. Circ. Res. 1995, 76, 1049–1056. [Google Scholar] [CrossRef]
- Zhu, J.; Ferrier, G.R. Ischemic preconditioning: Antiarrhythmic effects and electrophysiological mechanisms in isolated ven-tricle. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H66–H75. [Google Scholar] [CrossRef]
- Salie, R.; Lochner, A.; Loubser, D.J. The significance of the washout period in preconditioning. Cardiovasc. Ther. 2017, 35, e12252. [Google Scholar] [CrossRef]
- Pickard, J.M.J.; Burke, N.; Davidson, S.M.; Yellon, D.M. Intrinsic cardiac ganglia and acetylcholine are important in the mechanism of ischaemic preconditioning. Basic Res. Cardiol. 2017, 112, 11. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Kessebohm, K.; Kronsbein, K.; Lohse, M.J.; Richardt, G. Activation of β-adrenergic receptor kinase during myo-cardial ischemia. Circ. Res. 1996, 79, 455–460. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xu, C.; Liu, Y.; Wang, H.; Han, H.; Wang, Z. Choline produces cytoprotective effects against ischemic my-ocardial injuries: Evidence for the role of cardiac m3 subtype muscarinic acetylcholine receptors. Cell Physiol. Biochem. 2005, 16, 163–174. [Google Scholar] [CrossRef]
- Maslov, L.N.; Khaliulin, I.; Oeltgen, P.R.; Naryzhnaya, N.V.; Pei, J.M.; Brown, S.A.; Lishmanov, Y.B.; Downey, J.M. Pro-spects for creation of cardioprotective and antiarrhythmic drugs based on opioid receptor agonists. Med. Res. Rev. 2016, 36, 871–923. [Google Scholar] [CrossRef] [PubMed]
- Vegh, A.; Szekeres, L.; Parratt, J.R. Protective effects of preconditioning of the ischaemic myocardium involve cyclo-oxygenase products. Cardiovasc. Res. 1990, 24, 1020–1023. [Google Scholar] [CrossRef]
- Vegh, A.; Szekeres, L.; Parratt, J. Preconditioning of the ischaemic myocardium; involvement of the l-arginine nitric oxide pathway. Br. J. Pharmacol. 1992, 107, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Thornton, J.; van Winkle, D.; Stanley, A.; Olsson, R.; Downey, J. Protection against infarction afforded by precondi-tioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 1991, 84, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia–reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the Mitochondrial Permeability Transition in Myocardial Disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef]
- Argaud, L.; Gateau-Roesch, O.; Raisky, O.; Loufouat, J.; Robert, D.; Ovize, M. Postconditioning inhibits mitochondrial per-meability transition. Circulation 2005, 111, 194–197. [Google Scholar] [CrossRef]
- Heusch, G.; Boengler, K.; Schulz, R. Inhibition of Mitochondrial Permeability Transition Pore Opening: The Holy Grail of Cardioprotection; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Halestrap, A.P. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of low pH against chemical and hypoxic cell damage. Biochem. J. 1991, 278, 715–719. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Chiara, F.; Castellaro, D.; Marin, O.; Petronilli, V.; Brusilow, W.S.; Juhaszova, M.; Sollott, S.J.; Forte, M.; Bernardi, P.; Rasola, A. Hexokinase II Detachment from Mitochondria Triggers Apoptosis through the Permeability Transition Pore Independent of Voltage-Dependent Anion Channels. PLoS ONE 2008, 3, e1852. [Google Scholar] [CrossRef] [PubMed]
- Purves, G.I.; Kamishima, T.; Davies, L.M.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels. J. Physiol. 2009, 587, 3639–3650. [Google Scholar] [CrossRef]
- Bos, J.L. Epac: A new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003, 4, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.K.; Teigen, K.; Kopperud, R.; Hodneland, E.; Schwede, F.; Christensen, A.E.; Martinez, A.; Døskeland, S.O. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct struc-tural features and cyclic nucleotide recognition. J. Biol. Chem. 2006, 281, 21500–21511. [Google Scholar] [CrossRef]
- Baillie, G.S.; Houslay, M.D. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr. Opin. Cell Biol. 2005, 17, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Bylund, D. Adrenergic receptors. In Encyclopedia of Biological Chemistry; Lennarz, W.J., Lane, M.D., Eds.; Elsevier BV: Amsterdam, The Netherlands, 2013; pp. 57–60. [Google Scholar]
- El-Armouche, A.; Zolk, O.; Rau, T.; Eschenhagen, T. Inhibitory G-proteins and their role in desensitization of the adenylyl cyclase pathway in heart failure. Cardiovasc. Res. 2003, 60, 478–487. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial Binding of Hexokinase II Inhibits Bax-induced Cytochrome c Release and Apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [PubMed]
- da-Silva, W.S.; Gómez-Puyou, A.; de Gómez-Puyou, M.T.; Moreno-Sanchez, R.; de Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia–reperfusion injury. Cardiovasc. Res. 2003, 60, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.C.; Lee, L.; Rawlings, N.; Ouwendijk, J.; Parker, J.E.; Andrienko, T.N.; Henley, J.M.; Halestrap, A.P. Hexokinase II dissociation alone cannot account for changes in heart mitochondrial function, morphology and sensitivity to permeability transition pore opening following ischemia. PLoS ONE 2020, 15, e0234653. [Google Scholar] [CrossRef] [PubMed]
- Ludman, A.J.; Yellon, D.M.; Hausenloy, D.J. Cardiac preconditioning for ischaemia: Lost in translation. Dis. Model. Mech. 2010, 3, 35–38. [Google Scholar] [CrossRef][Green Version]
- Dixon, J.A.; Spinale, F.G. Large animal models of heart failure. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Alvino, V.V.; Fernández-Jiménez, R.; Rodriguez-Arabaolaza, I.; Slater, S.; Mangialardi, G.; Avolio, E.; Spencer, H.; Culliford, L.; Hassan, S.; Ballesteros, L.S.; et al. Transplantation of Allogeneic Pericytes Improves Myocardial Vascularization and Reduces Interstitial Fibrosis in a Swine Model of Reperfused Acute Myocardial Infarction. J. Am. Hear. Assoc. 2018, 7, 006727. [Google Scholar] [CrossRef] [PubMed]
- Gadeberg, H.C.; Bond, R.C.; Kong, C.H.T.; Chanoit, G.P.; Ascione, R.; Cannell, M.B.; James, A.F. Heterogeneity of T-Tubules in Pig Hearts. PLoS ONE 2016, 11, e0156862. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | C-Pre (n = 7) | 8-Br-Pre (n = 5) | C-Post (n = 6) | 8-Br-Post (n = 7) | |
|---|---|---|---|---|---|
| Regional Ischaemia | Number of VPBs | 830 ± 194 | 401 ± 126 | 738 ± 193 | 856 ± 267 |
| Number of VT/VF | 38 ± 15 | 3 ± 1 * | 77 ± 27 | 39 ± 15 | |
| Arrhythmia Score | 6.8 ± 0.2 | 5.0 ± 0.8 * | 6.8 ± 0.2 | 6.7 ± 0.3 | |
| Reperfusion | Number of VPBs | 409 ± 117 | 160 ± 62 | 211 ± 77 | 249 ± 62 |
| Number of VT/VF | 10 ± 2 | 3 ± 1 * | 9 ± 2 | 3 ± 1 * | |
| Arrhythmia Score | 6.3 ± 0.2 | 4.8 ± 0.4* | 6.4 ± 0.3 | 4.7 ± 0.6 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaliulin, I.; Ascione, R.; Maslov, L.N.; Amal, H.; Suleiman, M.S. Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition. Cells 2021, 10, 1223. https://doi.org/10.3390/cells10051223
Khaliulin I, Ascione R, Maslov LN, Amal H, Suleiman MS. Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition. Cells. 2021; 10(5):1223. https://doi.org/10.3390/cells10051223
Chicago/Turabian StyleKhaliulin, Igor, Raimondo Ascione, Leonid N. Maslov, Haitham Amal, and M. Saadeh Suleiman. 2021. "Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition" Cells 10, no. 5: 1223. https://doi.org/10.3390/cells10051223
APA StyleKhaliulin, I., Ascione, R., Maslov, L. N., Amal, H., & Suleiman, M. S. (2021). Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition. Cells, 10(5), 1223. https://doi.org/10.3390/cells10051223