
Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities

Abstract
1. Introduction
2. ROS Sources and Effects
3. Effects of ROS on the Functions of HSCs
4. Redox Regulation in Normal HSCs
5. The Role of Redox Dyshomeostasis in the Occurrence of ALL
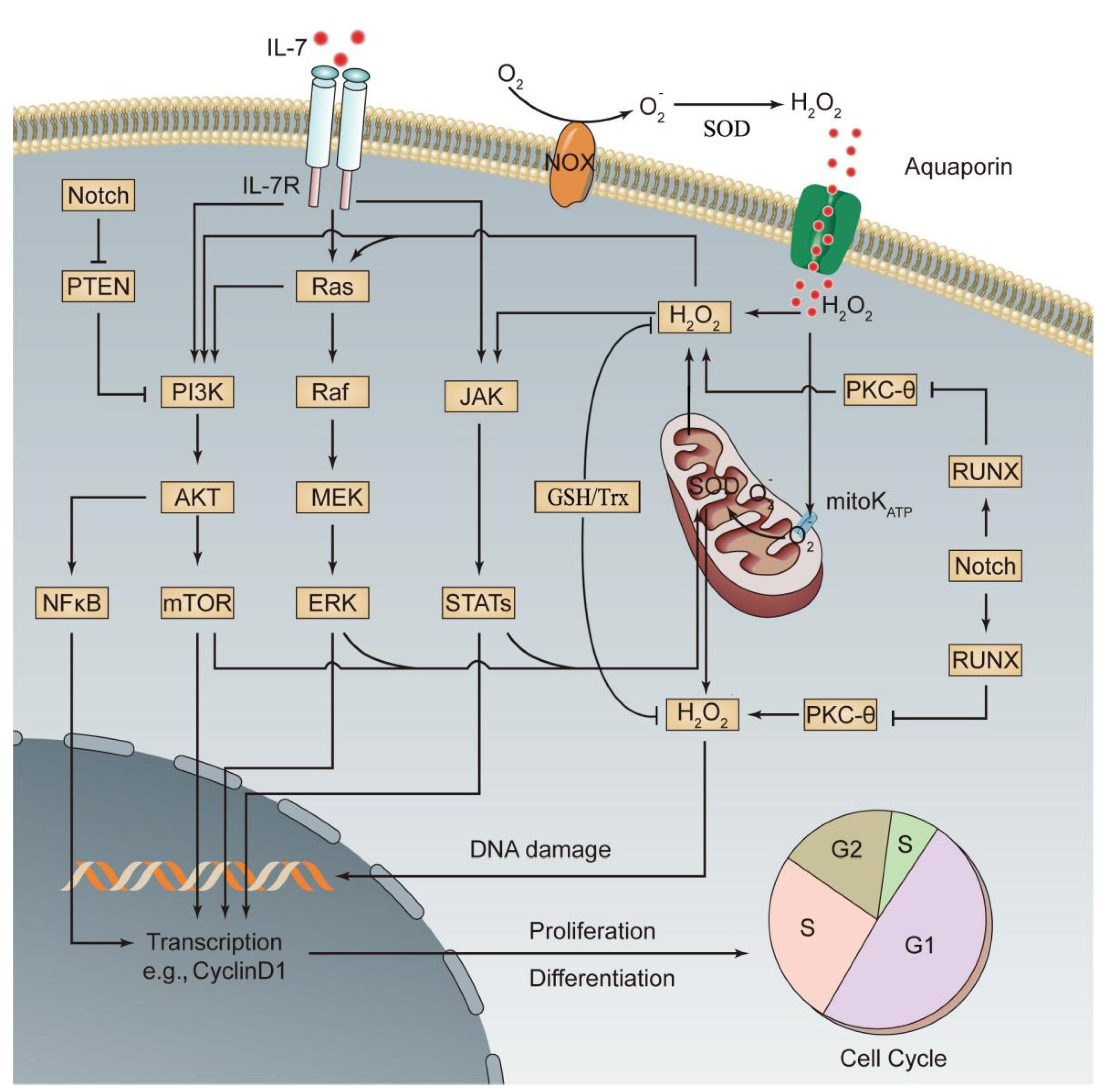
6. Redox Regulation in Leukemia
7. Targeting ROS in ALL Treatment
8. Impact of Pro-Oxidant Therapy on Normal HSCs
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ehrenfeld, V.; Fulda, S. Thioredoxin inhibitor PX-12 induces mitochondria-mediated apoptosis in acute lymphoblastic leukemia cells. Biol. Chem. 2020, 401, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Masurekar, A.; Johnson, S.; Chakraborty, S.; Griffiths, J.; Smith, D.; Alexander, S.; Dempsey, C.; Parker, C.; Harrison, S.; et al. Stromal cell-mediated mitochondrial redox adaptation regulates drug resistance in childhood acute lymphoblastic leukemia. Oncotarget 2015, 6, 43048–43064. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.F.; Liu, H.; Luo, X.J.; Zhao, Z.; Zou, Z.Y.; Li, J.; Lin, X.J.; Liang, Y. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit. Rev. Oncol. Hematol. 2017, 112, 21–30. [Google Scholar] [CrossRef]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Cao, H.; Wang, Y.; Wang, Q.; Wang, R.; Guo, S.; Zhao, X.; Zhang, Y.; Tong, D.; Yang, Z. Taxol prevents myocardial ischemia-reperfusion injury by inducing JNK-mediated HO-1 expression. Pharm. Biol. 2016, 54, 555–560. [Google Scholar] [PubMed]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, X.; Zou, Z.; Liang, Y. The Role of Reactive Oxygen Species in Tumor Treatment and its Impact on Bone Marrow Hematopoiesis. Curr. Drug Targets 2020, 21, 477–498. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, L.; Guo, Q. Oxidative stress and liver disease. Hepatol. Res. 2012, 42, 741–749. [Google Scholar] [CrossRef]
- Bump, E.A.; Taylor, Y.C.; Brown, J.M. Role of glutathione in the hypoxic cell cytotoxicity of misonidazole. Cancer Res. 1983, 43, 997–1002. [Google Scholar] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zou, Z.; Wu, Z.; Zhao, Z.; Luo, X.; Xie, C.; Liang, Y. TNF-α-induced programmed cell death in the pathogenesis of acquired aplastic anemia. Expert Rev. Hematol. 2015, 8, 515–526. [Google Scholar] [CrossRef]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef]
- Bowie, M.B.; McKnight, K.D.; Kent, D.G.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Investig. 2006, 116, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; He, S.; Ashkenazi, R.; Gentry, S.N.; Teta, M.; Kushner, J.A.; Jackson, T.L.; Morrison, S.J. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 2007, 449, 238–242. [Google Scholar] [CrossRef]
- Filippi, M.D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial Role in Stemness and Differentiation of Hematopoietic Stem Cells. Stem Cells Int. 2019, 2019, 4067162. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Ghaffari, S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid. Redox Signal. 2008, 10, 1923–1940. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef]
- Chen, Y.F.; Zhao, Z.Q.; Wu, Z.M.; Zou, Z.Y.; Luo, X.J.; Li, J.; Xie, C.; Liang, Y. The role of RIP1 and RIP3 in the development of aplastic anemia induced by cyclophosphamide and busulphan in mice. Int. J. Clin. Exp. Pathol. 2014, 7, 8411–8420. [Google Scholar]
- Samimi, A.; Kalantari, H.; Lorestani, M.Z.; Shirzad, R.; Saki, N. Oxidative stress in normal hematopoietic stem cells and leukemia. APMIS 2018, 126, 284–294. [Google Scholar] [CrossRef]
- Bottero, V.; Withoff, S.; Verma, I.M. NF-kappaB and the regulation of hematopoiesis. Cell Death Differ. 2006, 13, 785–797. [Google Scholar] [CrossRef]
- Nakata, S.; Matsumura, I.; Tanaka, H.; Ezoe, S.; Satoh, Y.; Ishikawa, J.; Era, T.; Kanakura, Y. NF-kappaB family proteins participate in multiple steps of hematopoiesis through elimination of reactive oxygen species. J. Biol. Chem. 2004, 279, 55578–55586. [Google Scholar] [CrossRef]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef] [PubMed]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Muraguchi, T.; Hoshii, T.; Hirao, A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid. Redox Signal. 2008, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Li, H.; Pazhanisamy, S.K.; Meng, A.; Wang, Y.; Zhou, D. Reactive oxygen species and hematopoietic stem cell senescence. Int. J. Hematol. 2011, 94, 24–32. [Google Scholar] [CrossRef]
- Hosokawa, K.; Arai, F.; Yoshihara, H.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Miyamoto, K.; Shima, H.; Ito, K.; Suda, T. Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction. Biochem. Biophys. Res. Commun. 2007, 363, 578–583. [Google Scholar] [CrossRef]
- He, N.; Zhang, L.; Cui, J.; Li, Z. Bone marrow vascular niche: Home for hematopoietic stem cells. Bone Marrow Res. 2014, 2014, 128436. [Google Scholar] [CrossRef]
- Zhang, Y.; Dépond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Perry, J.M.; Tao, F.; Roy, A.; Lin, T.; He, X.C.; Chen, S.; Lu, X.; Nemechek, J.; Ruan, L.; Yu, X.; et al. Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol. 2020, 22, 689–700. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative resistance of leukemic stem cells and oxidative damage to hematopoietic stem cells under pro-oxidative therapy. Cell Death Dis. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Sahin, E.; Jiang, S.; Sanchez-Aguilera, A.; Scott, K.L.; Chin, L.; Williams, D.A.; Kwiatkowski, D.J.; DePinho, R.A. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc. Natl. Acad. Sci. USA 2008, 105, 19384–19389. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Wu, Z.M.; Luo, X.J.; Bai, S.; Zhao, L.D. Effect of the conditional knockout of bone marrow specific RIPK3 gene on bone marrow hematopoiesis in mice. Int. J. Clin. Exp. Pathol. 2018, 11, 568–576. [Google Scholar]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Ludin, A.; Itkin, T.; Gur-Cohen, S.; Mildner, A.; Shezen, E.; Golan, K.; Kollet, O.; Kalinkovich, A.; Porat, Z.; D’Uva, G.; et al. Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat. Immunol. 2012, 13, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi Ishikawa, E.; Gonzalez-Nieto, D.; Ghiaur, G.; Dunn, S.K.; Ficker, A.M.; Murali, B.; Madhu, M.; Gutstein, D.E.; Fishman, G.I.; Barrio, L.C.; et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9071–9076. [Google Scholar] [CrossRef] [PubMed]
- Khatri, R.; Krishnan, S.; Roy, S.; Chattopadhyay, S.; Kumar, V.; Mukhopadhyay, A. Reactive Oxygen Species Limit the Ability of Bone Marrow Stromal Cells to Support Hematopoietic Reconstitution in Aging Mice. Stem Cells Dev. 2016, 25, 948–958. [Google Scholar] [CrossRef]
- Mrózek, K.; Harper, D.P.; Aplan, P.D. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 991–1010. [Google Scholar] [CrossRef]
- Greaves, M.F. Biological models for leukaemia and lymphoma. IARC Sci. Publ. 2004, 157, 351–372. [Google Scholar]
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E.; et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J. Hematol. Oncol. 2017, 10, 148. [Google Scholar] [CrossRef]
- Kantner, H.P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia 2013, 15, 1292–1300. [Google Scholar] [CrossRef]
- Juric, D.; Lacayo, N.J.; Ramsey, M.C.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Goldstone, A.H.; O’Dwyer, P.J.; Paietta, E.; Sikic, B.I. Differential gene expression patterns and interaction networks in BCR-ABL-positive and -negative adult acute lymphoblastic leukemias. J. Clin. Oncol. 2007, 25, 1341–1349. [Google Scholar] [CrossRef]
- Parada, Y.; Banerji, L.; Glassford, J.; Lea, N.C.; Collado, M.; Rivas, C.; Lewis, J.L.; Gordon, M.Y.; Thomas, N.S.; Lam, E.W. BCR-ABL and interleukin 3 promote haematopoietic cell proliferation and survival through modulation of cyclin D2 and p27Kip1 expression. J. Biol. Chem. 2001, 276, 23572–23580. [Google Scholar] [CrossRef] [PubMed]
- Bäsecke, J.; Griesinger, F.; Trümper, L.; Brittinger, G. Leukemia- and lymphoma-associated genetic aberrations in healthy individuals. Ann. Hematol. 2002, 81, 64–75. [Google Scholar] [CrossRef]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox control of leukemia: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Batista, C.R.; de Oliveira, B.R.; Creighton, R.; Ferguson, J.; Clemmer, K.; Knight, D.; Iansavitchous, J.; Mahmood, D.; Avino, M.; et al. Janus Kinase Mutations in Mice Lacking PU.1 and Spi-B Drive B Cell Leukemia through Reactive Oxygen Species-Induced DNA Damage. Mol. Cell Biol. 2020, 40, e00189-20. [Google Scholar] [CrossRef]
- Steeghs, E.M.P.; Jerchel, I.S.; de Goffau-Nobel, W.; Hoogkamer, A.Q.; Boer, J.M.; Boeree, A.; van de Ven, C.; Koudijs, M.J.; Besselink, N.J.M.; de Groot-Kruseman, H.A.; et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget 2017, 8, 89923–89938. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, K. The making of a lymphocyte: The choice among disparate cell fates and the IKAROS enigma. Genes Dev. 2017, 31, 439–450. [Google Scholar] [CrossRef]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef]
- Rasool, M.; Farooq, S.; Malik, A.; Shaukat, A.; Manan, A.; Asif, M.; Sani, S.; Qazi, M.H.; Kamal, M.A.; Iqbal, Z.; et al. Assessment of circulating biochemical markers and antioxidative status in acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) patients. Saudi. J. Biol. Sci. 2015, 22, 106–111. [Google Scholar] [CrossRef]
- Almondes, K.G.; de Oliveira, T.F.; Siviero-Miachon, A.A.; Lee, M.L.; Rondó, P.H.; Loureiro, A.P.; Spinola-Castro, A.M.; Cozzolino, S.M. Selenium inadequacy is not associated with oxidative stress in child and adolescent acute lymphocytic leukemia survivors. Nutrition 2014, 30, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tian, Y.; Yan, C.; Jin, X.; Tang, J.; Shen, X. Determinants of urinary 8-hydroxy-2′-deoxyguanosine in Chinese children with acute leukemia. Environ. Toxicol. 2009, 24, 446–452. [Google Scholar] [CrossRef]
- Dincer, Y.; Yüksel, S.; Batar, B.; Güven, M.; Onaran, I.; Celkan, T. DNA Repair Gene Polymorphisms and Their Relation with DNA Damage, DNA Repair, and Total Antioxidant Capacity in Childhood Acute Lymphoblastic Leukemia Survivors. J. Pediatr. Hematol. Oncol. 2015, 37, 344–350. [Google Scholar] [CrossRef]
- Sentürker, S.; Karahalil, B.; Inal, M.; Yilmaz, H.; Müslümanoglu, H.; Gedikoglu, G.; Dizdaroglu, M. Oxidative DNA base damage and antioxidant enzyme levels in childhood acute lymphoblastic leukemia. FEBS Lett. 1997, 416, 286–290. [Google Scholar] [CrossRef]
- Olinski, R.; Styczynski, J.; Olinska, E.; Gackowski, D. Viral infection-oxidative stress/DNA damage-aberrant DNA methylation: Separate or interrelated events responsible for genetic instability and childhood ALL development? Biochim. Biophys. Acta 2014, 1846, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. The axis of mTOR-mitochondria-ROS and stemness of the hematopoietic stem cells. Cell Cycle 2009, 8, 1158–1160. [Google Scholar] [CrossRef]
- Chan, S.M.; Weng, A.P.; Tibshirani, R.; Aster, J.C.; Utz, P.J. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007, 110, 278–286. [Google Scholar] [CrossRef]
- Hales, E.C.; Taub, J.W.; Matherly, L.H. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: Targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal 2014, 26, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Yellon, D.M. PTEN, the Achilles’ heel of myocardial ischaemia/reperfusion injury? Br. J. Pharmacol. 2007, 150, 833–838. [Google Scholar] [CrossRef]
- Hole, P.S.; Pearn, L.; Tonks, A.J.; James, P.E.; Burnett, A.K.; Darley, R.L.; Tonks, A. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34+ hematopoietic progenitor cells. Blood 2010, 115, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2017, 18, 1904. [Google Scholar] [CrossRef]
- Oliveira, M.L.; Akkapeddi, P.; Ribeiro, D.; Melão, A.; Barata, J.T. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: An update. Adv. Biol. Regul. 2019, 71, 88–96. [Google Scholar] [CrossRef]
- Silva, A.; Gírio, A.; Cebola, I.; Santos, C.I.; Antunes, F.; Barata, J.T. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 2011, 25, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Thompson, J.E.; Wang, E.S.; Wetzler, M. Philadelphia chromosome-positive acute lymphoblastic leukemia: Current treatment and future perspectives. Cancer 2011, 117, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Paolini, S.; Martinelli, G. Tyrosine kinase inhibitors for the treatment of Philadelphia chromosome-positive adult acute lymphoblastic leukemia. Cancer 2007, 110, 1178–1186. [Google Scholar] [CrossRef]
- Varallo-Rodriguez, C.; Freyer, C.W., Jr.; Ontiveros, E.P.; Griffiths, E.A.; Wang, E.S.; Wetzler, M. Bosutinib for the Treatment of Philadelphia Chromosome-Positive Leukemias. Expert Opin. Orphan Drugs 2015, 3, 599–608. [Google Scholar] [CrossRef]
- Sanford, D.S.; Kantarjian, H.; O’Brien, S.; Jabbour, E.; Cortes, J.; Ravandi, F. The role of ponatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia. Expert Rev. Anticancer Ther. 2015, 15, 365–373. [Google Scholar] [CrossRef]
- Knoechel, B.; Bhatt, A.; Pan, L.; Pedamallu, C.S.; Severson, E.; Gutierrez, A.; Dorfman, D.M.; Kuo, F.C.; Kluk, M.; Kung, A.L.; et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: Genetic and epigenetic findings in an outlier case. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000539. [Google Scholar] [CrossRef]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef]
- Rosin, N.Y.; Koehrer, S.; Kim, E.; O’Brien, S.; Wierda, W.G.; Thomas, D.A.; Estrov, Z.; Kantarjian, H.M.; Lannutti, B.J.; Burger, J.A. In Vitro Effects of PI3Kδ Inhibitor GS-1101 (Cal-101) in Acute Lymphoblastic Leukemia (ALL). Blood 2012, 120, 3534. [Google Scholar] [CrossRef]
- Pereira, J.K.; Machado-Neto, J.A.; Lopes, M.R.; Morini, B.C.; Traina, F.; Costa, F.F.; Saad, S.T.; Favaro, P. Molecular effects of the phosphatidylinositol-3-kinase inhibitor NVP-BKM120 on T and B-cell acute lymphoblastic leukaemia. Eur. J. Cancer 2015, 51, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Naderali, E.; Valipour, B.; Khaki, A.A.; Soleymani Rad, J.; Alihemmati, A.; Rahmati, M.; Nozad Charoudeh, H. Positive Effects of PI3K/Akt Signaling Inhibition on PTEN and P53 in Prevention of Acute Lymphoblastic Leukemia Tumor Cells. Adv. Pharm. Bull. 2019, 9, 470–480. [Google Scholar] [CrossRef]
- Chiarini, F.; Falà, F.; Tazzari, P.L.; Ricci, F.; Astolfi, A.; Pession, A.; Pagliaro, P.; McCubrey, J.A.; Martelli, A.M. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009, 69, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens, M.; Driessen, E.M.; Willekes, M.; Pinhanços, S.S.; Schneider, P.; Pieters, R.; Stam, R.W. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget 2017, 8, 14835–14846. [Google Scholar] [CrossRef]
- Moses, B.S.; Slone, W.L.; Thomas, P.; Evans, R.; Piktel, D.; Angel, P.M.; Walsh, C.M.; Cantrell, P.S.; Rellick, S.L.; Martin, K.H.; et al. Bone marrow microenvironment modulation of acute lymphoblastic leukemia phenotype. Exp. Hematol. 2016, 44, 50–59.e1-2. [Google Scholar] [CrossRef]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hébert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Giambra, V.; Jenkins, C.R.; Wang, H.; Lam, S.H.; Shevchuk, O.O.; Nemirovsky, O.; Wai, C.; Gusscott, S.; Chiang, M.Y.; Aster, J.C.; et al. NOTCH1 promotes T cell leukemia-initiating activity by RUNX-mediated regulation of PKC-θ and reactive oxygen species. Nat. Med. 2012, 18, 1693–1698. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Irwin, M.E.; Gao, Y.; Ban, K.; Shi, P.; Arlinghaus, R.B.; Amin, H.M.; Chandra, J. Inhibition of the NADPH oxidase regulates heme oxygenase 1 expression in chronic myeloid leukemia. Cancer 2012, 118, 3433–3445. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.M.; Zhao, S.G.; Huang, G.Y.; Liu, Q. NADPH oxidase-mediated generation of reactive oxygen species is critically required for survival of undifferentiated human promyelocytic leukemia cell line HL-60. Free Radic. Res. 2004, 38, 629–637. [Google Scholar] [CrossRef]
- Zhang, Z.; Blake, D.R.; Stevens, C.R.; Kanczler, J.M.; Winyard, P.G.; Symons, M.C.; Benboubetra, M.; Harrison, R. A reappraisal of xanthine dehydrogenase and oxidase in hypoxic reperfusion injury: The role of NADH as an electron donor. Free Radic. Res. 1998, 28, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Maders, L.D.; Bagatini, M.D.; Santos, K.F.; Spanevello, R.M.; Maldonado, P.A.; Brulé, A.O.; Araújo Mdo, C.; Schetinger, M.R.; Morsch, V.M. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin. Biochem. 2008, 41, 511–518. [Google Scholar] [CrossRef]
- Gaman, A.M.; Buga, A.M.; Gaman, M.A.; Popa-Wagner, A. The role of oxidative stress and the effects of antioxidants on the incidence of infectious complications of chronic lymphocytic leukemia. Oxid. Med. Cell Longev. 2014, 2014, 158135. [Google Scholar] [CrossRef]
- Tahir, I.M.; Iqbal, T.; Jamil, A.; Saqib, M. Association of BCL-2 with oxidative stress and total antioxidant status in pediatric acute lymphoblastic leukemia. J. Biol. Regul. Homeost. Agents 2017, 31, 1023–1027. [Google Scholar]
- Ben Mahmoud, L.; Mdhaffar, M.; Ghozzi, H.; Ammar, M.; Hakim, A.; Atheymen, R.; Sahnoun, Z.; Elloumi, M.; Zeghal, K. Oxidative Stress in Tunisian Patients with Acute Lymphoblastic Leukemia and Its Involvement in Leukemic Relapse. J. Pediatr. Hematol. Oncol. 2017, 39, e124–e130. [Google Scholar] [CrossRef]
- Nishiura, T.; Suzuki, K.; Kawaguchi, T.; Nakao, H.; Kawamura, N.; Taniguchi, M.; Kanayama, Y.; Yonezawa, T.; Iizuka, S.; Taniguchi, N. Elevated serum manganese superoxide dismutase in acute leukemias. Cancer Lett. 1992, 62, 211–215. [Google Scholar] [CrossRef]
- Schoeneberger, H.; Belz, K.; Schenk, B.; Fulda, S. Impairment of antioxidant defense via glutathione depletion sensitizes acute lymphoblastic leukemia cells for Smac mimetic-induced cell death. Oncogene 2015, 34, 4032–4043. [Google Scholar] [CrossRef][Green Version]
- Silic-Benussi, M.; Scattolin, G.; Cavallari, I.; Minuzzo, S.; Del Bianco, P.; Francescato, S.; Basso, G.; Indraccolo, S.; D’Agostino, D.M.; Ciminale, V. Selective killing of human T-ALL cells: An integrated approach targeting redox homeostasis and the OMA1/OPA1 axis. Cell Death Dis. 2018, 9, 822. [Google Scholar] [CrossRef]
- Jasek-Gajda, E.; Jurkowska, H.; Jasińska, M.; Lis, G.J. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants 2020, 9, 633. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Groninger, E.; Meeuwsen-De Boer, G.J.; De Graaf, S.S.; Kamps, W.A.; De Bont, E.S. Vincristine induced apoptosis in acute lymphoblastic leukaemia cells: A mitochondrial controlled pathway regulated by reactive oxygen species? Int. J. Oncol. 2002, 21, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, H.; Tada-Oikawa, S.; Hiraku, Y.; Kojima, M.; Kawanishi, S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005, 76, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Ban, K.; Dujka, M.E.; McConkey, D.J.; Munsell, M.; Palladino, M.; Chandra, J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007, 110, 267–277. [Google Scholar] [CrossRef]
- Nguyen, T.; Dai, Y.; Attkisson, E.; Kramer, L.; Jordan, N.; Nguyen, N.; Kolluri, N.; Muschen, M.; Grant, S. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin. Cancer Res. 2011, 17, 3219–3232. [Google Scholar] [CrossRef]
- Brem, R.; Karran, P. Oxidation-mediated DNA cross-linking contributes to the toxicity of 6-thioguanine in human cells. Cancer Res. 2012, 72, 4787–4795. [Google Scholar] [CrossRef]
- Zhang, F.; Fu, L.; Wang, Y. 6-thioguanine induces mitochondrial dysfunction and oxidative DNA damage in acute lymphoblastic leukemia cells. Mol. Cell Proteomics. 2013, 12, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; Appell, M.L. Interconnections between apoptotic and autophagic pathways during thiopurine-induced toxicityin cancer cells: The role of reactive oxygen species. Oncotarget 2016, 7, 75616–75634. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin inhibits the production of reactive oxygen species from NADH: Ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef]
- Mendivil-Perez, M.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Response to rotenone is glucose-sensitive in a model of human acute lymphoblastic leukemia: Involvement of oxidative stress mechanism, DJ-1, Parkin, and PINK-1 proteins. Oxid. Med. Cell Longev. 2014, 2014, 457154. [Google Scholar] [CrossRef]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting mitochondrial respiration selectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am. J. Cancer Res. 2017, 7, 2395–2405. [Google Scholar] [PubMed]
- Chandra, J.; Tracy, J.; Loegering, D.; Flatten, K.; Verstovsek, S.; Beran, M.; Gorre, M.; Estrov, Z.; Donato, N.; Talpaz, M.; et al. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood 2006, 107, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Le, S.B.; Hailer, M.K.; Buhrow, S.; Wang, Q.; Flatten, K.; Pediaditakis, P.; Bible, K.C.; Lewis, L.D.; Sausville, E.A.; Pang, Y.P.; et al. Inhibition of mitochondrial respiration as a source of adaphostin-induced reactive oxygen species and cytotoxicity. J. Biol. Chem. 2007, 282, 8860–8872. [Google Scholar] [CrossRef]
- Haß, C.; Belz, K.; Schoeneberger, H.; Fulda, S. Sensitization of acute lymphoblastic leukemia cells for LCL161-induced cell death by targeting redox homeostasis. Biochem. Pharmacol. 2016, 105, 14–22. [Google Scholar] [CrossRef]
- Lee, K.; Briehl, M.M.; Mazar, A.P.; Batinic-Haberle, I.; Reboucas, J.S.; Glinsmann-Gibson, B.; Rimsza, L.M.; Tome, M.E. The copper chelator ATN-224 induces peroxynitrite-dependent cell death in hematological malignancies. Free Radic. Biol. 2013, 60, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Gerby, B.; Veiga, D.F.; Krosl, J.; Nourreddine, S.; Ouellette, J.; Haman, A.; Lavoie, G.; Fares, I.; Tremblay, M.; Litalien, V.; et al. High-throughput screening in niche-based assay identifies compounds to target preleukemic stem cells. J. Clin. Investig. 2016, 126, 4569–4584. [Google Scholar] [CrossRef]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 2016, 1863, 449–463. [Google Scholar] [CrossRef]
- Chen, Y.; Gibson, S.B. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 2008, 4, 246–248. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS…thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Jing, B.; Jin, J.; Xiang, R.; Liu, M.; Yang, L.; Tong, Y.; Xiao, X.; Lei, H.; Liu, W.; Xu, H.; et al. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis. 2018, 9, 589. [Google Scholar] [CrossRef]
- Yami, A.; Hamzeloo-Moghadam, M.; Darbandi, A.; Karami, A.; Mashati, P.; Takhviji, V.; Gharehbaghian, A. Ergolide, a potent sesquiterpene lactone induces cell cycle arrest along with ROS-dependent apoptosis and potentiates vincristine cytotoxicity in ALL cell lines. J. Ethnopharmacol. 2020, 253, 112504. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Li, M.H.; Chen, Y.L.; Sun, H.Y.; Liu, S.L.; Zheng, W.W.; Zhang, M.Y.; Li, H.; Fu, W.; Zhang, W.J.; et al. Chemotherapy-induced niche perturbs hematopoietic reconstitution in B-cell acute lymphoblastic leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 204. [Google Scholar] [CrossRef] [PubMed]
- Yahata, T.; Takanashi, T.; Muguruma, Y.; Ibrahim, A.A.; Matsuzawa, H.; Uno, T.; Sheng, Y.; Onizuka, M.; Ito, M.; Kato, S.; et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011, 118, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Althoff, M.J.; Cancelas, J.A. Signaling Pathways Regulating Hematopoietic Stem Cell and Progenitor Aging. Curr. Stem Cell Rep. 2018, 4, 166–181. [Google Scholar] [CrossRef]
- Gao, N.; Rahmani, M.; Dent, P.; Grant, S. 2-Methoxyestradiol-induced apoptosis in human leukemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene 2005, 24, 3797–3809. [Google Scholar] [CrossRef]
- Fidyt, K.; Pastorczak, A.; Goral, A.; Szczygiel, K.; Fendler, W.; Muchowicz, A.; Bartlomiejczyk, M.A.; Madzio, J.; Cyran, J.; Graczyk-Jarzynka, A.; et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol. Oncol. 2019, 13, 1180–1195. [Google Scholar] [CrossRef]
- Zhang, L.W.; Chen, Q.X.; Wang, J.Y. Advances in reactive oxygen species responsive anti-cancer prodrugs. Acta Chim. Sinica 2020, 78, 642–656. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv. Sci. 2016, 4, 1600124. [Google Scholar] [CrossRef]
- Cadahía, J.P.; Previtali, V.; Troelsen, N.S.; Clausen, M.H. Prodrug strategies for targeted therapy triggered by reactive oxygen species. Medchemcomm 2019, 10, 1531–1549. [Google Scholar] [CrossRef]
- Lin, M.; Guo, W.; Zhang, Z.; Zhou, Y.; Chen, J.; Wang, T.; Zhong, X.; Lu, Y.; Yang, Q.; Wei, Q.; et al. Reduced Toxicity of Liposomal Nitrogen Mustard Prodrug Formulation Activated by an Intracellular ROS Feedback Mechanism in Hematological Neoplasm Models. Mol. Pharm. 2020, 17, 499–506. [Google Scholar] [CrossRef]
- Crater, J.; Kannan, S. Molecular mechanism of nitrogen mustard induced leukocyte(s) chemotaxis. Med. Hypotheses 2007, 68, 318–319. [Google Scholar] [CrossRef]
- Chen, W.; Fan, H.; Balakrishnan, K.; Wang, Y.; Sun, H.; Fan, Y.; Gandhi, V.; Arnold, L.A.; Peng, X. Discovery and Optimization of Novel Hydrogen Peroxide Activated Aromatic Nitrogen Mustard Derivatives as Highly Potent Anticancer Agents. J. Med. Chem. 2018, 61, 9132–9145. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Xu, L.; Ou, S.; Edwards, H.; Luedtke, D.; Ge, Y.; Qin, Z. H2O2/Peroxynitrite-Activated Hydroxamic Acid HDAC Inhibitor Prodrugs Show Antileukemic Activities against AML Cells. ACS Med. Chem. Lett. 2018, 9, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Hagen, H.; Marzenell, P.; Jentzsch, E.; Wenz, F.; Veldwijk, M.R.; Mokhir, A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Marzenell, P.; Hagen, H.; Sellner, L.; Zenz, T.; Grinyte, R.; Pavlov, V.; Daum, S.; Mokhir, A. Aminoferrocene-based prodrugs and their effects on human normal and cancer cells as well as bacterial cells. J. Med. Chem. 2013, 56, 6935–6944. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Chekhun, V.F.; Todor, I.N.; Lukianova, N.Y.; Shvets, Y.V.; Sellner, L.; Putzker, K.; Lewis, J.; Zenz, T.; de Graaf, I.A.; et al. Improved synthesis of N-benzylaminoferrocene-based prodrugs and evaluation of their toxicity and antileukemic activity. J. Med. Chem. 2015, 58, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Schikora, M.; Reznikov, A.; Chaykovskaya, L.; Sachinska, O.; Polyakova, L.; Mokhir, A. Activity of aminoferrocene-based prodrugs against prostate cancer. Bioorg. Med. Chem. Lett. 2015, 25, 3447–3450. [Google Scholar] [CrossRef]
- Tao, W.; He, Z. ROS-responsive drug delivery systems for biomedical applications. Asian J. Pharm. Sci. 2018, 13, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Bae, B.C.; Park, S.J.; Na, K. Reactive oxygen species responsive drug releasing nanoparticle based on chondroitin sulfate-anthocyanin nanocomplex for efficient tumor therapy. J. Control. Release 2016, 222, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.W.; Han, M. Progress in ROS sensitive Nano-drug delivery system. Chin. J. Mod. Appl. Pharm. 2017, 34, 766–770. [Google Scholar]
- Liu, L.C.; Rui, L.L.; Gao, Y.; Zhang, W.A. Self-assembly and disassembly of a redox-responsive ferrocene-containing amphiphilic block copolymer for controlled release. Polym. Chem. 2015, 6, 1817–1829. [Google Scholar] [CrossRef]
- Kwon, J.; Kim, J.; Park, S.; Khang, G.; Kang, P.M.; Lee, D. Inflammation-responsive antioxidant nanoparticles based on a polymeric prodrug of vanillin. Biomacromolecules 2013, 14, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.K.; Paul, M.; Paul, S. Curcumin induces caspase mediated apoptosis in JURKAT cells by disrupting the redox balance. Asian Pac. J. Cancer Prev. 2014, 15, 93–100. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Semkova, S.; Aoki, I.; Bakalova, R. Resveratrol Modulates the Redox-status and Cytotoxicity of Anticancer Drugs by Sensitizing Leukemic Lymphocytes and Protecting Normal Lymphocytes. Anticancer Res. 2019, 39, 3745–3755. [Google Scholar] [CrossRef]
- Ede, B.C.; Asmaro, R.R.; Moppett, J.P.; Diamanti, P.; Blair, A. Investigating chemoresistance to improve sensitivity of childhood T-cell acute lymphoblastic leukemia to parthenolide. Haematologica 2018, 103, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Han, S.; Kamberos, N.L. Piperlongumine inhibits the proliferation and survival of B-cell acute lymphoblastic leukemia cell lines irrespective of glucocorticoid resistance. Biochem. Biophys. Res. Commun. 2014, 452, 669–675. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Ndontsa, B.L.; Kuete, V.; Nguekeu, Y.M.M.; Çelik, İ.; Mbouangouere, R.; Tane, P.; Efferth, T. A naturally occuring triterpene saponin ardisiacrispin B displayed cytotoxic effects in multi-factorial drug resistant cancer cells via ferroptotic and apoptotic cell death. Phytomedicine 2018, 43, 78–85. [Google Scholar] [CrossRef]
- Efferth, T.; Giaisi, M.; Merling, A.; Krammer, P.H.; Li-Weber, M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS ONE 2007, 2, e693. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Nakamura, H.; Masutani, H.; Sasada, T.; Takabayashi, A.; Yamaoka, Y.; Yodoi, J. Baicalin induces apoptosis via mitochondrial pathway as prooxidant. Mol. Immunol. 2002, 38, 781–791. [Google Scholar] [CrossRef]
- Mezencev, R.; Updegrove, T.; Kutschy, P.; Repovská, M.; McDonald, J.F. Camalexin induces apoptosis in T-leukemia Jurkat cells by increased concentration of reactive oxygen species and activation of caspase-8 and caspase-9. J. Nat. Med. 2011, 65, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Khan, A.Q.; Ahmed, E.I.; Akhtar, S.; Ali, T.A.; Merhi, M.; Dermime, S.; Steinhoff, M.; et al. Curcumin Induces Apoptotic Cell Death via Inhibition of PI3-Kinase/AKT Pathway in B-Precursor Acute Lymphoblastic Leukemia. Front Oncol. 2019, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Ni, H.M.; Wang, S.Y.; Tourkova, I.L.; Shurin, M.R.; Harada, H.; Yin, X.M. Cyanidin-3-rutinoside, a natural polyphenol antioxidant, selectively kills leukemic cells by induction of oxidative stress. J. Biol. Chem. 2007, 282, 13468–13476. [Google Scholar] [CrossRef]
- Aghvami, M.; Ebrahimi, F.; Zarei, M.H.; Salimi, A.; Pourahmad, J.R.; Pourahmad, J. Matrine Induction of ROS Mediated Apoptosis in Human ALL B-lymphocytes Via Mitochondrial Targeting. Asian Pac. J. Cancer Prev. 2018, 19, 555–560. [Google Scholar]
- Zunino, S.J.; Ducore, J.M.; Storms, D.H. Parthenolide induces significant apoptosis and production of reactive oxygen species in high-risk pre-B leukemia cells. Cancer Lett. 2007, 254, 119–127. [Google Scholar] [CrossRef]
- Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Khan, A.Q.; Akhtar, S.; Mateo, J.M.; Merhi, M.; Taha, R.; Omri, H.E.; Mraiche, F.; et al. Sanguinarine suppresses growth and induces apoptosis in childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 782–794. [Google Scholar] [CrossRef]
- Kannappan, V.; Wang, W.; Li, P.; Xu, B. Low dose triptolide reverses chemoresistance in adult acute lymphoblastic leukemia cells via reactive oxygen species generation and DNA damage response disruption. Oncotarget 2016, 7, 85515–85528. [Google Scholar]



{kind=link}
{kind=link}
| Signaling Pathway | Targets | Representative Drugs | Antileukemic Effect | Refs |
|---|---|---|---|---|
| BCR/ABL | Tyrosine kinase inhibitor (TKI) | Imatinib | First-generation TKI that can block the ATP-binding sites of BCR-ABL and prevent activation of the conformation of oncogenic proteins | [85] |
| Nilotinib | Second-generation TKI and high-affinity aminopyrimidine-based ATP-competitive inhibitor with more specific inhibition of BCR/ABL activity | [86] | ||
| Dasatinib | Second-generation TKI that can bind to inactive and active BCR/ABL kinase and inhibit Src family kinases and c-Kit | [86] | ||
| Bosutinib | Third-generation TKI and potent dual inhibitor of Src and ABL kinases with longer-term safety than second-generation and other third-generation TKIs | [87] | ||
| Ponatinib | Third-generation TKI that is effective for known mutations in imatinib-resistant genes (including T315I) | [88] | ||
| Notch | γ-secretase inhibitor (GSIs) | BMS-906024 | Inhibits the activity of Notch signaling by downregulating the expression of multiple known target genes of Notch but has no marked effect on c-Myc | [89] |
| PF-03084014 | Downregulates the level of the Notch intracellular domain and the expression of Notch target genes Hes-1 and c-Myc and induces cell cycle arrest and apoptosis of T-ALL cells | [90] | ||
| PI3K/AKT/mTOR | PI3K-δ inhibitor | Idelalisib | Downregulates the level of AKT phosphorylation in B-ALL cells, inhibits cell proliferation, and blocks the homing of B-ALL cells into the bone marrow | [91] |
| NVP-BKM120 | Downregulates the phosphorylation levels of AKT and mTOR in T-ALL cells, inhibits cell cycle progression, and promotes apoptosis | [92] | ||
| AKT inhibitor | MK-2206 | Downregulates AKT phosphorylation levels in both T-ALL and B-ALL cell lines (it can also promote PTEN phosphorylation in B-ALL cell lines), inhibits cell proliferation, and promotes apoptosis | [93] | |
| PI3K/mTOR inhibitor | PI-103 | More potent than inhibitors that are selective only for PI3K or for mTOR and can effectively induce cell cycle arrest and apoptosis in T-ALL cells | [94] | |
| JAK/STAT | JAK inhibitor | Ruxolitinib | JAK1/2 inhibitor that can reduce ROS and ROS-induced gene expression signatures and inhibit the growth of leukemia cells | [66] |
| RAS | MEK inhibitor | Selumetinib Trametinib MEK162 | Reduce ERK phosphorylation and induce apoptosis in the RAS-mutant MLL-rearranged ALL cells | [95] |
| ROS-Responsive Linker | Activation and Active Drug Release Mechanisms under the Action of ROS | Refs |
|---|---|---|
| Alkyl thioether/selenide | In the presence of oxidative conditions, thioether-containing polymers undergo phase transition from hydrophobic sulfide to more hydrophilic sulfoxide or sulfone. The increased hydrophilicity promotes the hydrolysis of ester bonds, thereby accelerating drug release. | [140,151] |
| Aminoacrylate | Prodrugs formed from aminoacrylate, in which electron-rich alkenes are easily oxidized by ROS, undergo [2 + 2] cycloaddition reaction, thereby releasing molecular drugs via self-breakage. | [140,142] |
| Anthocyanin | Under oxidative stress, anthocyanins can undergo responsive breakage to release drugs. | [152,153] |
| Arylboronic acid/ester | B—C bonds are oxidized through coordination of ROS with boron atoms to form borate and arylphenols. The drugs are released through the self-breakage of arylphenols. | [142] |
| Ferrocene | Ferrocene has certain hydrophobicity and can form water-soluble salts after oxidation. When ferrocene is attached to the hydrophobic end of a copolymer, ROS-responsive drug release can be achieved. | [153,154] |
| Peroxalate ester | Peroxalate ester can be easily oxidized by ROS to generate the intermediate dioxetanedione, which is rapidly decomposed into carbon dioxide, and release drugs. | [151,155] |
| Poly(propylene sulfide) (PPS) | Under the oxidation of ROS, the sulfur in propylene sulfide is oxidized to form sulfur oxides, resulting in increased hydrophilicity and promoting drug release. | [153] |
| Thioketal | Thioketal can be rapidly cleaved by ROS species and degraded into acetone and thiols as byproducts to achieve drug release. | [151] |
| Natural Compound | Cell Type | Action | Possible Anti-Leukemia Mechanism | Refs |
|---|---|---|---|---|
| Ardisiacrispin B | CCRF-CEM human T-cell ALL cell line | Induces apoptosis | Activates caspases 8 and 9 and caspase 3/7 and increases ROS production | [160] |
| Artesunate | Jurkat, CEM, and Molt-4 human T-cell ALL cell lines | Induces apoptosis | ROS-dependent mitochondria-mediated pathway | [161] |
| Baicalin | Jurkat human T-cell ALL cell line, human peripheral blood mononuclear cells (PBMCs) isolated from blood of healthy volunteers | Induces apoptosis in Jurkat cells while having little cytotoxicity on PBMCs | ROS-dependent mitochondria-mediated apoptotic pathway | [162] |
| Camalexin | Jurkat human T-cell ALL cell line, human lymphoblasts, and primary fibroblasts | In the micromolar range, camalexin exhibits time- and concentration-dependent cytotoxicity to Jurkat cells but has little cytotoxicity to normal cells. | ROS-dependent mitochondria-mediated apoptotic pathway | [163] |
| Curcumin | 697, REH, RS4;11, and SupB15 human B-cell precursor ALL cell lines | Induces apoptosis | ROS-dependent mitochondria-mediated intrinsic pathway | [164] |
| Cyanidin-3-rutinoside | HL-60 human promyelocytic leukemia cell line, CCRF-CEM and Molt-4 human T-cell ALL cell lines, human PBMCs isolated from healthy donors | Induces apoptosis in leukemia cell lines while having little cytotoxicity to normal PBMCs | ROS-dependent activation of p38 MAPK and JNK | [165] |
| Matrine | Human ALL B-lymphocytes | Promotes apoptosis in ALL cells | Promotes ROS generation, upregulates Bax, and downregulates Bcl-2 | [166] |
| Parthenolide | SEM and RS4;11 pre-B ALL cell lines, human T-cell ALL cells | Induces rapid apoptotic cell death | Promotes ROS generation | [158,167] |
| Piperlongumine | GC-resistant B-ALL cell lines and GC-sensitive B-ALL cell lines, human PBMCs B cells | Regardless of GC-resistance, piperlongumine inhibits the proliferation of all B-ALL cell lines but not normal B cells and induces apoptosis | Promotes ROS generation | [159] |
| Resveratrol | Jurkat human T-cell ALL cell line, normal lymphocytes | Resveratrol synergizes with barasertib or everolimus to enhance the cytotoxic effect on ALL cells without affecting normal lymphocytes | Promotes ROS generation | [157] |
| Sanguinarine | 697, REH, RS4;11, and SupB15 human B-cell precursor ALL cell lines | Promotes ALL cell apoptosis | Promotes ROS generation, upregulates Bax, and downregulates Bcl-2 | [168] |
| Triptolide | ALL cell line (NALM-6/R) with cross-resistance to cytarabine (araC) and doxorubicin (ADM) | Reversal of the drug resistance of ALL cells inhibits cell proliferation, induces apoptosis in vitro, and inhibits tumor growth in a mouse xenograft model | Impairs mitochondrial membrane potential and increases ROS production | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Li, J.; Zhao, Z. Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities. Cells 2021, 10, 1218. https://doi.org/10.3390/cells10051218
Chen Y, Li J, Zhao Z. Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities. Cells. 2021; 10(5):1218. https://doi.org/10.3390/cells10051218
Chicago/Turabian StyleChen, Yongfeng, Jing Li, and Zhiqiang Zhao. 2021. "Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities" Cells 10, no. 5: 1218. https://doi.org/10.3390/cells10051218
APA StyleChen, Y., Li, J., & Zhao, Z. (2021). Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities. Cells, 10(5), 1218. https://doi.org/10.3390/cells10051218