
Cancer Stem Cells and Neovascularization


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
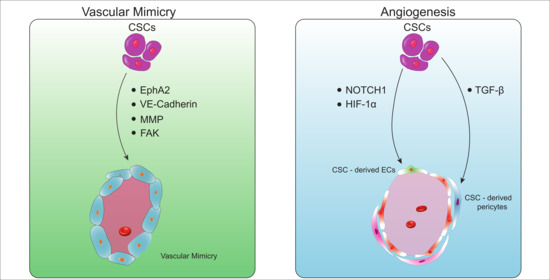
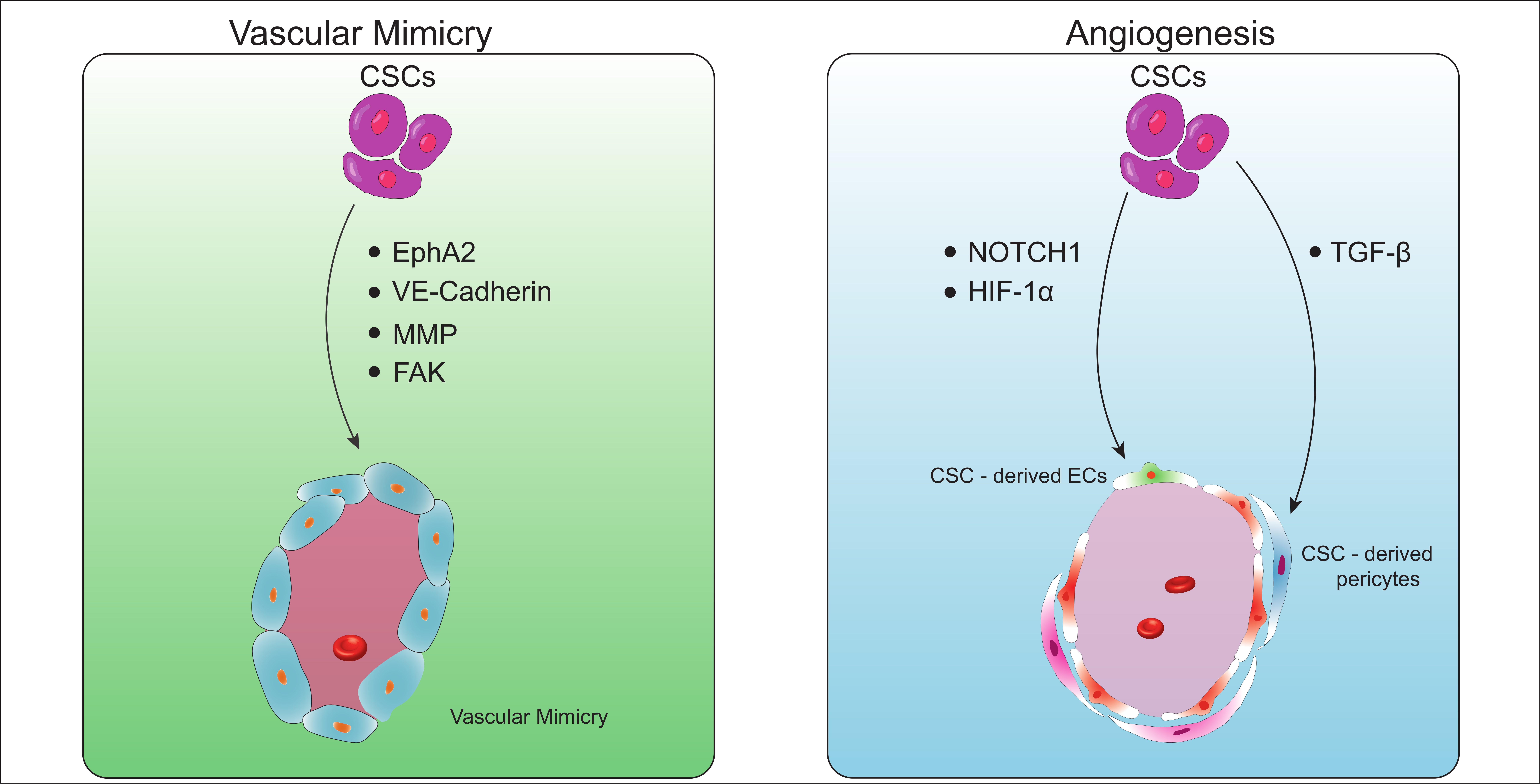
2. Vascular Mimicry
2.1. Molecular Determinants Regulating the Formation of Vascular Mimicry
2.2. The Role of CSCs in Vascular Mimicry Formation
2.2.1. CD44+ CSCs
2.2.2. ALDH+ CSCs
2.2.3. CD133+ CSCs
2.3. Therapeutically Targeting Vascular Mimicry
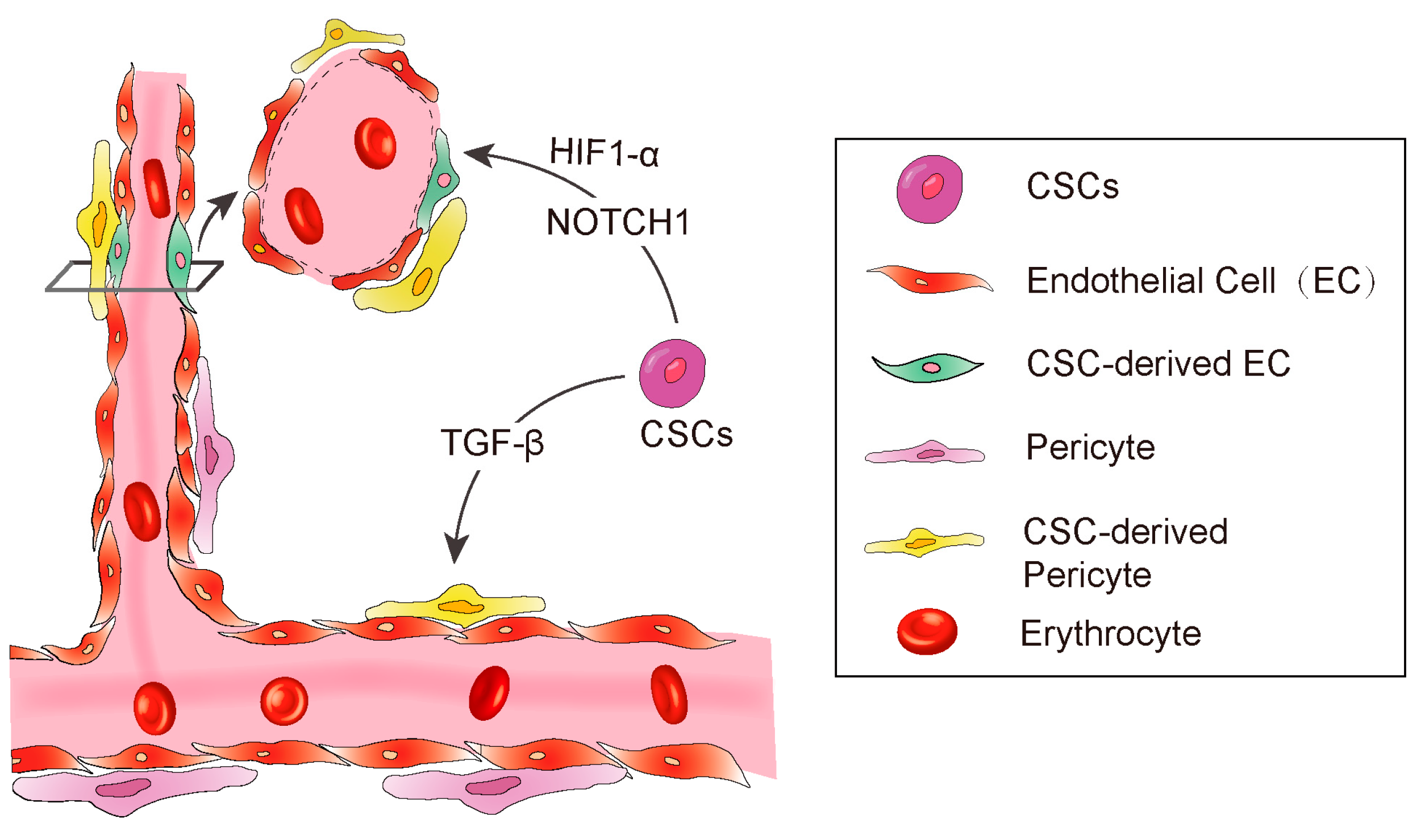
3. CSC-Derived Endothelial Cells
3.1. CSCs Transdifferentiate into Endothelial Cells
3.1.1. Glioblastoma
3.1.2. Renal Carcinoma
3.1.3. Breast Cancer
3.2. CSCs Transdifferentiate into Pericytes
3.3. The Factors Affecting CSC Transdifferentiation
3.3.1. Hypoxia
3.3.2. TGF-β
3.3.3. NOTCH1
4. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ribatti, D.; Vacca, A.; Dammacco, F. The Role of the Vascular Phase in Solid Tumor Growth: A Historical Review. Neoplasia 1999, 1, 293–302. [Google Scholar] [CrossRef]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The Intestinal Stem Cell Signature Identifies Colorectal Cancer Stem Cells and Predicts Disease Relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Seftor, E.A.; Meltzer, P.S.; Schatteman, G.C.; Gruman, L.M.; Hess, A.R.; Kirschmann, D.A.; Seftor, R.E.; Hendrix, M.J. Expression of multiple molecular phenotypes by aggressive melanoma tumor cells: Role in vasculogenic mimicry. Crit. Rev. Oncol./Hematol. 2002, 44, 17–27. [Google Scholar] [CrossRef]
- Suzuki, S.; Sano, K.; Tanihara, H. Diversity of the cadherin family: Evidence for eight new cadherins in nervous tissue. Cell Regul. 1991, 2, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.-G.; Resnati, M.; Raiteri, M.; Pigott, R.; Pisacane, A.; Houen, G.; Ruco, L.; Dejana, E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J. Cell Biol. 1992, 118, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.R.; Seftor, E.A.; Gardner, L.M.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.; Kinch, M.S.; Hendrix, M.J. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001, 61, 3250–3255. [Google Scholar] [PubMed]
- Hess, A.R.; Seftor, E.A.; Gruman, L.M.; Kinch, M.S.; Seftor, R.E.; Hendrix, M.J. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: Implications for vasculogenic mimicry. Cancer Biol. Ther. 2006, 5, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.B.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.G.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J.C. Cooperative Interactions of Laminin 5 γ2 Chain, Matrix Metalloproteinase-2, and Membrane Type-1-Matrix/Metalloproteinase Are Required for Mimicry of Embryonic Vasculogenesis by Aggressive Melanoma. Cancer Res. 2001, 61, 6322. [Google Scholar]
- Hess, A.R.; Postovit, L.-M.; Margaryan, N.V.; Seftor, E.A.; Schneider, G.B.; Seftor, R.E.; Nickoloff, B.J.; Hendrix, M.J. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 2005, 65, 9851–9860. [Google Scholar] [CrossRef]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, B.; Sun, D.; Liu, T.; Che, N.; Gu, Q.; Dong, X.; Li, R.; Liu, Y.; Li, J. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol. Rep. 2015, 33, 149–156. [Google Scholar] [CrossRef]
- Ladeda, V.; Ghiso, J.A.A.; de Kier Joffé, E.B. Function and expression of CD44 during spreading, migration, and invasion of murine carcinoma cells. Exp. Cell Res. 1998, 242, 515–527. [Google Scholar] [CrossRef]
- Hiraga, T.; Ito, S.; Nakamura, H. Cancer stem–like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013, 73, 4112–4122. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fu, C.; Bai, H.; Song, E.; Song, Y. CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells. FEBS Lett. 2014, 588, 4573–4582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, B.; Zhao, X.; Liu, Z.; Wang, X.; Yao, X.; Dong, X.; Chi, J. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J. Surg. Oncol. 2013, 108, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Paulis, Y.W.; Huijbers, E.J.; van der Schaft, D.W.; Soetekouw, P.M.; Pauwels, P.; Tjan-Heijnen, V.C.; Griffioen, A.W. CD44 enhances tumor aggressiveness by promoting tumor cell plasticity. Oncotarget 2015, 6, 19634. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.; Dehghan, A. The expression and functional significance of vascular endothelial-cadherin, CD44, and vimentin in oral squamous cell carcinoma. J. Int. Soc. Prev. Community Dent. 2018, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Toledo-Guzmán, M.E.; Hernandez, M.I.; Gómez-Gallegos, Á.A.; Ortiz-Sánchez, E. ALDH as a Stem Cell Marker in Solid Tumors. Curr. Stem Cell Res. Ther. 2019, 14, 375. [Google Scholar] [CrossRef]
- Zhu, B.; Zhou, L.; Yu, L.; Wu, S.; Song, W.; Gong, X.; Wang, D. Evaluation of the correlation of vasculogenic mimicry, ALDH1, KAI1 and microvessel density in the prediction of metastasis and prognosis in colorectal carcinoma. BMC Surg. 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Xing, P.; Dong, H.; Liu, Q.; Zhao, T.; Yao, F.; Xu, Y.; Chen, B.; Zheng, X.; Wu, Y.; Jin, F. ALDH1 expression and vasculogenic mimicry are positively associated with poor prognosis in patients with breast cancer. Cell. Physiol. Biochem. 2018, 49, 961–970. [Google Scholar] [CrossRef]
- Izawa, Y.; Kashii-Magaribuchi, K.; Yoshida, K.; Nosaka, M.; Tsuji, N.; Yamamoto, A.; Kuroyanagi, K.; Tono, K.; Tanihata, M.; Imanishi, M. Stem-like human breast cancer cells initiate vasculogenic mimicry on matrigel. Acta Histochem. Cytochem. 2018, 51, 173–183. [Google Scholar] [CrossRef]
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef]
- Mirshahi, P.; Rafii, A.; Vincent, L.; Berthaut, A.; Varin, R.; Kalantar, G.; Marzac, C.; Calandini, O.; Marie, J.; Soria, C. Vasculogenic mimicry of acute leukemic bone marrow stromal cells. Leukemia 2009, 23, 1039–1048. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, T.; Sun, B.; Zhao, X.; Zhao, X.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yao, N.; Cheng, S.; Li, L.; Liu, S.; Yang, Z.; Shang, G.; Zhang, D.; Yao, Z. Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer. Cancer Biol. Med. 2019, 16, 299. [Google Scholar] [PubMed]
- Gong, W.; Sun, B.; Zhao, X.; Zhang, D.; Sun, J.; Liu, T.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y. Nodal signaling promotes vasculogenic mimicry formation in breast cancer via the Smad2/3 pathway. Oncotarget 2016, 7, 70152. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Sun, B.; Sun, H.; Zhao, X.; Zhang, D.; Liu, T.; Zhao, N.; Gu, Q.; Dong, X.; Liu, F. Nodal signaling activates the Smad2/3 pathway to regulate stem cell-like properties in breast cancer cells. Am. J. Cancer Res. 2017, 7, 503. [Google Scholar]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Wang, J.Y.; Sun, T.; Zhao, X.L.; Zhang, S.W.; Zhang, D.F.; Gu, Q.; Wang, X.H.; Zhao, N.; Qie, S.; Sun, B.C. Functional significance of VEGF-a in human ovarian carcinoma: Role in vasculogenic mimicry. Cancer Biol. Ther. 2008, 7, 758–766. [Google Scholar] [CrossRef]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef]
- van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Zhao, W.; Ma, Y.; Yang, X. Demonstration of vasculogenic mimicry in astrocytomas and effects of Endostar on U251 cells. Pathol. Res. Pract. 2011, 207, 645–651. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Q.; Li, X.-Y.; Yang, Q.-Y.; Xu, W.-W.; Liu, G.-L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 16. [Google Scholar] [CrossRef]
- Zeng, F.; Ju, R.-J.; Liu, L.; Xie, H.-J.; Mu, L.-M.; Zhao, Y.; Yan, Y.; Hu, Y.-J.; Wu, J.-S.; Lu, W.-L. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer. Oncotarget 2015, 6, 36625. [Google Scholar] [CrossRef]
- Tu, D.G.; Yu, Y.; Lee, C.H.; Kuo, Y.L.; Lu, Y.C.; Tu, C.W.; Chang, W.W. Hinokitiol inhibits vasculogenic mimicry activity of breast cancer stem/progenitor cells through proteasome-mediated degradation of epidermal growth factor receptor. Oncol. Lett. 2016, 11, 2934–2940. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, P.; Zhang, M.; Ma, F. Brucine suppresses breast cancer metastasis via inhibiting epithelial mesenchymal transition and matrix metalloproteinases expressions. Chin. J. Integr. Med. 2018, 24, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Petrova, T.V. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 2000, 19, 5598–5605. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell–like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Hao, M.; Ouyang, Y.; Zheng, J.; Chen, D. CD133+ cancer stem cells promoted by VEGF accelerate the recurrence of hepatocellular carcinoma. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood J. Am. Soc. Hematol. 2011, 118, 2906–2917. [Google Scholar] [CrossRef]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef]
- Wenger, R.H.; Gassmann, M. Oxygen (es) and the hypoxia-inducible factor-1. Biol. Chem. 1997, 378, 609–616. [Google Scholar] [PubMed]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, D.; Kim, K.; Noha, M.; Teramoto, A. Hypoxia inducible factor 1-α regulates of platelet derived growth factor-B in human glioblastoma cells. J. Neuro-Oncol. 2006, 76, 13–21. [Google Scholar] [CrossRef]
- Wang, M.-K.; Sun, H.-Q.; Xiang, Y.-C.; Jiang, F.; Su, Y.-P.; Zou, Z.-M. Different roles of TGF-β in the multi-lineage differentiation of stem cells. World J. Stem Cells 2012, 4, 28. [Google Scholar] [CrossRef]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef]
- Rao, S.; Zaidi, S.; Banerjee, J.; Jogunoori, W.; Sebastian, R.; Mishra, B.; Nguyen, B.N.; Wu, R.C.; White, J.; Deng, C. Transforming growth factor-β in liver cancer stem cells and regeneration. Hepatol. Commun. 2017, 1, 477–493. [Google Scholar] [CrossRef]
- Tang, B.; Yoo, N.; Vu, M.; Mamura, M.; Nam, J.-S.; Ooshima, A.; Du, Z.; Desprez, P.-Y.; Anver, M.R.; Michalowska, A.M.; et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007, 67, 8643–8652. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Batlle, R.; Andrés, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal–endothelial transition by p38α through TGF-β and JNK signaling. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Kulla, A.; Burkhardt, K.; Meyer-Puttlitz, B.; Teesalu, T.; Asser, T.; Wiestler, O.D.; Becker, A.J. Analysis of the TP53 gene in laser-microdissected glioblastoma vasculature. Acta Neuropathol. 2003, 105, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Orr, B.A.; Ligon, K.L.; Eberhart, C.G. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 2012, 3, 98. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhao, D.; Dai, X.; Chen, J.; Rong, X.; Wang, H.; Wang, A.; Li, M.; Dong, J.; Huang, Q. Fusion of cancer stem cells and mesenchymal stem cells contributes to glioma neovascularization. Oncol. Rep. 2015, 34, 2022–2030. [Google Scholar] [CrossRef]
- Antoszczak, M.; Huczyński, A. Anticancer activity of polyether ionophore-salinomycin. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2015, 15, 575–591. [Google Scholar] [CrossRef]
- Dewangan, J.; Srivastava, S.; Rath, S.K. Salinomycin: A new paradigm in cancer therapy. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Dilokthornsakul, P.; Chaiyakunapruk, N.; Termrungruanglert, W.; Pratoomsoot, C.; Saokeaw, S.; Sruamsiri, R. The effects of metformin on ovarian cancer: A systematic review. Int. J. Gynecol. Cancer 2013, 23, 1544–1551. [Google Scholar] [CrossRef]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Xu, J.; Liu, S. Cancer Stem Cells and Neovascularization. Cells 2021, 10, 1070. https://doi.org/10.3390/cells10051070
Li F, Xu J, Liu S. Cancer Stem Cells and Neovascularization. Cells. 2021; 10(5):1070. https://doi.org/10.3390/cells10051070
Chicago/Turabian StyleLi, Fengkai, Jiahui Xu, and Suling Liu. 2021. "Cancer Stem Cells and Neovascularization" Cells 10, no. 5: 1070. https://doi.org/10.3390/cells10051070
APA StyleLi, F., Xu, J., & Liu, S. (2021). Cancer Stem Cells and Neovascularization. Cells, 10(5), 1070. https://doi.org/10.3390/cells10051070