
Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy



Abstract
1. Introduction
2. NK Cell Biology
2.1. NK Cells in Innate and Adaptive Immunity
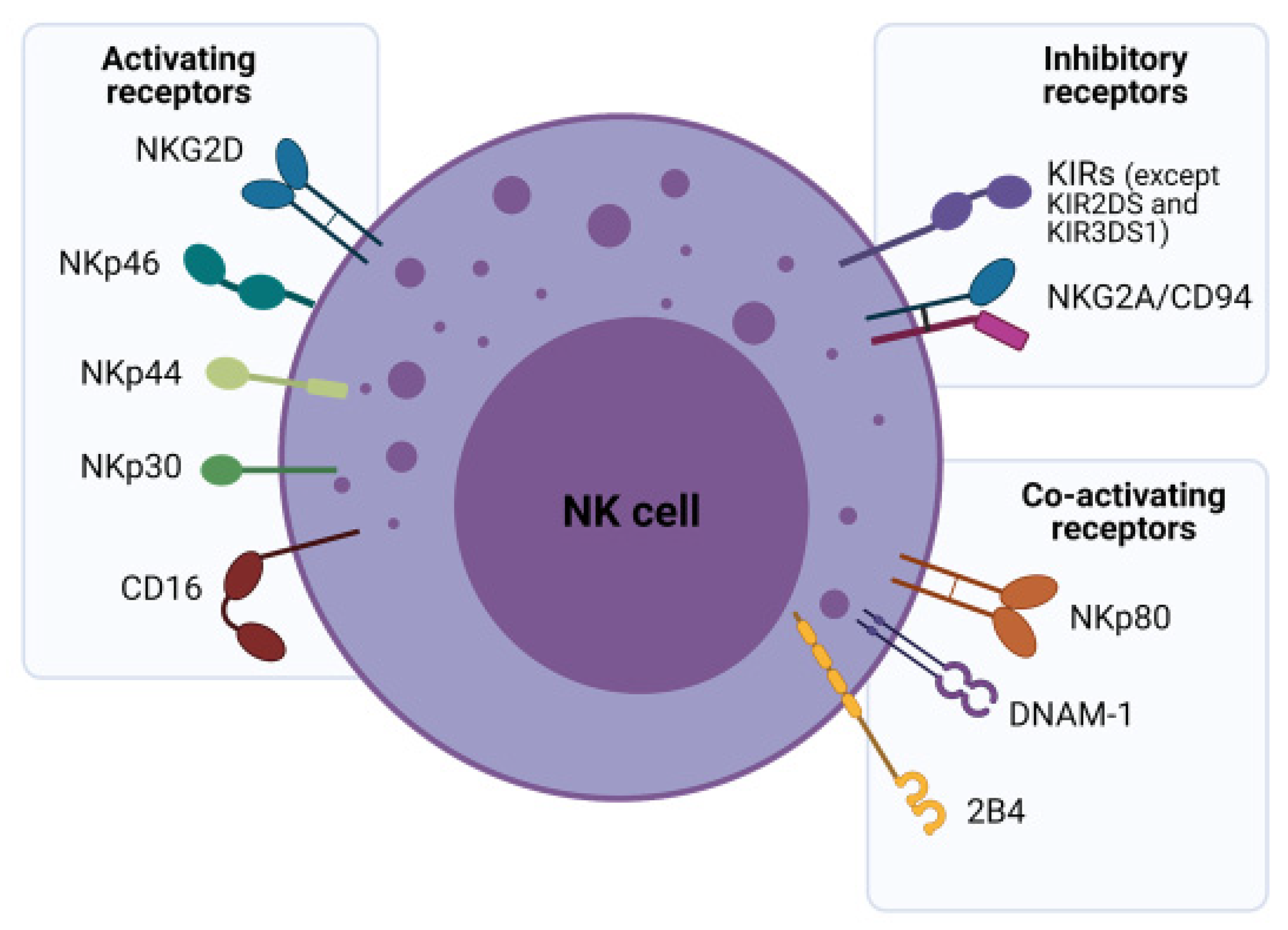
2.2. NK Cell Receptors and Their Role in Tumor Surveillance
3. Modifying NK Cells to Be Better Effector Cells
3.1. Cytokine-Based Cell Expansion, Propagation and Therapies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Company | Description |
|---|---|---|
| GoCAR™ | Bellicum Pharmaceuticals | CD123 or HER2 CAR-NK cells with rimiducid-inducible iMC and autocrine IL-15 exhibit enhanced persistence and antitumor activity in CD123+ AML and HER2+ solid tumor models. |
| FT596 | Fate Therapeutics | CD19 CAR with CD16 Fc receptor and IL-15 receptor fusion protein with a flexible IL-15 increases NK cell activation and targets both CD19 and CD20 in B cell malignancy. |
| NKX-101 | Nkarta | NKG2D CAR-NK cells and mbIL-15 for further activation as well. NKG2D ligands are expressed in many tumor cells. |
| TAK-007 | Takeda | CD19 CAR-NK cells with a CD28 costimulatory domain, IL-15 and an inducible caspase 9 suicide gene. |
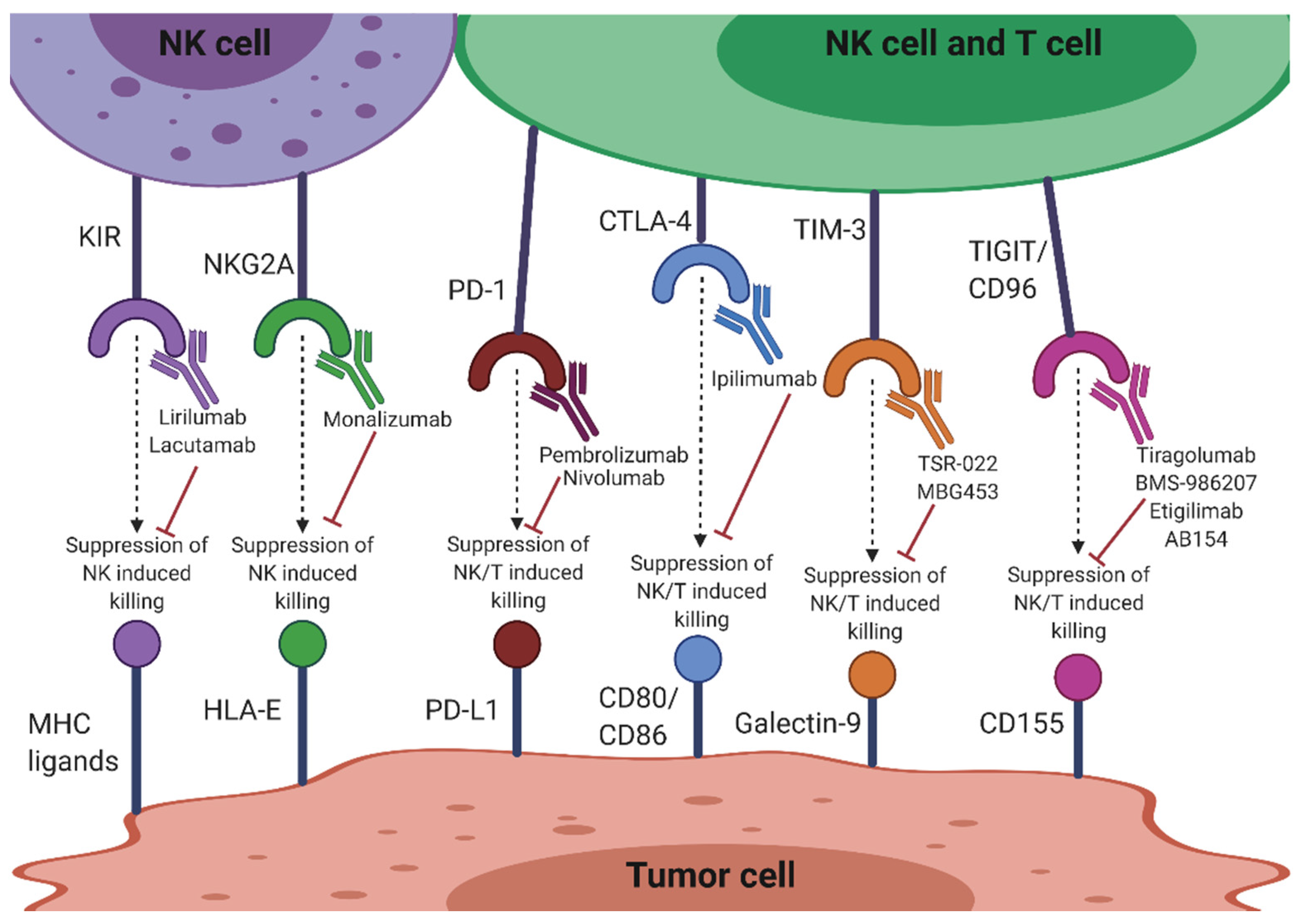
3.2. Therapeutic Approaches to Enhancement of NK Cell Function
3.2.1. KIR
3.2.2. NKG2A
3.2.3. CTLA-4
3.2.4. PD-1/PD-L1
3.2.5. TIGIT and CD96
3.2.6. TIM-3
3.2.7. CD200R
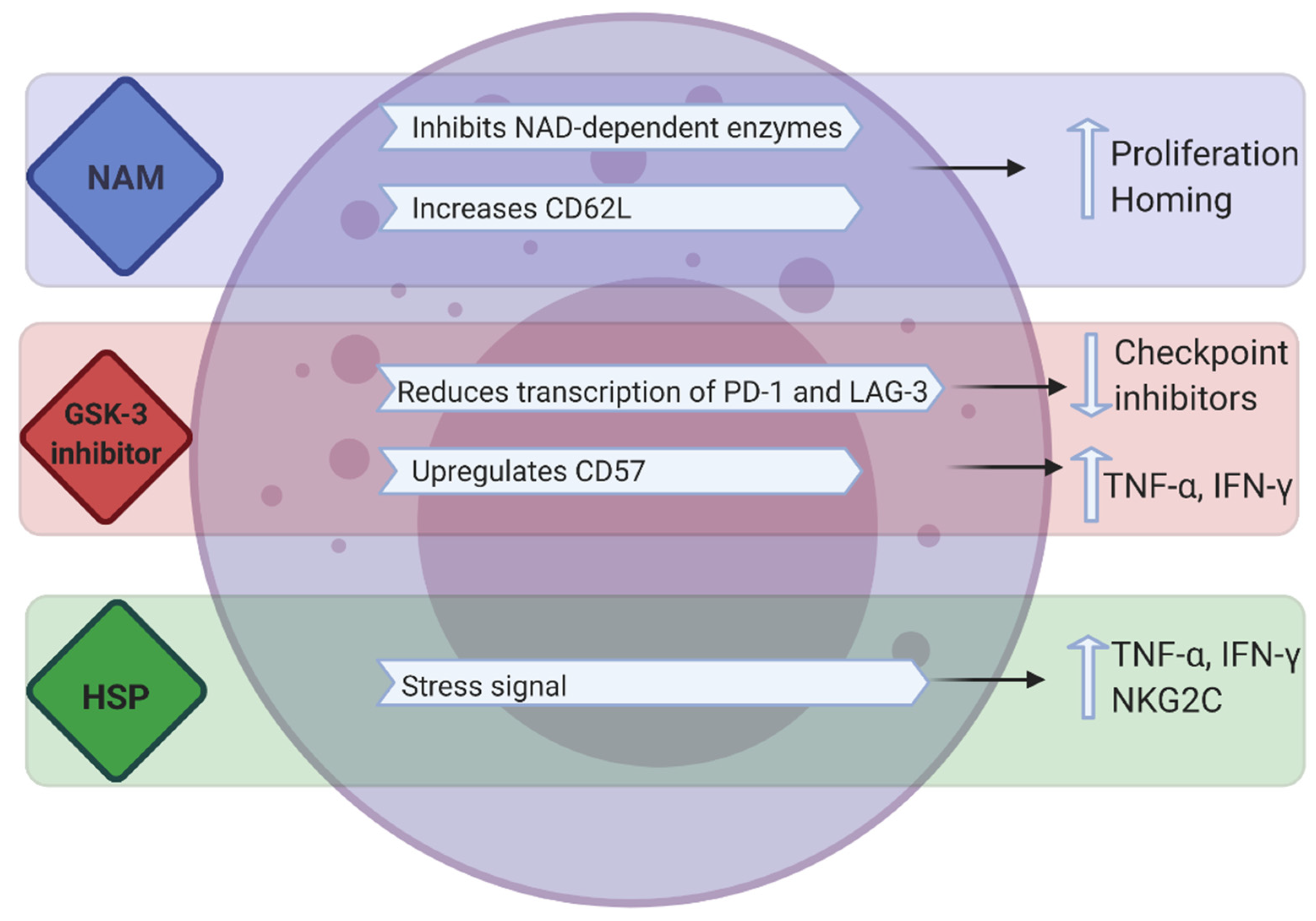
3.3. Synthetic Compounds and Recombinant Proteins
3.3.1. NAM
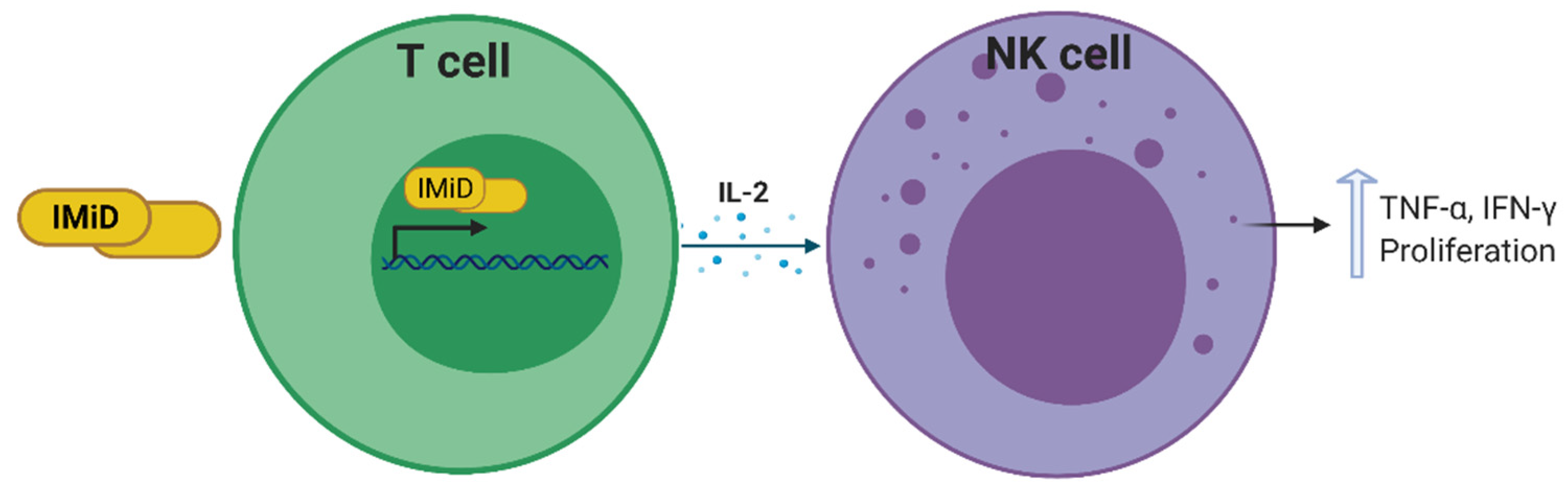
3.3.2. Thalidomide and Thalidomide Derivatives
3.3.3. GSK Inhibitors
3.3.4. Hsp70
3.4. CARs
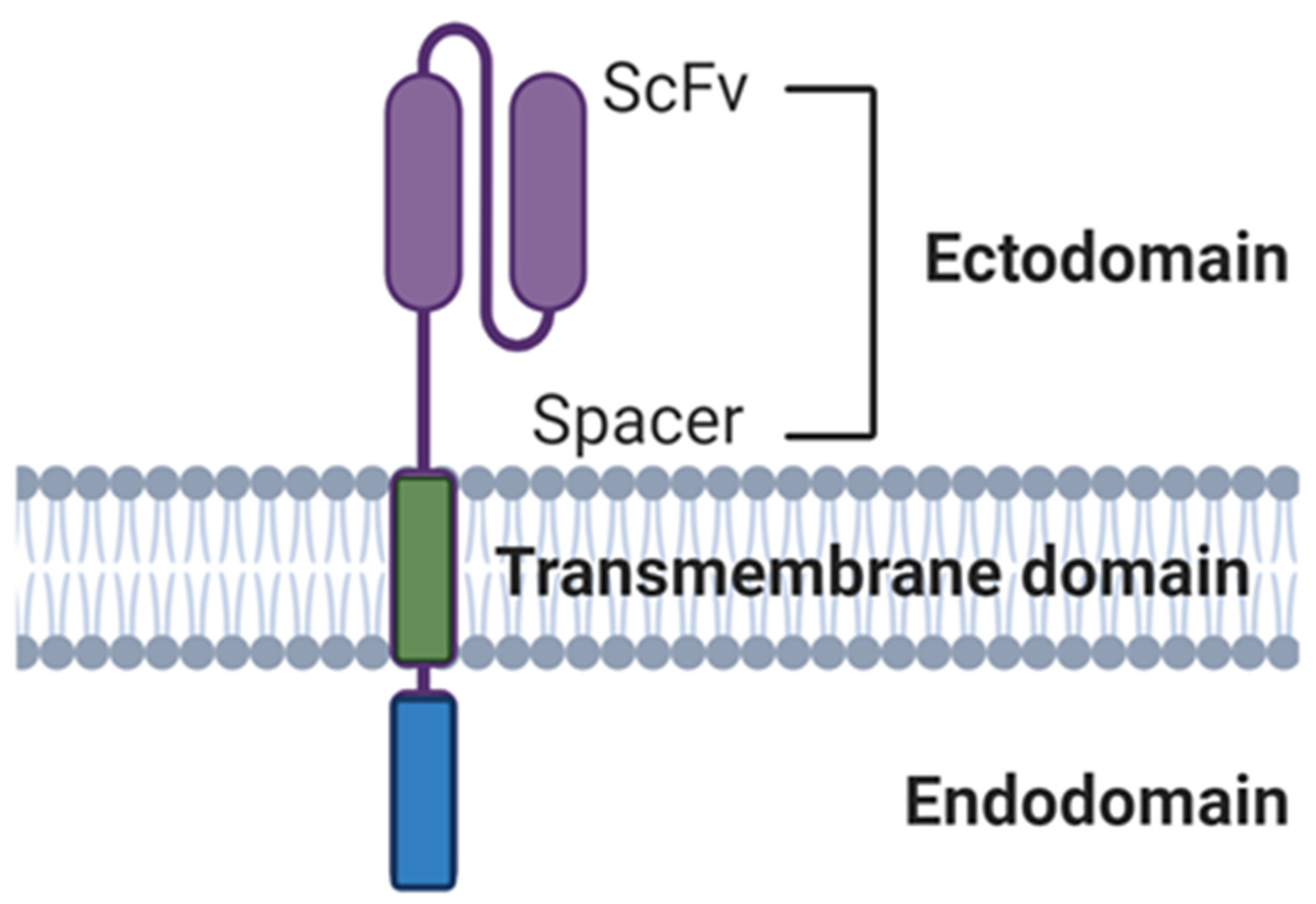
3.4.1. Ectodomains
3.4.2. Endodomains
3.4.3. Safety Features
3.4.4. iCasp9 and Other Inducible Safety Features
3.4.5. Masked CARs
3.4.6. Oxygen-Sensitive CARs
3.4.7. Combinatorial Targeting
3.5. Genetic Ablation to Improve Function
3.5.1. CIS
3.5.2. CDK8
3.5.3. DGK
4. Off-the-Shelf NK Cells for Cancer Immunotherapy
Sources of NK Cells
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lai, Y.S.; Li, Y.; Blum, R.H.; Kaufman, D.S. Concise review: Human pluripotent stem cells to produce cell-based cancer immunotherapy. Stem Cells 2018, 36, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Buning, H.; Sauer, M.; Schambach, A. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef]
- Carotta, S. Targeting NK cells for anticancer immunotherapy: Clinical and preclinical approaches. Front. Immunol. 2016, 7, 152. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering natural killer cells for cancer immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Suen, W.C.; Lee, W.Y.; Leung, K.T.; Pan, X.H.; Li, G. Natural killer cell-based cancer immunotherapy: A review on 10 years completed clinical trials. Cancer Investig. 2018, 36, 431–457. [Google Scholar] [CrossRef]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef]
- Burns, L.J.; Weisdorf, D.J.; DeFor, T.E.; Vesole, D.H.; Repka, T.L.; Blazar, B.R.; Burger, S.R.; Panoskaltsis-Mortari, A.; Keever-Taylor, C.A.; Zhang, M.J.; et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: A phase I/II trial. Bone Marrow. Transplant. 2003, 32, 177–186. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Gasteiger, G.; Hemmers, S.; Firth, M.A.; Le Floc’h, A.; Huse, M.; Sun, J.C.; Rudensky, A.Y. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J. Exp. Med. 2013, 210, 1167–1178. [Google Scholar] [CrossRef]
- Smyth, M.J.; Teng, M.W.; Swann, J.; Kyparissoudis, K.; Godfrey, D.I.; Hayakawa, Y. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J. Immunol. 2006, 176, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Fan, J.; Wei, H.; Zhang, C.; Tian, Z. Use of interleukin-15 for preparation of adherent NK cells from human peripheral blood: Comparison with interleukin-2. J. Immunol. Methods 2003, 279, 79–90. [Google Scholar] [CrossRef]
- Evans, R.; Fuller, J.A.; Christianson, G.; Krupke, D.M.; Troutt, A.B. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: The potential role of NK cell subpopulations. Cell Immunol. 1997, 179, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Troutt, A.B.; Rustum, Y.M. Interleukin 15 protects against toxicity and potentiates antitumor activity of 5-fluorouracil alone and in combination with leucovorin in rats bearing colorectal cancer. Cancer Res. 1998, 58, 1695–1699. [Google Scholar] [PubMed]
- Decot, V.; Voillard, L.; Latger-Cannard, V.; Aissi-Rothe, L.; Perrier, P.; Stoltz, J.F.; Bensoussan, D. Natural-killer cell amplification for adoptive leukemia relapse immunotherapy: Comparison of three cytokines, IL-2, IL-15, or IL-7 and impact on NKG2D, KIR2DL1, and KIR2DL2 expression. Exp. Hematol. 2010, 38, 351–362. [Google Scholar] [CrossRef]
- Imamichi, H.; Sereti, I.; Lane, H.C. IL-15 acts as a potent inducer of CD4+CD25hi cells expressing FOXP3. Eur. J. Immunol. 2008, 38, 1621–1630. [Google Scholar] [CrossRef]
- Cooley, S.; Verneris, M.R.; Curtsinger, J.; McKenna, D.; Weisdorf, D.J.; Blazar, B.R.; Waldmann, T.A.; Miller, J.S. Recombinant human IL-15 promotes in vivo expansion of adoptively transferred NK cells in a first-in-human phase I dose escalation study in patients with AML. Blood 2012, 120, 894-894. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, A.; Fernandez, L.; Valentin, J.; Martinez-Romera, I.; Corral, M.D.; Ramirez, M.; Abad, L.; Santamaria, S.; Gonzalez-Vicent, M.; Sirvent, S.; et al. A phase I/II trial of interleukin-15--stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy 2015, 17, 1594–1603. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Han, K.P.; Zhu, X.; Liu, B.; Jeng, E.; Kong, L.; Yovandich, J.L.; Vyas, V.V.; Marcus, W.D.; Chavaillaz, P.A.; Romero, C.A.; et al. IL-15:IL-15 receptor alpha superagonist complex: High-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011, 56, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ye, X.; Qu, X.; Cui, D.; Zhang, L.; Xu, Z.; Wan, H.; Zhang, L.; Tao, W. Discovery of a novel IL-15 based protein with improved developability and efficacy for cancer immunotherapy. Sci. Rep. 2018, 8, 7675. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Kivimäe, S.; Hennessy, M.; Pena, R.; Quach, P.; Moffet, A.; Nieves, W.; Zhang, P.; Marcondes, M.Q.; Madakamutil, L.; et al. NKTR-255, a polymer-conjugated IL-15 enhances antibody-dependent cellular cytotoxicity mediated By NK cells in a B cell lymphoma model. Blood 2019, 134, 5302-5302. [Google Scholar] [CrossRef]
- Rautela, J.; Huntington, N.D. IL-15 signaling in NK cell cancer immunotherapy. Curr. Opin Immunol. 2017, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Chu, S.; Kodal, B.; Bendzick, L.; Ryan, C.; Lenvik, A.J.; Boylan, K.L.M.; Wong, H.C.; Skubitz, A.P.N.; Miller, J.S.; et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol. Oncol. 2017, 145, 453–461. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I trial of ALT-803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Wagner, J.; Pfannenstiel, V.; Waldmann, A.; Bergs, J.W.J.; Brill, B.; Huenecke, S.; Klingebiel, T.; Rodel, F.; Buchholz, C.J.; Wels, W.S.; et al. A two-phase expansion protocol combining interleukin (IL)-15 and IL-21 improves natural killer cell proliferation and cytotoxicity against rhabdomyosarcoma. Front. Immunol. 2017, 8, 676. [Google Scholar] [CrossRef]
- Heinze, A.; Grebe, B.; Bremm, M.; Huenecke, S.; Munir, T.A.; Graafen, L.; Frueh, J.T.; Merker, M.; Rettinger, E.; Soerensen, J.; et al. The synergistic use of IL-15 and IL-21 for the generation of NK cells from CD3/CD19-depleted grafts improves their ex vivo expansion and cytotoxic potential against neuroblastoma: Perspective for optimized immunotherapy post haploidentical stem cell transplantation. Front. Immunol. 2019, 10, 2816. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, P.; Kamenjarin, K.; Ossa, J.F.V.; Uherek, B.; Bonig, H.; Wels, W.S. Directed differentiation of mobilized hematopoietic stem and progenitor cells into functional NK cells with enhanced antitumor activity. Cells 2020, 9, 811. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Curti, B.D.; Redman, B.G.; Bhatia, S.; Weber, J.S.; Agarwala, S.S.; Sievers, E.L.; Hughes, S.D.; DeVries, T.A.; Hausman, D.F. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J. Clin. Oncol. 2008, 26, 2034–2039. [Google Scholar] [CrossRef]
- Petrella, T.M.; Tozer, R.; Belanger, K.; Savage, K.J.; Wong, R.; Smylie, M.; Kamel-Reid, S.; Tron, V.; Chen, B.E.; Hunder, N.N.; et al. Interleukin-21 has activity in patients with metastatic melanoma: A phase II study. J. Clin. Oncol. 2012, 30, 3396–3401. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Y.; Liu, L.L.; Lundqvist, A. Strategies to augment natural killer (NK) cell activity against solid tumors. Cancers 2019, 11, 1040. [Google Scholar] [CrossRef]
- Sahm, C.; Schonfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Shook, D.; Kamiya, T.; Shimasaki, N.; Chai, S.M.; Coustan-Smith, E.; Imai, C.; Campana, D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood 2014, 124, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Terren, I.; Mikelez, I.; Odriozola, I.; Gredilla, A.; Gonzalez, J.; Orrantia, A.; Vitalle, J.; Zenarruzabeitia, O.; Borrego, F. Implication of interleukin-12/15/18 and ruxolitinib in the phenotype, proliferation, and polyfunctionality of human cytokine-preactivated natural killer cells. Front. Immunol. 2018, 9, 737. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, D.H.; Choi, W.S.; Yi, E.; Kim, H.; Kim, J.M.; Jin, H.S.; Kim, H.S. Harnessing NK cells for cancer immunotherapy: Immune checkpoint receptors and chimeric antigen receptors. BMB Rep. 2021, 54, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front. Immunol. 2019, 10, 2354. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar] [CrossRef]
- Vey, N.; Bourhis, J.H.; Boissel, N.; Bordessoule, D.; Prebet, T.; Charbonnier, A.; Etienne, A.; Andre, P.; Romagne, F.; Benson, D.; et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood 2012, 120, 4317–4323. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell dysfunction and checkpoint immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef]
- Van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Segal, N.H.; Naidoo, J.; Curigliano, G.; Patel, S.; Sahebjam, S.; Papadopoulos, K.P.; Gordon, M.S.; Wang, D.; Rueda, A.G.; Song, X.; et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J. Clin. Oncol. 2018, 36, 3540. [Google Scholar] [CrossRef]
- Cohen, R.B.; Bauman, J.R.; Salas, S.; Colevas, A.D.; Even, C.; Cupissol, D.; Posner, M.R.; Lefebvre, G.; Saada-Bouzid, E.; Bernadach, M.; et al. Combination of monalizumab and cetuximab in recurrent or metastatic head and neck cancer patients previously treated with platinum-based chemotherapy and PD-(L)1 inhibitors. J. Clin. Oncol. 2020, 38, 6516-6516. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti-CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Ralpha complexes enhances tumor control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef]
- Laurent, S.; Queirolo, P.; Boero, S.; Salvi, S.; Piccioli, P.; Boccardo, S.; Minghelli, S.; Morabito, A.; Fontana, V.; Pietra, G.; et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J. Transl. Med. 2013, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Frankel, T.L.; Burns, W.; Royal, R.E. Regression of pancreatic cancer from ipilimumab (anti-CTLA-4) mediated by an NK-cell subset (CD56brightCD16dim). J. Am. Collage Surg. 2009, 209, S120. [Google Scholar] [CrossRef]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Lanuza, P.M.; Pesini, C.; Arias, M.A.; Calvo, C.; Ramirez-Labrada, A.; Pardo, J. Recalling the biological significance of immune checkpoints on NK cells: A chance to overcome LAG3, PD1, and CTLA4 inhibitory pathways by adoptive NK cell transfer? Front. Immunol. 2019, 10, 3010. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e333. [Google Scholar] [CrossRef] [PubMed]
- Julia, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef]
- Hicks, K.C.; Fantini, M.; Donahue, R.N.; Schwab, A.; Knudson, K.M.; Tritsch, S.R.; Jochems, C.; Clavijo, P.E.; Allen, C.T.; Hodge, J.W.; et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology 2018, 7, e1466018. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.P.; Xi, C.H.; Zhu, Y.; Yin, L.D.; Wei, J.S.; Zhang, J.J.; Liu, X.C.; Guo, S.; Fu, Y.; Miao, Y. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget 2016, 7, 66586–66594. [Google Scholar] [CrossRef]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: Novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Goncalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine 2017, 22, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Coles, S.J.; Wang, E.C.; Man, S.; Hills, R.K.; Burnett, A.K.; Tonks, A.; Darley, R.L. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011, 25, 792–799. [Google Scholar] [CrossRef]
- Wright, G.J.; Cherwinski, H.; Foster-Cuevas, M.; Brooke, G.; Puklavec, M.J.; Bigler, M.; Song, Y.; Jenmalm, M.; Gorman, D.; McClanahan, T.; et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J. Immunol. 2003, 171, 3034–3046. [Google Scholar] [CrossRef]
- Rijkers, E.S.; de Ruiter, T.; Baridi, A.; Veninga, H.; Hoek, R.M.; Meyaard, L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol. Immunol. 2008, 45, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Ahl, P.J.; Bijin, V.A.; Kaliaperumal, N.; Lim, S.G.; Wang, C.-I.; Fairhurst, A.-M.; Connolly, J.E. LAG3 is a central regulator of NK cell cytokine production. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nuebling, T.; Schumacher, C.E.; Hofmann, M.; Hagelstein, I.; Schmiedel, B.J.; Maurer, S.; Federmann, B.; Rothfelder, K.; Roerden, M.; Dorfel, D.; et al. The immune checkpoint modulator OX40 and its ligand OX40L in NK-cell immunosurveillance and acute myeloid leukemia. Cancer Immunol. Res. 2018, 6, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popovic, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- Chan, I.S.; Knutsdottir, H.; Ramakrishnan, G.; Padmanaban, V.; Warrier, M.; Ramirez, J.C.; Dunworth, M.; Zhang, H.; Jaffee, E.M.; Bader, J.S.; et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum. Vaccin. Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Frei, G.M.; Persi, N.; Lador, C.; Peled, A.; Cohen, Y.C.; Nagler, A.; Peled, T. Nicotinamide, a form of vitamin B3, promotes expansion of natural killer cells that display increased in vivo survival and cytotoxic activity. Blood 2011, 118, 4035-4035. [Google Scholar] [CrossRef]
- Frei, G.M.; Berg, M.; Peled, T.; Reger, R.N.; Kotecha, R.; Onishi, T.; Espinoza-Calderon, L.; Persi, N.; Lador, C.; Peled, A.; et al. Improved homing to bone marrow, spleen and lung of adoptively infused NK cells expanded ex vivo with the small molecule nicotinamide using feeder-free conditions. Blood 2013, 122, 897-897. [Google Scholar] [CrossRef]
- Bachanova, V.; McKenna, D.H.; Luo, X.; Defor, T.E.; Cooley, S.; Warlick, E.; Weisdorf, D.J.; Brachya, G.; Peled, T.; Miller, J.S. First-in-human phase I study of nicotinamide-expanded related donor natural killer cells for the treatment of relapsed/refractory non-hodgkin lymphoma and multiple myeloma. Biol. Blood Marrow Transplant. 2019, 25, S175–S176. [Google Scholar] [CrossRef]
- Hayashi, T.; Hideshima, T.; Akiyama, M.; Podar, K.; Yasui, H.; Raje, N.; Kumar, S.; Chauhan, D.; Treon, S.P.; Richardson, P.; et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: Clinical application. Br. J. Haematol. 2005, 128, 192–203. [Google Scholar] [CrossRef]
- Childs, R.W.; Carlsten, M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: The force awakens. Nat. Rev. Drug Discov. 2015, 14, 487–498. [Google Scholar] [CrossRef]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxid. Med. Cell Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef]
- Rudd, C.E.; Chanthong, K.; Taylor, A. Small Molecule Inhibition of GSK-3 Specifically Inhibits the Transcription of Inhibitory Co-receptor LAG-3 for Enhanced Anti-tumor Immunity. Cell Rep. 2020, 30, 2075–2082.e2074. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Kwon, S.J.; Lee, H.; Park, H.R.; Choi, G.E.; Kang, S.W.; Kwon, S.W.; Kim, N.; Lee, S.Y.; Ryu, S.; et al. NK cell function triggered by multiple activating receptors is negatively regulated by glycogen synthase kinase-3beta. Cell Signal. 2015, 27, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, M.; Pfister, K.; Jordan, A.; Scholz, R.; Andreesen, R.; Schmitz, G.; Schmetzer, H.; Hiddemann, W.; Multhoff, G. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones 2000, 5, 438–442. [Google Scholar] [CrossRef]
- Multhoff, G.; Pfister, K.; Gehrmann, M.; Hantschel, M.; Gross, C.; Hafner, M.; Hiddemann, W. A 14-mer Hsp70 peptide stimulates natural killer (NK) cell activity. Cell Stress Chaperones 2001, 6, 337–344. [Google Scholar] [CrossRef]
- Gastpar, R.; Gross, C.; Rossbacher, L.; Ellwart, J.; Riegger, J.; Multhoff, G. The cell surface-localized heat shock protein 70 epitope TKD induces migration and cytolytic activity selectively in human NK cells. J. Immunol. 2004, 172, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase i trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef]
- Seimetz, D.; Heller, K.; Richter, J. Approval of first CAR-Ts: Have we solved all hurdles for ATMPs? Cell Med. 2019, 11, 2155179018822781. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Diao, Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef]
- Wang, L.; Dou, M.; Ma, Q.; Yao, R.; Liu, J. Chimeric antigen receptor (CAR)-modified NK cells against cancer: Opportunities and challenges. Int. Immunopharmacol. 2019, 74, 105695. [Google Scholar] [CrossRef] [PubMed]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef]
- Oberschmidt, O.; Kloess, S.; Koehl, U. Redirected primary human chimeric antigen receptor natural killer cells as an “off-the-shelf immunotherapy” for improvement in cancer treatment. Front. Immunol. 2017, 8, 654. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W., Jr.; Chatterjee, S.K. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Rynda, A.; Sentman, C.L. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J. Immunol. 2009, 183, 6939–6947. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e185. [Google Scholar] [CrossRef]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef]
- Pfefferle, A.; Huntington, N.D. You have got a fast CAR: Chimeric antigen receptor NK cells in cancer therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T cell malignancies using CAR-based immunotherapy: Challenges and potential solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Marin, V.; Cribioli, E.; Philip, B.; Tettamanti, S.; Pizzitola, I.; Biondi, A.; Biagi, E.; Pule, M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum. Gene Ther. Methods 2012, 23, 376–386. [Google Scholar] [CrossRef]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 254. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.C.; Pule, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Bryson, P.D.; Zhao, Y.; Cinay, G.E.; Li, S.; Guo, Y.; Siriwon, N.; Wang, P. Masked chimeric antigen receptor for tumor-specific activation. Mol. Ther. 2017, 25, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef]
- Charrot, S.; Hallam, S. CAR-T Cells: Future Perspectives. Hemasphere 2019, 3, e188. [Google Scholar] [CrossRef]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 2016, 167, 419–432. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Ren, Y.B.; Sun, S.J.; Han, S.Y. Safety strategies of genetically engineered T cells in cancer immunotherapy. Curr. Pharm. Des. 2018, 24, 78–83. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.S. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front. Immunol. 2018, 9, 2041. [Google Scholar] [CrossRef] [PubMed]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 2017, 6, e1267892. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell 2020, 27, 224–237. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Gotthardt, D.; Prchal-Murphy, M.; Didara, Z.; Menzl, I.; Prinz, D.; Edlinger, L.; Putz, E.M.; Sexl, V. NK cell-specific CDK8 deletion enhances antitumor responses. Cancer Immunol. Res. 2018, 6, 458–466. [Google Scholar] [CrossRef]
- Putz, E.M.; Gotthardt, D.; Hoermann, G.; Csiszar, A.; Wirth, S.; Berger, A.; Straka, E.; Rigler, D.; Wallner, B.; Jamieson, A.M.; et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 2013, 4, 437–444. [Google Scholar] [CrossRef]
- Noessner, E. DGK-alpha: A checkpoint in cancer-mediated immuno-inhibition and target for immunotherapy. Front. Cell Dev. Biol. 2017, 5, 16. [Google Scholar] [CrossRef]
- Singh, B.K.; Kambayashi, T. The immunomodulatory functions of diacylglycerol kinase zeta. Front. Cell Dev. Biol. 2016, 4, 96. [Google Scholar] [CrossRef]
- Kobayashi, N.; Hozumi, Y.; Ito, T.; Hosoya, T.; Kondo, H.; Goto, K. Differential subcellular targeting and activity-dependent subcellular localization of diacylglycerol kinase isozymes in transfected cells. Eur. J. Cell Biol. 2007, 86, 433–444. [Google Scholar] [CrossRef]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Regulation and functions of diacylglycerol kinases. Chem. Rev. 2011, 111, 6186–6208. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed]
- Prinz, P.U.; Mendler, A.N.; Masouris, I.; Durner, L.; Oberneder, R.; Noessner, E. High DGK-alpha and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J. Immunol. 2012, 188, 5990–6000. [Google Scholar] [CrossRef]
- Prinz, P.U.; Mendler, A.N.; Brech, D.; Masouris, I.; Oberneder, R.; Noessner, E. NK-cell dysfunction in human renal carcinoma reveals diacylglycerol kinase as key regulator and target for therapeutic intervention. Int. J. Cancer 2014, 135, 1832–1841. [Google Scholar] [CrossRef]
- Baldanzi, G.; Ragnoli, B.; Malerba, M. Potential role of diacylglycerol kinases in immune-mediated diseases. Clin. Sci. 2020, 134, 1637–1658. [Google Scholar] [CrossRef]
- Sato, M.; Liu, K.; Sasaki, S.; Kunii, N.; Sakai, H.; Mizuno, H.; Saga, H.; Sakane, F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology 2013, 92, 99–107. [Google Scholar] [CrossRef]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGKalpha inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase alpha-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef]
- Riese, M.J.; Grewal, J.; Das, J.; Zou, T.; Patil, V.; Chakraborty, A.K.; Koretzky, G.A. Decreased diacylglycerol metabolism enhances ERK activation and augments CD8+ T cell functional responses. J. Biol. Chem. 2011, 286, 5254–5265. [Google Scholar] [CrossRef] [PubMed]
- Wesley, E.M.; Xin, G.; McAllister, D.; Malarkannan, S.; Newman, D.K.; Dwinell, M.B.; Cui, W.; Johnson, B.D.; Riese, M.J. Diacylglycerol kinase zeta (DGKzeta) and Casitas b-lineage proto-oncogene b-deficient mice have similar functional outcomes in T cells but DGKzeta-deficient mice have increased T cell activation and tumor clearance. Immunohorizons 2018, 2, 107–118. [Google Scholar] [CrossRef]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 2003, 4, 882–890. [Google Scholar] [CrossRef]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.P. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 2006, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Zhong, X. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33, 97–118. [Google Scholar] [CrossRef]
- Joshi, R.P.; Schmidt, A.M.; Das, J.; Pytel, D.; Riese, M.J.; Lester, M.; Diehl, J.A.; Behrens, E.M.; Kambayashi, T.; Koretzky, G.A. The zeta isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci. Signal. 2013, 6, ra102. [Google Scholar] [CrossRef]
- Jung, I.Y.; Kim, Y.Y.; Yu, H.S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Singh, B.K.; Paustian, A.M.; Kambayashi, T. Diacylglycerol kinase zeta is a target to enhance NK cell function. J. Immunol. 2016, 197, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy—Advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Yilmaz, A.; Cui, H.; Caligiuri, M.A.; Yu, J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 168. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Mizuno, S.I.; Harada, M.; Nagafuji, K.; Miyamoto, T.; Iwasaki, H.; Fujisaki, T.; Kubota, A.; Ohno, Y.; Arima, F.; et al. Generation of human natural killer cells from peripheral blood CD34+ cells mobilized by granulocyte colony-stimulating factor. Br. J. Haematol. 1996, 92, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Sconocchia, G.; Provenzano, M.; Rezvani, K.; Li, J.; Melenhorst, J.; Hensel, N.; Barrett, A.J. CD34+ cells cultured in stem cell factor and interleukin-2 generate CD56+ cells with antiproliferative effects on tumor cell lines. J. Transl Med. 2005, 3, 15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giuliani, M.; Giron-Michel, J.; Negrini, S.; Vacca, P.; Durali, D.; Caignard, A.; Le Bousse-Kerdiles, C.; Chouaib, S.; Devocelle, A.; Bahri, R.; et al. Generation of a novel regulatory NK cell subset from peripheral blood CD34+ progenitors promoted by membrane-bound IL-15. PLoS ONE 2008, 3, e2241. [Google Scholar] [CrossRef]
- Moretta, F.; Petronelli, F.; Lucarelli, B.; Pitisci, A.; Bertaina, A.; Locatelli, F.; Mingari, M.C.; Moretta, L.; Montaldo, E. The generation of human innate lymphoid cells is influenced by the source of hematopoietic stem cells and by the use of G-CSF. Eur. J. Immunol. 2016, 46, 1271–1278. [Google Scholar] [CrossRef]
- Miller, J.S.; Prosper, F.; McCullar, V. Natural killer (NK) cells are functionally abnormal and NK cell progenitors are diminished in granulocyte colony-stimulating factor-mobilized peripheral blood progenitor cell collections. Blood 1997, 90, 3098–3105. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Bendzick, L.; Kaufman, D.S. Mouse xenograft model for intraperitoneal administration of NK cell immunotherapy for ovarian cancer. Methods Mol. Biol. 2016, 1441, 277–284. [Google Scholar] [CrossRef]
- Spanholtz, J.; Tordoir, M.; Eissens, D.; Preijers, F.; van der Meer, A.; Joosten, I.; Schaap, N.; de Witte, T.M.; Dolstra, H. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS ONE 2010, 5, e9221. [Google Scholar] [CrossRef]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef]
- Matsubara, H.; Niwa, A.; Nakahata, T.; Saito, M.K. Induction of human pluripotent stem cell-derived natural killer cells for immunotherapy under chemically defined conditions. Biochem. Biophys. Res. Commun. 2019, 515, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “off-the-shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.H.; Zhu, H.; Wang, Y.M.; Heragu, N.; Bernareggi, D.; Ruiz-Cisneros, A.; Bahena, A.; Ask, E.H.; Hoel, H.J.; Malmberg, K.J.; et al. Umbilical cord blood and iPSC-derived natural killer cells demonstrate key differences in cytotoxic activity and KIR profiles. Front. Immunol. 2020, 11, 561553. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.T.; Carlsten, M.; Sohlberg, E.; Liu, L.L.; Clancy, T.; Karimi, M.; Cooley, S.; Miller, J.S.; Klimkowska, M.; Schaffer, M.; et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 2018, 24, 1834–1844. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Dolatshad, H.; Trikha, P.; Hussain, S.A.; Elmas, E.; Foltz, J.A.; Moseman, J.E.; Thakkar, A.; Nakkula, R.J.; Lamb, M.; et al. Generation of knock-out primary and expanded human NK cells using Cas9 ribonucleoproteins. J. Vis. Exp. 2018, 136, e58237. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef]
- Hoogstad-van Evert, J.S.; Cany, J.; van den Brand, D.; Oudenampsen, M.; Brock, R.; Torensma, R.; Bekkers, R.L.; Jansen, J.H.; Massuger, L.F.; Dolstra, H. Umbilical cord blood CD34(+) progenitor-derived NK cells efficiently kill ovarian cancer spheroids and intraperitoneal tumors in NOD/SCID/IL2Rg(null) mice. Oncoimmunology 2017, 6, e1320630. [Google Scholar] [CrossRef]





| Antibody | Company | Clinical Study Progress | Combination Therapy |
|---|---|---|---|
| Tiragolumab | Genentech/Roche | Preclinical studies | In combination with antibodies to PD-1 |
| BMS-986207 | Bristol-Myers Squibb | Phase I/II study NCT02913313 | Monotherapy or in combination with nivolumab (anti-PD-1) in advanced solid tumors |
| Etigilimab | OncoMed Pharmaceuticals | Phase I clinical trial NCT031119428 | Monotherapy or in combination with nivolumab (anti-PD-1) in advanced solid tumors |
| AB154 | Arcus Biosciences | Phase I clinical trial NCT03628677 | Monotherapy or combined with AB122 (anti-PD-1) in advanced solid tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, R.; Pupovac, A.; Evtimov, V.; Boyd, N.; Shu, R.; Boyd, R.; Trounson, A. Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells 2021, 10, 1058. https://doi.org/10.3390/cells10051058
Islam R, Pupovac A, Evtimov V, Boyd N, Shu R, Boyd R, Trounson A. Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells. 2021; 10(5):1058. https://doi.org/10.3390/cells10051058
Chicago/Turabian StyleIslam, Rasa, Aleta Pupovac, Vera Evtimov, Nicholas Boyd, Runzhe Shu, Richard Boyd, and Alan Trounson. 2021. "Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy" Cells 10, no. 5: 1058. https://doi.org/10.3390/cells10051058
APA StyleIslam, R., Pupovac, A., Evtimov, V., Boyd, N., Shu, R., Boyd, R., & Trounson, A. (2021). Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells, 10(5), 1058. https://doi.org/10.3390/cells10051058