
‘Off-the-Shelf’ Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
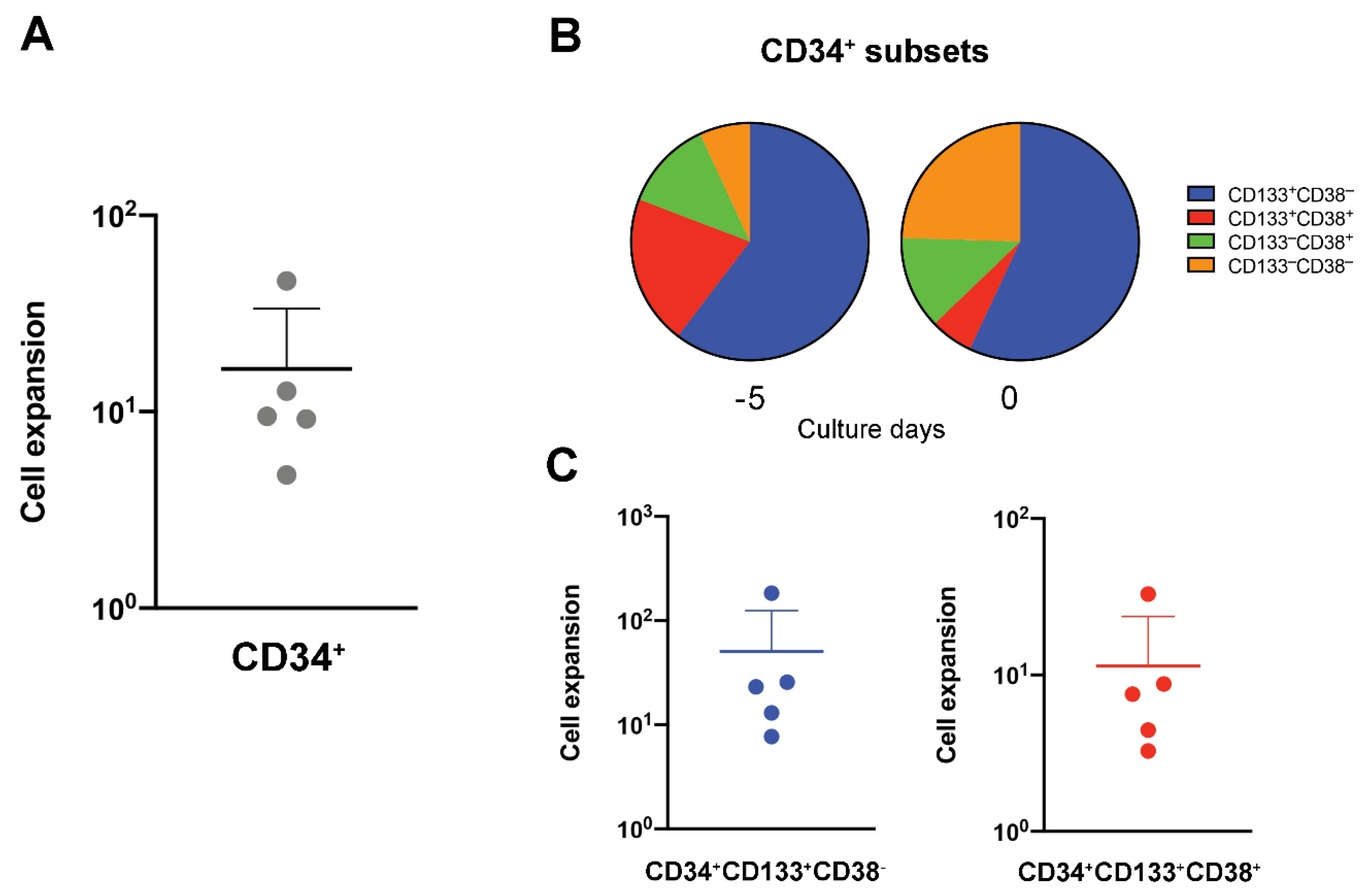
2.1. CD34+ Cell Preparation and Expansion from UCBs
2.2. T Cell Differentiation Assay
2.3. Cell Surface Marker Expression on Differentiated T Cells
2.4. CBMC-Derived T Cells
2.5. Cell Lines
2.6. In Vitro Cytotoxicity Assay
2.7. Statistical Analysis
3. Results
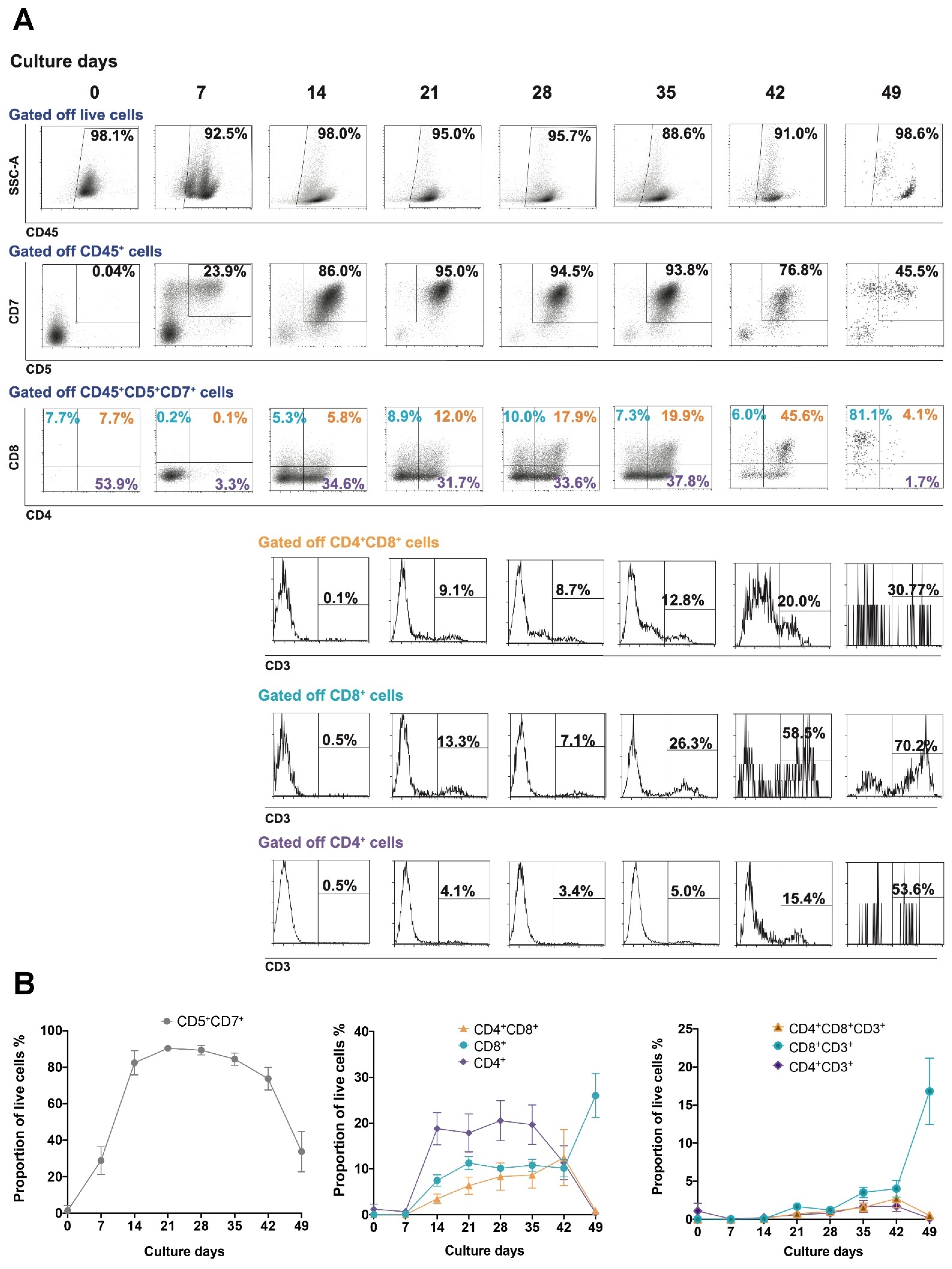
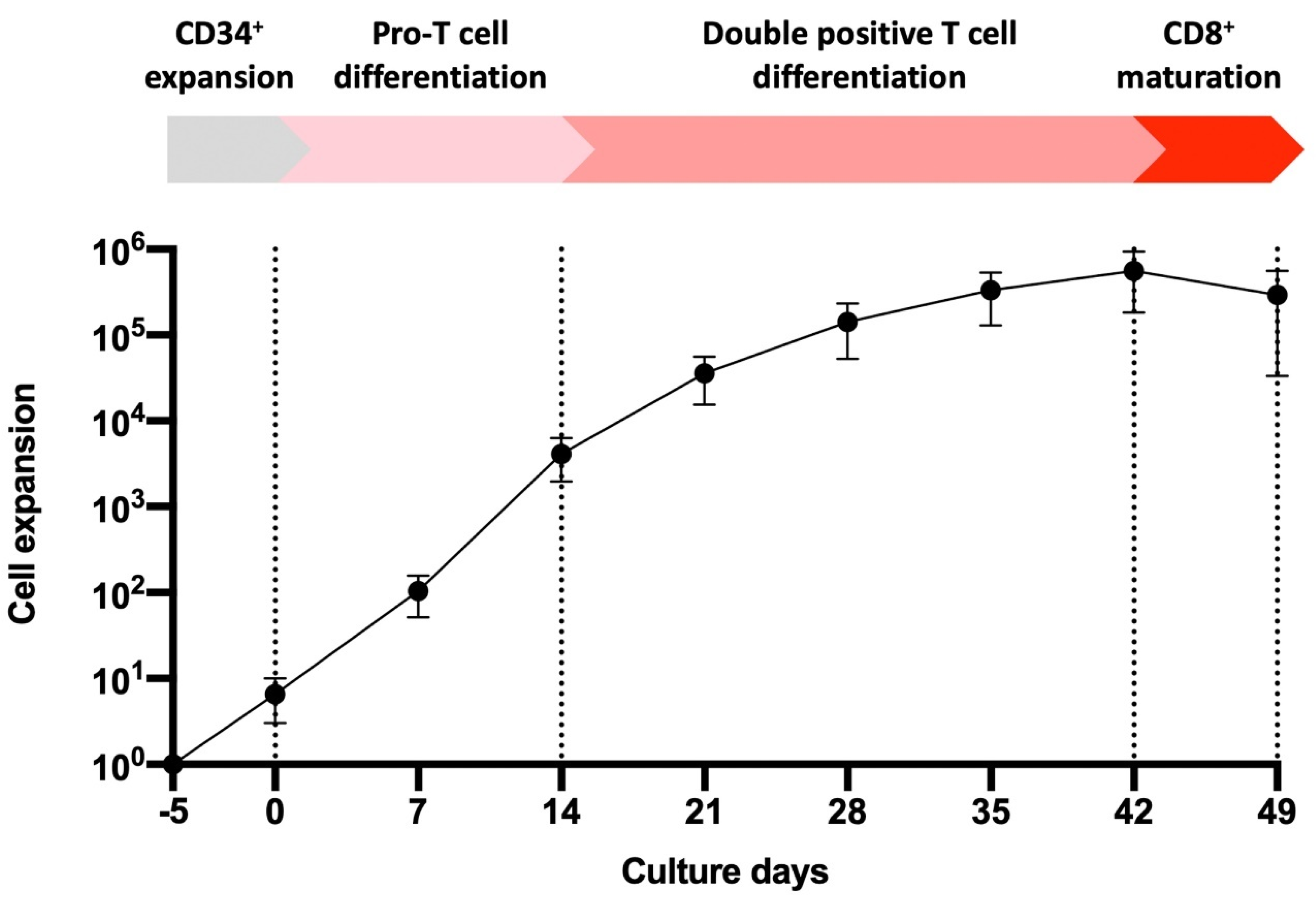
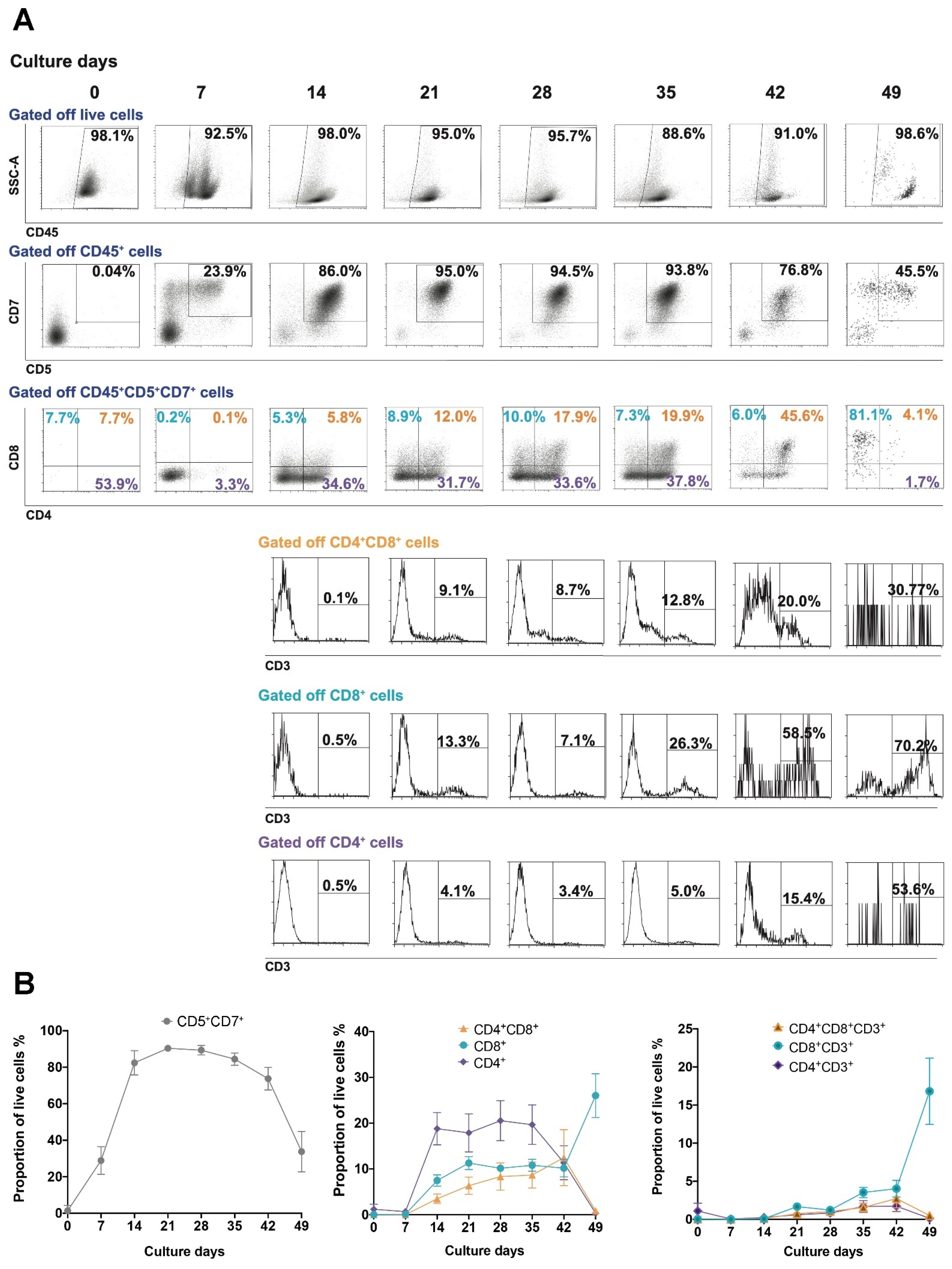
3.1. UCB-Derived CD34+ Expansion and Differentiation to T Cells
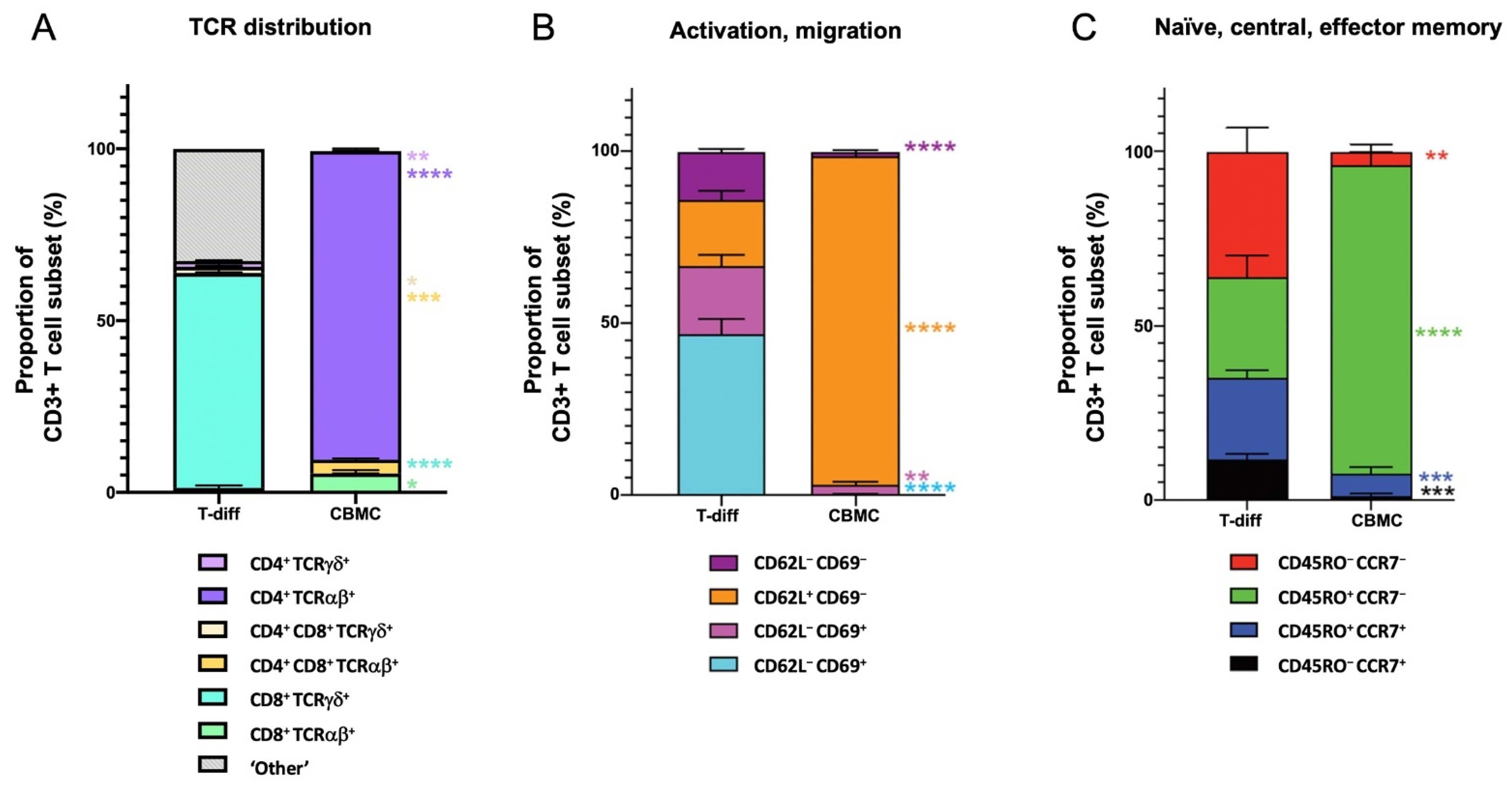
3.2. Maturation State of T Cells Differentiated from HSCs In Vitro
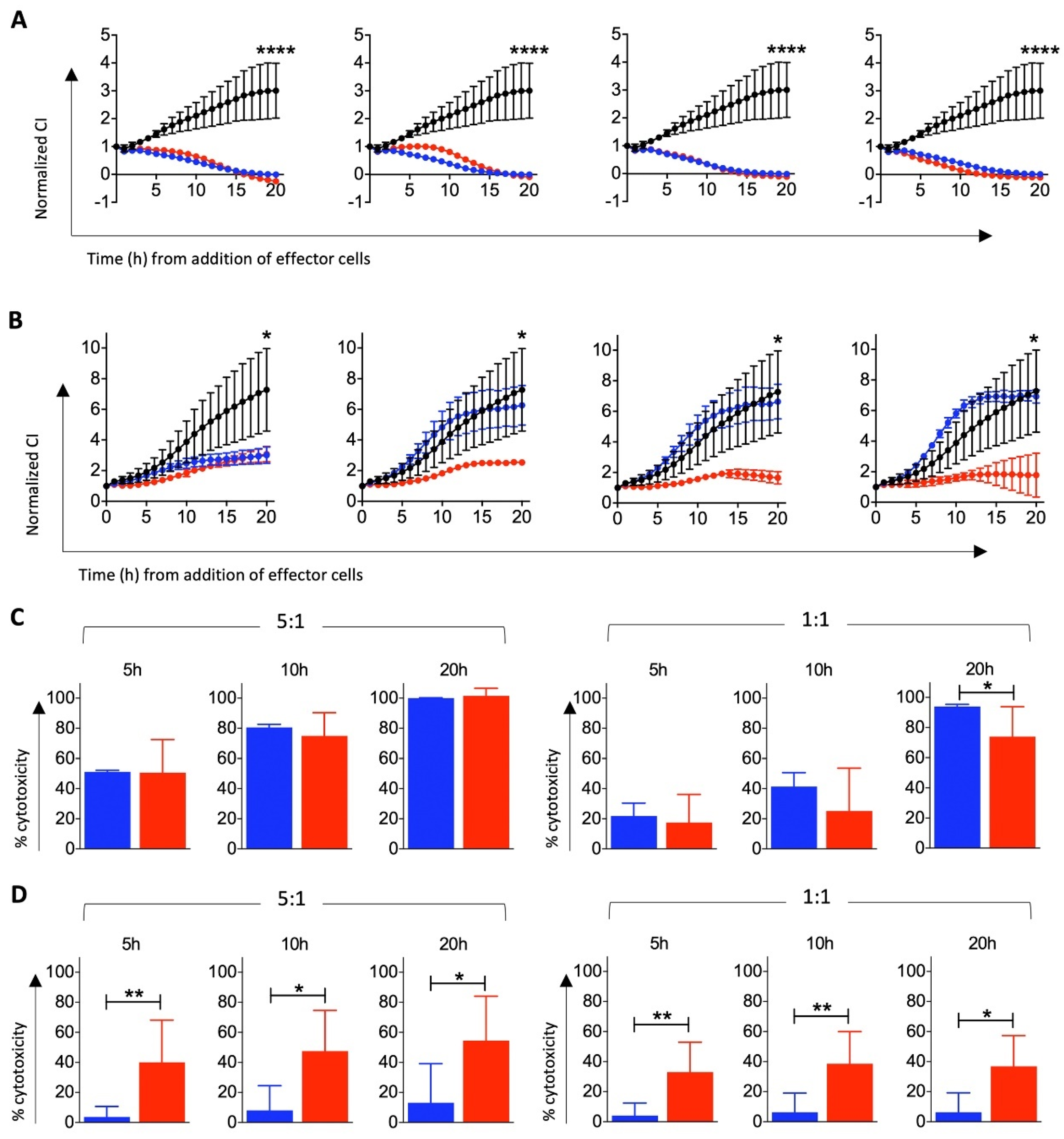
3.3. Cytotoxic Function of T Cells Differentiated from HSCs In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first CAR T therapy. Nat. Rev. Drug Discov. 2017, 16, 669. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, M.M.; Stamatis, C.; Lakelin, M.; Farid, S.; Titchener-Hooker, N.; Shah, N. Autologous CAR T-cell therapies supply chain: Challenges and opportunities? Cancer Gene Ther. 2020, 27, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Salam, N.; Rane, S.; Das, R.; Faulkner, M.; Gund, R.; Kandpal, U.; Lewis, V.; Mattoo, H.; Prabhu, S.; Ranganathan, V.; et al. T cell ageing: Effects of age on development, survival & function. Indian J. Med. Res. 2013, 138, 595–608. [Google Scholar]
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, J. The thymus gland in secondary immunodeficiency. Arch. Pathol. Lab. Med. 1987, 111, 1118–1122. [Google Scholar]
- Mehta, R.S.; Rezvani, K. Immune reconstitution post allogeneic transplant and the impact of immune recovery on the risk of infection. Virulence 2016, 7, 901–916. [Google Scholar] [CrossRef] [Green Version]
- Wils, E.J.; van der Holt, B.; Broers, A.E.; Posthumus-van Sluijs, S.J.; Gratama, J.W.; Braakman, E.; Cornelissen, J.J. Insufficient recovery of thymopoiesis predicts for opportunistic infections in allogeneic hematopoietic stem cell transplant recipients. Haematologica 2011, 96, 1846–1854. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef]
- Nussbaumer, O.; Koslowski, M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncol. Technol. 2019, 1, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Awong, G.; Herer, E.; Surh, C.D.; Dick, J.E.; La Motte-Mohs, R.N.; Zuniga-Pflucker, J.C. Characterization in vitro and engraftment potential in vivo of human progenitor T cells generated from hematopoietic stem cells. Blood 2009, 114, 972–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zuniga-Pflucker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.W.; Lai, Y.S.; Lamb, L.S., Jr.; Townes, T.M. Broad T-cell receptor repertoire in T-lymphocytes derived from human induced pluripotent stem cells. PLoS ONE 2014, 9, e97335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dervovic, D.D.; Ciofani, M.; Kianizad, K.; Zuniga-Pflucker, J.C. Comparative and functional evaluation of in vitro generated to ex vivo CD8 T cells. J. Immunol. 2012, 189, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- La Motte-Mohs, R.N.; Herer, E.; Zuniga-Pflucker, J.C. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 2005, 105, 1431–1439. [Google Scholar] [CrossRef]
- Schmitt, T.M.; de Pooter, R.F.; Gronski, M.A.; Cho, S.K.; Ohashi, P.S.; Zuniga-Pflucker, J.C. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat. Immunol. 2004, 5, 410–417. [Google Scholar] [CrossRef]
- Van Coppernolle, S.; Verstichel, G.; Timmermans, F.; Velghe, I.; Vermijlen, D.; De Smedt, M.; Leclercq, G.; Plum, J.; Taghon, T.; Vandekerckhove, B.; et al. Functionally mature CD4 and CD8 TCRalphabeta cells are generated in OP9-DL1 cultures from human CD34+ hematopoietic cells. J. Immunol. 2009, 183, 4859–4870. [Google Scholar] [CrossRef] [Green Version]
- Vodyanik, M.A.; Bork, J.A.; Thomson, J.A.; Slukvin, I.I. Human embryonic stem cell-derived CD34+ cells: Efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005, 105, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywacz, B.; Kataria, N.; Sikora, M.; Oostendorp, R.A.; Dzierzak, E.A.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood 2006, 108, 3824–3833. [Google Scholar] [CrossRef] [Green Version]
- McCullar, V.; Oostendorp, R.; Panoskaltsis-Mortari, A.; Yun, G.; Lutz, C.T.; Wagner, J.E.; Miller, J.S. Mouse fetal and embryonic liver cells differentiate human umbilical cord blood progenitors into CD56-negative natural killer cell precursors in the absence of interleukin-15. Exp. Hematol. 2008, 36, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Haddad, R.; Guardiola, P.; Izac, B.; Thibault, C.; Radich, J.; Delezoide, A.L.; Baillou, C.; Lemoine, F.M.; Gluckman, J.C.; Pflumio, F.; et al. Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood 2004, 104, 3918–3926. [Google Scholar] [CrossRef]
- Martin, C.H.; Woll, P.S.; Ni, Z.; Zuniga-Pflucker, J.C.; Kaufman, D.S. Differences in lymphocyte developmental potential between human embryonic stem cell and umbilical cord blood-derived hematopoietic progenitor cells. Blood 2008, 112, 2730–2737. [Google Scholar] [CrossRef]
- Knapp, D.J.; Hammond, C.A.; Miller, P.H.; Rabu, G.M.; Beer, P.A.; Ricicova, M.; Lecault, V.; Da Costa, D.; VanInsberghe, M.; Cheung, A.M.; et al. Dissociation of Survival, Proliferation, and State Control in Human Hematopoietic Stem Cells. Stem Cell Rep. 2017, 8, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Fares, I.; Chagraoui, J.; Gareau, Y.; Gingras, S.; Ruel, R.; Mayotte, N.; Csaszar, E.; Knapp, D.J.; Miller, P.; Ngom, M.; et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 2014, 345, 1509–1512. [Google Scholar] [CrossRef] [Green Version]
- Chagraoui, J.; Lehnertz, B.; Girard, S.; Spinella, J.F.; Fares, I.; Tomellini, E.; Mayotte, N.; Corneau, S.; MacRae, T.; Simon, L.; et al. UM171 induces a homeostatic inflammatory-detoxification response supporting human HSC self-renewal. PLoS ONE 2019, 14, e0224900. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.E., Jr.; Brunstein, C.G.; Boitano, A.E.; DeFor, T.E.; McKenna, D.; Sumstad, D.; Blazar, B.R.; Tolar, J.; Le, C.; Jones, J.; et al. Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell 2016, 18, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzensperger, R.; Kadakia, T.; Tai, X.; Alag, A.; Guinter, T.I.; Egawa, T.A.-O.; Erman, B.; Singer, A. Identification of lineage-specifying cytokines that signal all CD8(+)-cytotoxic-lineage-fate ‘decisions’ in the thymus. Nat. Immunol. 2017, 18, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerignoli, F.; Abassi, Y.A.; Lamarche, B.J.; Guenther, G.; Santa Ana, D.; Guimet, D.; Zhang, W.; Zhang, J.; Xi, B. In vitro immunotherapy potency assays using real-time cell analysis. PLoS ONE 2018, 13, e0193498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galy, A.; Verma, S.; Barcena, A.; Spits, H. Precursors of CD3+CD4+CD8+ cells in the human thymus are defined by expression of CD34. Delineation of early events in human thymic development. J. Exp. Med. 1993, 178, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, C.J.; Shpall, E.J.; Keller, G. Differential long-term and multilineage engraftment potential from subfractions of human CD34+ cord blood cells transplanted into NOD/SCID mice. Proc. Natl. Acad. Sci. USA 2002, 99, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Gorgens, A.; Radtke, S.; Horn, P.A.; Giebel, B. New relationships of human hematopoietic lineages facilitate detection of multipotent hematopoietic stem and progenitor cells. Cell Cycle 2013, 12, 3478–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.; Anderson, G. Thymic Epithelial Cells. Annu. Rev. Immunol. 2017, 35, 85–118. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J. Immunol. 2009, 182, 6879–6888. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, T.M.; Zuniga-Pflucker, J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef] [Green Version]
- de Pooter, R.; Zuniga-Pflucker, J.C. T-cell potential and development in vitro: The OP9-DL1 approach. Curr. Opin. Immunol. 2007, 19, 163–168. [Google Scholar] [CrossRef]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Reimann, C.; Six, E.; Dal-Cortivo, L.; Schiavo, A.; Appourchaux, K.; Lagresle-Peyrou, C.; de Chappedelaine, C.; Ternaux, B.; Coulombel, L.; Beldjord, K.; et al. Human T-lymphoid progenitors generated in a feeder-cell-free Delta-like-4 culture system promote T-cell reconstitution in NOD/SCID/gammac(-/-) mice. Stem Cells 2012, 30, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Langley, M.A.; Singh, J.; Edgar, J.M.; Mohtashami, M.; Zuniga-Pflucker, J.C.; Zandstra, P.W. Progenitor T-cell differentiation from hematopoietic stem cells using Delta-like-4 and VCAM-1. Nat. Methods 2017, 14, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Huijskens, M.J.; Walczak, M.; Koller, N.; Briede, J.J.; Senden-Gijsbers, B.L.; Schnijderberg, M.C.; Bos, G.M.; Germeraad, W.T. Technical advance: Ascorbic acid induces development of double-positive T cells from human hematopoietic stem cells in the absence of stromal cells. J. Leukoc. Biol. 2014, 96, 1165–1175. [Google Scholar] [CrossRef]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Seet, C.S.; Li, S.; Chick, B.; Zhu, Y.; Chang, P.; Tsai, S.; Sun, V.; Lopez, S.; Chen, H.C.; et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 376–389 e378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montel-Hagen, A.; Crooks, G.M. From pluripotent stem cells to T cells. Exp. Hematol. 2019, 71, 24–31. [Google Scholar] [CrossRef]
- Singh, J.; Zuniga-Pflucker, J.C. Producing proT cells to promote immunotherapies. Int. Immunol. 2018, 30, 541–550. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef]
- Nomura, A.; Taniuchi, I. The Role of CD8 Downregulation during Thymocyte Differentiation. Trends Immunol. 2020, 41, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.; Adoro, S.; Park, J.H. Lineage fate and intense debate: Myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol. 2008, 8, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Shilpi; Lal, G. Role of gamma-delta (gammadelta) T cells in autoimmunity. J. Leukoc. Biol. 2015, 97, 259–271. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Function of gammadelta T cells in tumor immunology and their application to cancer therapy. Exp. Mol. Med. 2021, 53, 318–327. [Google Scholar] [CrossRef]
- Lamb, L.S., Jr.; Lopez, R.D. Gammadelta T cells: A new frontier for immunotherapy? Biol. Blood Marrow Transplant. 2005, 11, 161–168. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Therapy 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vgamma9Vdelta2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell. Mol. Immunol. 2021, 18, 427–439. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyd, N.; Cartledge, K.; Cao, H.; Evtimov, V.; Pupovac, A.; Trounson, A.; Boyd, R. ‘Off-the-Shelf’ Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells. Cells 2021, 10, 2631. https://doi.org/10.3390/cells10102631
Boyd N, Cartledge K, Cao H, Evtimov V, Pupovac A, Trounson A, Boyd R. ‘Off-the-Shelf’ Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells. Cells. 2021; 10(10):2631. https://doi.org/10.3390/cells10102631
Chicago/Turabian StyleBoyd, Nicholas, Kellie Cartledge, Huimin Cao, Vera Evtimov, Aleta Pupovac, Alan Trounson, and Richard Boyd. 2021. "‘Off-the-Shelf’ Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells" Cells 10, no. 10: 2631. https://doi.org/10.3390/cells10102631
APA StyleBoyd, N., Cartledge, K., Cao, H., Evtimov, V., Pupovac, A., Trounson, A., & Boyd, R. (2021). ‘Off-the-Shelf’ Immunotherapy: Manufacture of CD8+ T Cells Derived from Hematopoietic Stem Cells. Cells, 10(10), 2631. https://doi.org/10.3390/cells10102631