
Next Generation Therapeutics for the Treatment of Myelofibrosis

Abstract
1. Introduction
2. JAK Inhibitors
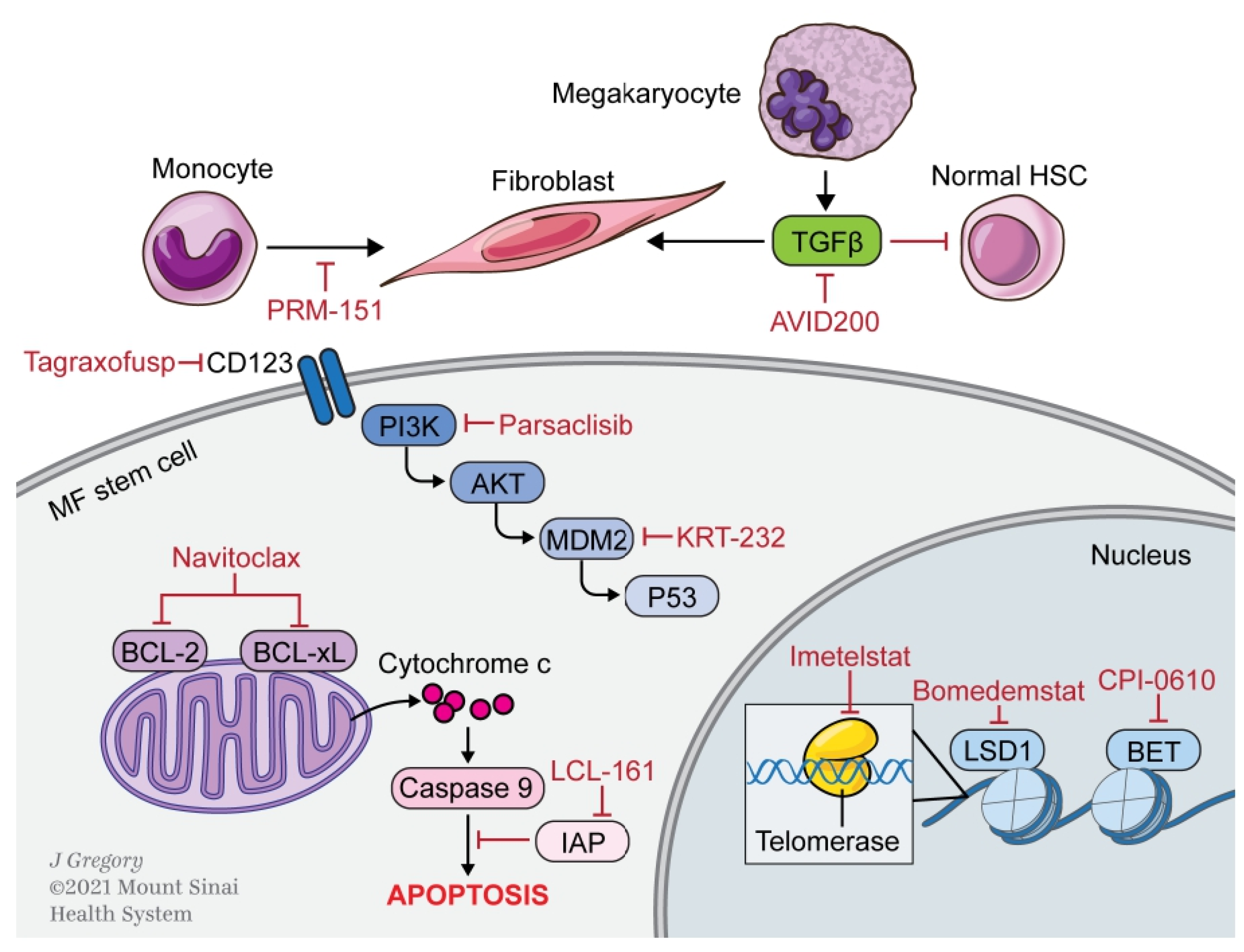
3. Novel Therapies
3.1. Apoptosis
3.1.1. BCL-2/xL
3.1.2. SMAC
3.1.3. MDM2
3.2. Epigenetic Modulation
3.2.1. Bromodomain and Extra-Terminal Domain
3.2.2. LSD1
3.3. Microenvironment
3.3.1. Pentraxin-2
3.3.2. TGFβ
3.3.3. Aurora Kinase
3.4. Signaling Pathways
3.5. Miscellaneous Targets
3.5.1. CD123
3.5.2. Telomerase

{kind=link}
| Drug | Mechanism | Population | Phase | N | Clinical Efficacy * | Ongoing Clinical Trial | |||
|---|---|---|---|---|---|---|---|---|---|
| SVR35% | TSS50% | BMF ≥ 1 Grade | Additional Efficacy Measures | ||||||
| Naïve to JAKi | |||||||||
| CPI-0610 + ruxoloitinib | BET inhibitor | JAKi naïve | 2 | 78 | 67% | 57% | 33% | NCT04603495 | |
| Inadequate response to JAKi | |||||||||
| Navitoclax + ruxolitinib | BCL-2/BCL-xL inhibitor | Ruxolitinib failure | 2 | 34 | 27% | 30% | 29% | NCT04472598 | |
| CPI-0610 + ruxolitinib | BET inhibitor | Ruxolitinib sub-optimal response TI | 2 | 26 | 22.2% | 36.8% | - | NCT04603495 | |
| Ruxolitinib sub-optimal response TD | 2 | 44 | 20.8% | 46.2% | - | 21.4% TD to TI | |||
| PRM-151 ± ruxolitinib | Recombinant Pentraxin-2 | Ruxolitinib inadequate response | 2 | 27 | - | - | 33.3% | Median SVR—26.1% Median TSS—64% | None |
| Parsaclisib + ruxolitinib | PI3Kδ inhibitor | Ruxolitinib inadequate response | 2 | 51 | - | - | - | In highest dose level: Median SVR—27.1% Median TSS—51.4% | NCT04551066 |
| Relapsed, refractory, or intolerant of JAKi | |||||||||
| Bomedemstat | LSD-1 inhibitor | JAKi R/R | 1/2 | 49 | 14% | 25% | - | NCT03136185 | |
| LCL-161 | SMAC mimetic | JAKi R/R | 2 | 47 | 2.1% | 23.4% | - | None | |
| CPI-0610 | BET inhibitor | Ruxolitinib failure TI | 2 | 27 | 23.8% | 47.4% | - | NCT04603495 | |
| Ruxolitinib failure TD | 2 | 16 | 0% | 8.3% | 34.4% TD to TI | ||||
| Tagraxofusp | CD123 directed ADC | JAKi R/R | 1/2 | 27 | 24% | 45% | - | NCT02268253 | |
| PRM-151 | Recombinant Pentraxin-2 | Ruxolitinib failure | 2 | 98 | - | - | 28.1% | 16% TD to TI | None |
| AVID-200 | TGFβ trap | Ruxolitinib R/R with 2/3 BMF | 1 | 12 | - | 41.7% | 0% | Median maximum platelet chage 48% | NCT03895112 |
| Imetelstat | Telomerase inhibitor | Ruxolitinib failure | 2 | 48 | 10% | 32.2% | 43.2% | NCT04576156 | |
| Alisertib | AURKA inhibitor | Ruxolitinib failure | 1 | 24 | - | 31.8% | - | 29% with SVR 50% by palpation | None |
| Response Categories. | Required Criteria (for All Response Categories, Benefit Must Last for ≥12 Weeks to Qualify as a Response) |
|---|---|
| CR | Bone marrow: * Age-adjusted normocellularity; <5% blasts; ≤grade 1 MF † and |
| Peripheral blood: Hemoglobin ≥ 100 g/L and <UNL; neutrophil count ≥ 1 × 109/L and <UNL; | |
| Platelet count ≥ 100 × 109/L and <UNL; <2% immature myeloid cells ‡ and | |
| Clinical: Resolution of disease symptoms; spleen and liver not palpable; no evidence of EMH | |
| PR | Peripheral blood: Hemoglobin ≥ 100 g/L and <UNL; neutrophil count ≥ 1 × 109/L and <UNL; platelet count ≥ 100 × 109/L and <UNL; <2% immature myeloid cells ‡ and |
| Clinical: Resolution of disease symptoms; spleen and liver not palpable; no evidence of EMH or | |
| Bone marrow: * Age-adjusted normocellularity; <5% blasts; ≤grade 1 MF †, and peripheral blood: Hemoglobin ≥ 85 but < 100 g/L and <UNL; neutrophil count ≥ 1 × 109/L and < UNL; platelet count ≥ 50, but < 100 × 109/L and <UNL; <2% immature myeloid cells ‡ and | |
| Clinical: Resolution of disease symptoms; spleen and liver not palpable; no evidence of EMH | |
| Clinical improvement (CI) | The achievement of anemia, spleen or symptoms response without progressive disease or increase in severity of anemia, thrombocytopenia, or neutropenia § |
| Anemia response | Transfusion-independent patients: a ≥20 g/L increase in hemoglobin level || |
| Transfusion-dependent patients: becoming transfusion-independent ¶ | |
| Spleen response # | A baseline splenomegaly that is palpable at 5–10 cm, below the LCM, becomes not palpable ** or |
| A baseline splenomegaly that is palpable at >10 cm, below the LCM, decreases by ≥50% ** | |
| A baseline splenomegaly that is palpable at <5 cm, below the LCM, is not eligible for spleen response | |
| A spleen response requires confirmation by MRI or computed tomography showing ≥35% spleen volume reduction | |
| Symptoms response | A ≥50% reduction in the MPN-SAF TSS †† |
| Progressive disease ‡‡ | Appearance of a new splenomegaly that is palpable at least 5 cm below the LCM or |
| A ≥100% increase in palpable distance, below LCM, for baseline splenomegaly of 5–10 cm or | |
| A 50% increase in palpable distance, below LCM, for baseline splenomegaly of >10 cm or | |
| Leukemic transformation confirmed by a bone marrow blast count of ≥20% or | |
| A peripheral blood blast content of ≥20% associated with an absolute blast count of ≥1 × 109/L that lasts for at least 2 weeks | |
| Stable disease | Belonging to none of the above listed response categories |
| Relapse | No longer meeting criteria for at least CI after achieving CR, PR, or CI, or |
| Loss of anemia response persisting for at least 1 month or | |
| Loss of spleen response persisting for at least 1 month |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef]
- Mesa, R.; Miller, C.B.; Thyne, M.; Mangan, J.; Goldberger, S.; Fazal, S.; Ma, X.; Wilson, W.; Paranagama, D.C.; Dubinski, D.G.; et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: The MPN Landmark survey. BMC Cancer 2016, 16, 167. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Harrison, C.N. Myelofibrosis: Clinicopathologic features, prognosis, and management. Clin. Adv. Hematol. Oncol. 2018, 16, 121–131. [Google Scholar] [PubMed]
- Sokol, K.; Tremblay, D.; Bhalla, S.; Rampal, R.; Mascarenhas, J.O. Implications of Mutation Profiling in Myeloid Malignancies-PART 2: Myeloproliferative Neoplasms and Other Myeloid Malignancies. Oncology 2018, 32, e45–e51. [Google Scholar]
- Lataillade, J.J.; Pierre-Louis, O.; Hasselbalch, H.C.; Uzan, G.; Jasmin, C.; Martyre, M.C.; Le Bousse-Kerdiles, M.C.; French INSERM; European EUMNET Networks on Myelofibrosis. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood 2008, 112, 3026–3035. [Google Scholar] [CrossRef]
- Deeg, H.J.; Bredeson, C.; Farnia, S.; Ballen, K.; Gupta, V.; Mesa, R.A.; Popat, U.; Hari, P.; Saber, W.; Seftel, M.; et al. Hematopoietic Cell Transplantation as Curative Therapy for Patients with Myelofibrosis: Long-Term Success in all Age Groups. Biol. Blood Marrow Transplant. 2015, 21, 1883–1887. [Google Scholar] [CrossRef][Green Version]
- United States Food and Drug Administration. JAKAFI (Ruxolitinib) Label; FDA: Silver Spring, MD, USA, 2011.
- United States Food and Drug Administration. INREBIC (Fedratinib) Label; FDA: Silver Spring, MD, USA, 2019.
- Wernig, G.; Kharas, M.G.; Okabe, R.; Moore, S.A.; Leeman, D.S.; Cullen, D.E.; Gozo, M.; McDowell, E.P.; Levine, R.L.; Doukas, J.; et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008, 13, 311–320. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Levine, R.L.; Pardanani, A.; Tefferi, A.; Gilliland, D.G. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 2007, 7, 673–683. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017, 10, 55. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 73. [Google Scholar] [CrossRef]
- Mesa, R.A.; Jamieson, C.; Bhatia, R.; Deininger, M.W.; Fletcher, C.D.; Gerds, A.T.; Gojo, I.; Gotlib, J.; Gundabolu, K.; Hobbs, G.; et al. NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2.2018. J. Natl. Compr. Cancer Netw. 2017, 15, 1193–1207. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Passamonti, F.; Zweegman, S.; Talpaz, M.; Verstovsek, S.; Rose, S.; Zhang, J.; et al. Fedratinib Induces Spleen Responses and Reduces Symptom Burden in Patients with Myeloproliferative Neoplasm (MPN)-Associated Myelofibrosis (MF) and Low Platelet Counts, who were Either Ruxolitinib-Naïve or were Previously Treated with Ruxolitinib. Blood 2019, 134, 668. [Google Scholar] [CrossRef]
- Bassiony, S.; Harrison, C.N.; McLornan, D.P. Evaluating the Safety, Efficacy, and Therapeutic Potential of Momelotinib in the Treatment of Intermediate/High-Risk Myelofibrosis: Evidence to Date. Ther. Clin. Risk Manag. 2020, 16, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef]
- Mesa, R.A.; Kiladjian, J.J.; Catalano, J.V.; Devos, T.; Egyed, M.; Hellmann, A.; McLornan, D.; Shimoda, K.; Winton, E.F.; Deng, W.; et al. SIMPLIFY-1: A Phase III Randomized Trial of Momelotinib Versus Ruxolitinib in Janus Kinase Inhibitor-Naive Patients With Myelofibrosis. J. Clin. Oncol. 2017, 35, 3844–3850. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Tremblay, D.; Mascarenhas, J. Pacritinib to treat myelofibrosis patients with thrombocytopenia. Expert Rev. Hematol. 2018, 11, 707–714. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018. [Google Scholar] [CrossRef]
- Gerds, A.T.; Savona, M.R.; Scott, B.L.; Talpaz, M.; Egyed, M.; Harrison, C.N.; Yacoub, A.; Vannucchi, A.; Mead, A.J.; Kiladjian, J.J.; et al. Determining the recommended dose of pacritinib: Results from the PAC203 dose-finding trial in advanced myelofibrosis. Blood Adv. 2020, 4, 5825–5835. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Mehra, M.; He, J.; Potluri, R.; Loefgren, C. Patient characteristics and outcomes after ruxolitinib discontinuation in patients with myelofibrosis. J. Med. Econ. 2020, 23, 721–727. [Google Scholar] [CrossRef]
- Kuykendall, A.T.; Shah, S.; Talati, C.; Al Ali, N.; Sweet, K.; Padron, E.; Sallman, D.A.; Lancet, J.E.; List, A.F.; Zuckerman, K.S.; et al. Between a rux and a hard place: Evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann. Hematol. 2018, 97, 435–441. [Google Scholar] [CrossRef]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef]
- Palandri, F.; Breccia, M.; Bonifacio, M.; Polverelli, N.; Elli, E.M.; Benevolo, G.; Tiribelli, M.; Abruzzese, E.; Iurlo, A.; Heidel, F.H.; et al. Life after ruxolitinib: Reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer 2020, 126, 1243–1252. [Google Scholar] [CrossRef]
- Korsmeyer, S.J. Regulators of cell death. TIG 1995, 11, 101–105. [Google Scholar] [CrossRef]
- Tognon, R.; Gasparotto, E.P.; Neves, R.P.; Nunes, N.S.; Ferreira, A.F.; Palma, P.V.; Kashima, S.; Covas, D.T.; Santana, M.; Souto, E.X.; et al. Deregulation of apoptosis-related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in Essential Thrombocythemia and Myelofibrosis. J. Hematol. Oncol. 2012, 5, 2. [Google Scholar] [CrossRef]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.; Bracken, L.S.; et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, J.; Li, Y.; Berenzon, D.; Wang, X.; Mascarenhas, J.; Xu, M.; Hoffman, R. Treatment with the Bcl-xL inhibitor ABT-737 in combination with interferon alpha specifically targets JAK2V617F-positive polycythemia vera hematopoietic progenitor cells. Blood 2010, 116, 4284–4287. [Google Scholar] [CrossRef]
- Petiti, J.; Lo Iacono, M.; Rosso, V.; Andreani, G.; Jovanovski, A.; Podesta, M.; Lame, D.; Gobbi, M.; Fava, C.; Saglio, G.; et al. Bcl-xL represents a therapeutic target in Philadelphia negative myeloproliferative neoplasms. J. Cell Mol. Med. 2020, 24, 10978–10986. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Garcia, J.; Potluri, J.; Holes, L.; Harb, J.; Jung, P.; Hutti, J.; Prchal, J.; Verstovsek, S.; Harrison, C. The Addition of Navitoclax to Ruxolitinib Demonstrates Efficacy within Different High-Risk Populations in Patients with Relapsed/Refractory Myelofibrosis. In Proceedings of the ASH Annual Meeting, San Diego, CA, USA, 5–8 December 2020. Abstract 52. [Google Scholar]
- Owens, T.W.; Gilmore, A.P.; Streuli, C.H.; Foster, F.M. Inhibitor of Apoptosis Proteins: Promising Targets for Cancer Therapy. J. Carcinog Mutagen. 2013, S14. [Google Scholar] [CrossRef]
- Craver, B.M.; Nguyen, T.K.; Nguyen, J.; Nguyen, H.; Huynh, C.; Morse, S.J.; Fleischman, A.G. The SMAC mimetic LCL-161 selectively targets JAK2(V617F) mutant cells. Exp. Hematol. Oncol. 2020, 9, 1. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Carter, B.Z.; Kantarjian, H.M.; Cortes, J.E.; Bose, P.; Kadia, T.M.; Garcia-Manero, G.; Bueso-Ramos, C.E.; DiNardo, C.D.; Bledsoe, S.; et al. Final Results of Phase 2 Clinical Trial of LCL161, a Novel Oral SMAC Mimetic/IAP Antagonist, for Patients with Intermediate to High Risk Myelofibrosis. Blood 2019, 134, 555. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, W.; Li, Y.; Berenzon, D.; Wang, X.; Wang, J.; Mascarenhas, J.; Xu, M.; Hoffman, R. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 2010, 38, 472–480. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Lu, M.; Wang, X.; Li, Y.; Tripodi, J.; Mosoyan, G.; Mascarenhas, J.; Kremyanskaya, M.; Najfeld, V.; Hoffman, R. Combination treatment in vitro with Nutlin, a small-molecule antagonist of MDM2, and pegylated interferon-alpha 2a specifically targets JAK2V617F-positive polycythemia vera cells. Blood 2012, 120, 3098–3105. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xia, L.; Li, Y.; Wang, X.; Hoffman, R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon alpha 2a target JAK2V617F-positive progenitor and stem cells. Blood 2014, 124, 771–779. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Lu, M.; Kosiorek, H.; Virtgaym, E.; Xia, L.; Sandy, L.; Mesa, R.; Petersen, B.; Farnoud, N.; Najfeld, V.; et al. Oral idasanutlin in patients with polycythemia vera. Blood 2019, 134, 525–533. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Higgins, B.; Anders, D.; Burbury, K.; El-Galaly, T.; Gerds, A.; Gupta, V.; Kovic, B.; Maffioli, M.; Mesa, R.; et al. Safety and Efficacy of Idasanutlin in Patients (pts) with Hydroxyurea (HU)-Resistant/Intolerant Polycythemia Vera (PV): Results of an International Phase II Study. In Proceedings of the ASH Annual Meeting, San Diego, CA, USA, 5–8 December 2020. Abstract 479. [Google Scholar]
- Al-Ali, H.; Delgado, R.; Lange, A.; Pluta, A.; McLornan, D.; Vachhani, P.; Damaj, G.; Jost, P.; Rejtő, L.; Hus, M.; et al. KRT-232, a first-in-class, murine double minute 2 inhibitor (MDM2I), for myelofibrosis (MF) relapsed or refractory (R/R) to janus-associated kinase inhibitor (JAKI) treatment (TX). In Proceedings of the EHA Annual Meeting, Virtual Conference, Held Online, 12 June 2020. Abstract S215. [Google Scholar]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Mathews Griner, L.A.; Guha, R.; Shinn, P.; Keller, J.M.; et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 29-43.e7. [Google Scholar] [CrossRef]
- Talpaz, M.; Rampal, R.; Verstovsek, S.; Harrison, C.; Drummond, M.W.; Kiladjian, J.; Vannucchi, A.; Kremyanskaya, M.; Schiller, G.; Patriarca, A.; et al. CPI-0610, a Bromodomain and Extraterminal Domain Protein (BET) Inhibitor, As Monotherapy in Advanced Myelofibrosis Patients Refractory/Intolerant to JAK Inhibitor: Update from Phase 2 MANIFEST Study. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 6 December 2020. Abstract 2163. [Google Scholar]
- Verstovsek, S.; Mascarenhas, J.; Kremyanskaya, M.; Hoffman, R.; Rampal, R.; Gupta, V.; Talpaz, M.; Granacher, N.; Leber, B.; Kiladjian, J.; et al. CPI-0610, Bromodomain and Extraterminal Domain Protein (BET) Inhibitor, As “Add-on” to Ruxolitinib, in Advanced Myelofibrosis Patients with Suboptimal Response: Update of MANIFEST Phase 2 Study. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 5 December 2020. Abstract 56. [Google Scholar]
- Mascarenhas, J.; Harrison, C.; Patriarca, A.; Devos, T.; Palandri, F.; Rampal, R.; Mead, A.; Kremyanskaya, M.; Somervaille, T.; Wondergem, M.; et al. CPI-0610, a Bromodomain and Extraterminal Domain Protein (BET) Inhibitor, in Combination with Ruxolitinib, in JAK-Inhibitor-Naïve Myelofibrosis Patients: Update of MANIFEST Phase 2 Study. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 5 December 2020. Abstract 55. [Google Scholar]
- Sprussel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Handschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; Konig, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y., Jr.; Levine, R.L.; et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. Hemasphere 2018, 2, e54. [Google Scholar] [CrossRef]
- Yacoub, A.; Pettit, K.M.; Bradley, T.J.; Gerds, A.; Tartaczuch, M.; Shortt, J.; Curtin, N.J.; Rossetti, J.M.; Burbury, K.; Mead, A.; et al. A Phase 2 Study of the LSD1 Inhibitor IMG7289 (bomedemstat) for the Treatment of Advanced Myelofibrosis. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 5 December 2020. [Google Scholar]
- Duffield, J.S.; Lupher, M.L., Jr. PRM-151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect 2010, 23, 305–315. [Google Scholar] [CrossRef]
- Pilling, D.; Buckley, C.D.; Salmon, M.; Gomer, R.H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 2003, 171, 5537–5546. [Google Scholar] [CrossRef]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, M.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients With Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Hasserjian, R.P.; Pozdnyakova, O.; Veletic, I.; Mesa, R.A.; Foltz, L.; Mascarenhas, J.; Ritchie, E.K.; Palmer, J.; Silver, R.T.; et al. PRM-151 in Myelofibrosis: Efficacy and Safety in an Open Label Extension Study. Blood 2018, 132, 686. [Google Scholar] [CrossRef]
- Verstovsek, S.; Talpaz, M.; Wadleigh, M.; Palmer, J.; Isidori, A.; te Boekhorst, P.; Savona, M.; Gotlib, J.; Hasserjian, R.; Pozdnyakova, O. A randomized, double blind phase 2 study of 3 different doses of prm-151 in patients with myelofibrosis who were previously treated with or ineligible for ruxolitinib: S828. In Proceedings of the EHA 2019, Amsterdam, The Netherlands, 15 June 2019. abstract 3. [Google Scholar]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.; Eto, K.; Ema, H.; Nakauchi, H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 2009, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Scandura, J.M.; Boccuni, P.; Massague, J.; Nimer, S.D. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 15231–15236. [Google Scholar] [CrossRef]
- Fortunel, N.O.; Hatzfeld, A.; Hatzfeld, J.A. Transforming growth factor-beta: Pleiotropic role in the regulation of hematopoiesis. Blood 2000, 96, 2022–2036. [Google Scholar] [CrossRef]
- Fortunel, N.; Hatzfeld, J.; Kisselev, S.; Monier, M.N.; Ducos, K.; Cardoso, A.; Batard, P.; Hatzfeld, A. Release from quiescence of primitive human hematopoietic stem/progenitor cells by blocking their cell-surface TGF-beta type II receptor in a short-term in vitro assay. Stem Cells 2000, 18, 102–111. [Google Scholar] [CrossRef]
- Langer, J.C.; Henckaerts, E.; Orenstein, J.; Snoeck, H.W. Quantitative trait analysis reveals transforming growth factor-beta2 as a positive regulator of early hematopoietic progenitor and stem cell function. J. Exp. Med. 2004, 199, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, L.; Mascarenhas, J.; Migliaccio, A.R.; O’Connor-McCourt, M.; Tremblay, G.; Denis, J.-F.; Iancu-Rubin, C.; Hoffman, R. AVID200, a Potent Trap for TGF-β Ligands Inhibits TGF-β1 Signaling in Human Myelofibrosis. Blood 2018, 132, 1791. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Kosiorek, H.; Varricchio, L.; Bhave, R.; Kuykendall, A.; Komrokji, R.; Gerds, A.; Palmer, J.; Gabler, A.; Sandy, L.; et al. Rationale for and Results of a Phase I Study of the TGF-β 1/3 Inhibitor AVID200 in Subjects with Myelofibrosis: MPN-RC 118 Trial. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 5 December 2020. Abstract 1254. [Google Scholar]
- Soderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann. N. Y. Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef]
- Maguer-Satta, V.; Bartholin, L.; Jeanpierre, S.; Ffrench, M.; Martel, S.; Magaud, J.P.; Rimokh, R. Regulation of human erythropoiesis by activin A, BMP2, and BMP4, members of the TGFbeta family. Exp. Cell Res. 2003, 282, 110–120. [Google Scholar] [CrossRef]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Gerds, A.T.; Vannucchi, A.M.; Passamonti, F.; Kremyanskaya, M.; Gotlib, J.R.; Palmer, J.M.; McCaul, K.; Ribrag, V.; Mead, A.J.; Harrison, C.N.; et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood 2019, 134, 557. [Google Scholar] [CrossRef]
- Bose, P.; Pemmaraju, N.; Masarova, L.; Bledsoe, S.D.; Daver, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.E.; Kornblau, S.M.; Andreeff, M.; et al. Sotatercept (ACE-011) for Anemia of Myelofibrosis: A Phase 2 Study. Blood 2020, 136, 10–11. [Google Scholar] [CrossRef]
- Villeval, J.L.; Cohen-Solal, K.; Tulliez, M.; Giraudier, S.; Guichard, J.; Burstein, S.A.; Cramer, E.M.; Vainchenker, W.; Wendling, F. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood 1997, 90, 4369–4383. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Bianchi, L.; Cellai, C.; Paoletti, F.; Rana, R.A.; Lorenzini, R.; Migliaccio, G.; Migliaccio, A.R. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood 2002, 100, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Geddis, A.E.; Kaushansky, K. Megakaryocytes express functional Aurora-B kinase in endomitosis. Blood 2004, 104, 1017–1024. [Google Scholar] [CrossRef]
- Wen, Q.J.; Yang, Q.; Goldenson, B.; Malinge, S.; Lasho, T.; Schneider, R.K.; Breyfogle, L.J.; Schultz, R.; Gilles, L.; Koppikar, P.; et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat. Med. 2015, 21, 1473–1480. [Google Scholar] [CrossRef]
- Gangat, N.; Marinaccio, C.; Swords, R.; Watts, J.M.; Gurbuxani, S.; Rademaker, A.; Fought, A.J.; Frankfurt, O.; Altman, J.K.; Wen, Q.J.; et al. Aurora Kinase A Inhibition Provides Clinical Benefit, Normalizes Megakaryocytes, and Reduces Bone Marrow Fibrosis in Patients with Myelofibrosis: A Phase I Trial. Clin. Cancer Res. 2019, 25, 4898–4906. [Google Scholar] [CrossRef] [PubMed]
- Bartalucci, N.; Guglielmelli, P.; Vannucchi, A.M. Rationale for targeting the PI3K/Akt/mTOR pathway in myeloproliferative neoplasms. Clin. Lymphoma Myeloma Leuk. 2013, 13 (Suppl. 2), S307–S309. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef]
- Grimwade, L.F.; Happerfield, L.; Tristram, C.; McIntosh, G.; Rees, M.; Bench, A.J.; Boyd, E.M.; Hall, M.; Quinn, A.; Piggott, N.; et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br. J. Haematol. 2009, 147, 495–506. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Bogani, C.; Bartalucci, N.; Martinelli, S.; Tozzi, L.; Guglielmelli, P.; Bosi, A.; Vannucchi, A.M.; on behalf of the Associazione Italiana per la Ricerca sul Cancro AGIMM Gruppo Italiano Malattie Mieloproliferative. mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS ONE 2013, 8, e54826. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Barosi, G.; Rambaldi, A.; Marchioli, R.; Masciulli, A.; Tozzi, L.; Biamonte, F.; Bartalucci, N.; Gattoni, E.; Lupo, M.L.; et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood 2011, 118, 2069–2076. [Google Scholar] [CrossRef]
- Durrant, S.T.; Nagler, A.; Guglielmelli, P.; Lavie, D.; le Coutre, P.; Gisslinger, H.; Chuah, C.; Maffioli, M.; Bharathy, S.; Dong, T.; et al. Results from HARMONY: An open-label, multicenter, 2-arm, phase 1b, dose-finding study assessing the safety and efficacy of the oral combination of ruxolitinib and buparlisib in patients with myelofibrosis. Haematologica 2019, 104, e551–e554. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3Kdelta inhibitors in cancer: Rationale and serendipity merge in the clinic. Cancer Discov. 2011, 1, 562–572. [Google Scholar] [CrossRef]
- Yacoub, A.; Wang, E.; Rampal, R.; Borate, U.; Kremyanskaya, M.; Ali, H.; Hobbs, G.; O’Connell, C.; Assad, A.; Erickson-Viitanen, S.; et al. Addition of parsaclisib, a pi3kdelta inhibitor, in patients (PTS) with suboptimal response to ruxolitinib (RUX): A Phase 2 study in pts with myelofibrosis (MF). In Proceedings of the EHA Annual Meeting, Virtual Conference, Held Online, 12 June 2020. Abstract S216. [Google Scholar]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef]
- Lasho, T.; Finke, C.; Kimlinger, T.K.; Zblewski, D.; Chen, D.; Patnaik, M.M.; Hanson, C.A.; Brooks, C.; Tefferi, A.; Pardanani, A. Expression of CD123 (IL-3R-alpha), a Therapeutic Target of SL-401, on Myeloproliferative Neoplasms. Blood 2014, 124, 5577. [Google Scholar] [CrossRef]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Gupta, V.; Ali, H.; Yacoub, A.; Wang, E.S.; Lee, S.; Schiller, G.J.; Sardone, M.; Wysowskyj, H.; Chen, J.; et al. Results from a Phase 1/2 Clinical Trial of Tagraxofusp (SL-401) in Patients with Intermediate, or High Risk, Relapsed/Refractory Myelofibrosis. Blood 2019, 134, 558. [Google Scholar] [CrossRef]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Salmoiraghi, S.; Risso, A.; Carobbio, A.; Buttiglieri, S.; Spatola, T.; Sivera, P.; Ricca, I.; Barbui, T.; Tarella, C.; et al. Telomere shortening in Ph-negative chronic myeloproliferative neoplasms: A biological marker of polycythemia vera and myelofibrosis, regardless of hydroxycarbamide therapy. Exp. Hematol. 2013, 41, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.S.; Gellert, G.C.; Hochreiter, A.; Pongracz, K.; Wright, W.E.; Zielinska, D.; Chin, A.C.; Harley, C.B.; Shay, J.W.; Gryaznov, S.M. Lipid modification of GRN163, an N3′-->P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 2005, 24, 5262–5268. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Harley, C.B.; Baerlocher, G.M. Imetelstat (GRN163L)--telomerase-based cancer therapy. Clin. Lymphoma Myeloma Leuk. 2010, 184, 221–234. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Komrokji, R.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.; Baer, M.; Hoffman, R.; Odenike, O.; et al. Favorable Overall Survival with Imetelstat Treatment Correlates with Other Clinical Benefits in Intermediate 2 or High Risk Myelofibrosis Relapsed/Refractory to Janus Kinase Inhibitor. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 6 December 2020. Abstract 53. [Google Scholar]
- Mascarenhas, J.; Komrokji, R.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.; Baer, M.; Hoffman, R.; Odenike, O.; et al. Correlation Analyses of Imetelstat Exposure with Pharmacodynamic Effect, Efficacy and Safety in a Phase 2 Study in Patients with Higher-Risk Myelofibrosis Refractory to Janus Kinase Inhibitor Identified an Optimal Dosing Regimen for Phase 3 Study. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 5 December 2020. Abstract 1283. [Google Scholar]
- Mascarenhas, J.; Komrokji, R.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.; Baer, M.; Hoffman, R.; Odenike, O.; et al. Telomerase Activity, Telomere Length and hTERT Expression Correlate with Clinical Outcomes in Higher-Risk Myelofibrosis (MF) Relapsed/Refractory (R/R) to Janus Kinase Inhibitor Treated with Imetelstat. In Proceedings of the ASH Annual Meeting, Virtual Conference, Held Online, 6 December 2020. Abstract 347. [Google Scholar]
- Masarova, L.; Alhuraiji, A.; Bose, P.; Daver, N.; Pemmaraju, N.; Cortes, J.; Pierce, S.; Kantarjian, H.; Verstovsek, S. Significance of thrombocytopenia in patients with primary and postessential thrombocythemia/polycythemia vera myelofibrosis. Eur. J. Haematol. 2018, 100, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Elena, C.; Passamonti, F.; Rumi, E.; Malcovati, L.; Arcaini, L.; Boveri, E.; Merli, M.; Pietra, D.; Pascutto, C.; Lazzarino, M. Red blood cell transfusion-dependency implies a poor survival in primary myelofibrosis irrespective of IPSS and DIPSS. Haematologica 2011, 96, 167–170. [Google Scholar] [CrossRef]
- Mesa, R.A.; Miller, C.B.; Thyne, M.; Mangan, J.; Goldberger, S.; Fazal, S.; Ma, X.; Wilson, W.; Paranagama, D.C.; Dubinski, D.G.; et al. Differences in treatment goals and perception of symptom burden between patients with myeloproliferative neoplasms (MPNs) and hematologists/oncologists in the United States: Findings from the MPN Landmark survey. Cancer 2017, 123, 449–458. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremblay, D.; Mascarenhas, J. Next Generation Therapeutics for the Treatment of Myelofibrosis. Cells 2021, 10, 1034. https://doi.org/10.3390/cells10051034
Tremblay D, Mascarenhas J. Next Generation Therapeutics for the Treatment of Myelofibrosis. Cells. 2021; 10(5):1034. https://doi.org/10.3390/cells10051034
Chicago/Turabian StyleTremblay, Douglas, and John Mascarenhas. 2021. "Next Generation Therapeutics for the Treatment of Myelofibrosis" Cells 10, no. 5: 1034. https://doi.org/10.3390/cells10051034
APA StyleTremblay, D., & Mascarenhas, J. (2021). Next Generation Therapeutics for the Treatment of Myelofibrosis. Cells, 10(5), 1034. https://doi.org/10.3390/cells10051034