
Coordinated and Independent Roles for MLH Subunits in DNA Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Coordinated Roles for the MLH Proteins in DNA Metabolism
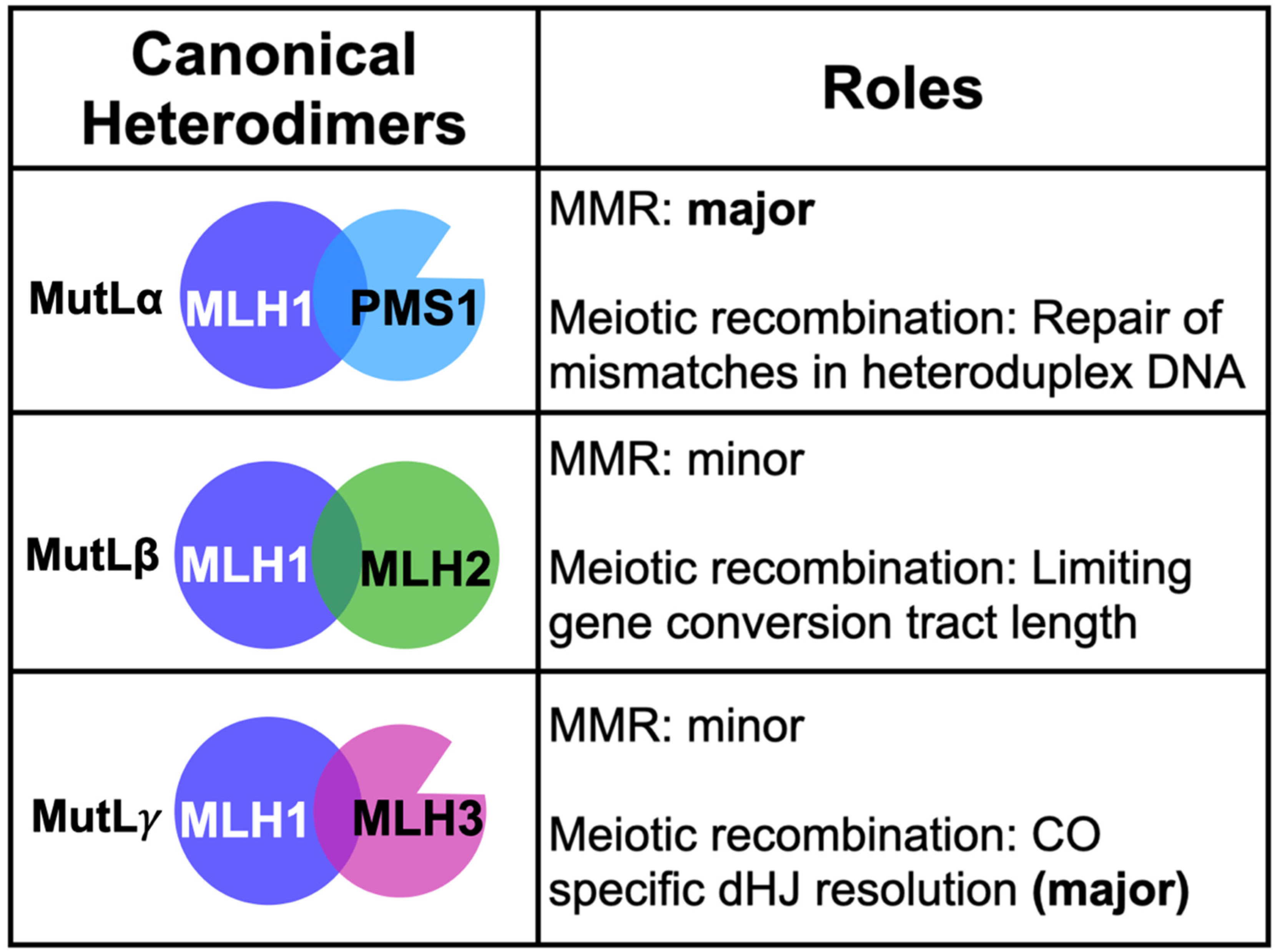
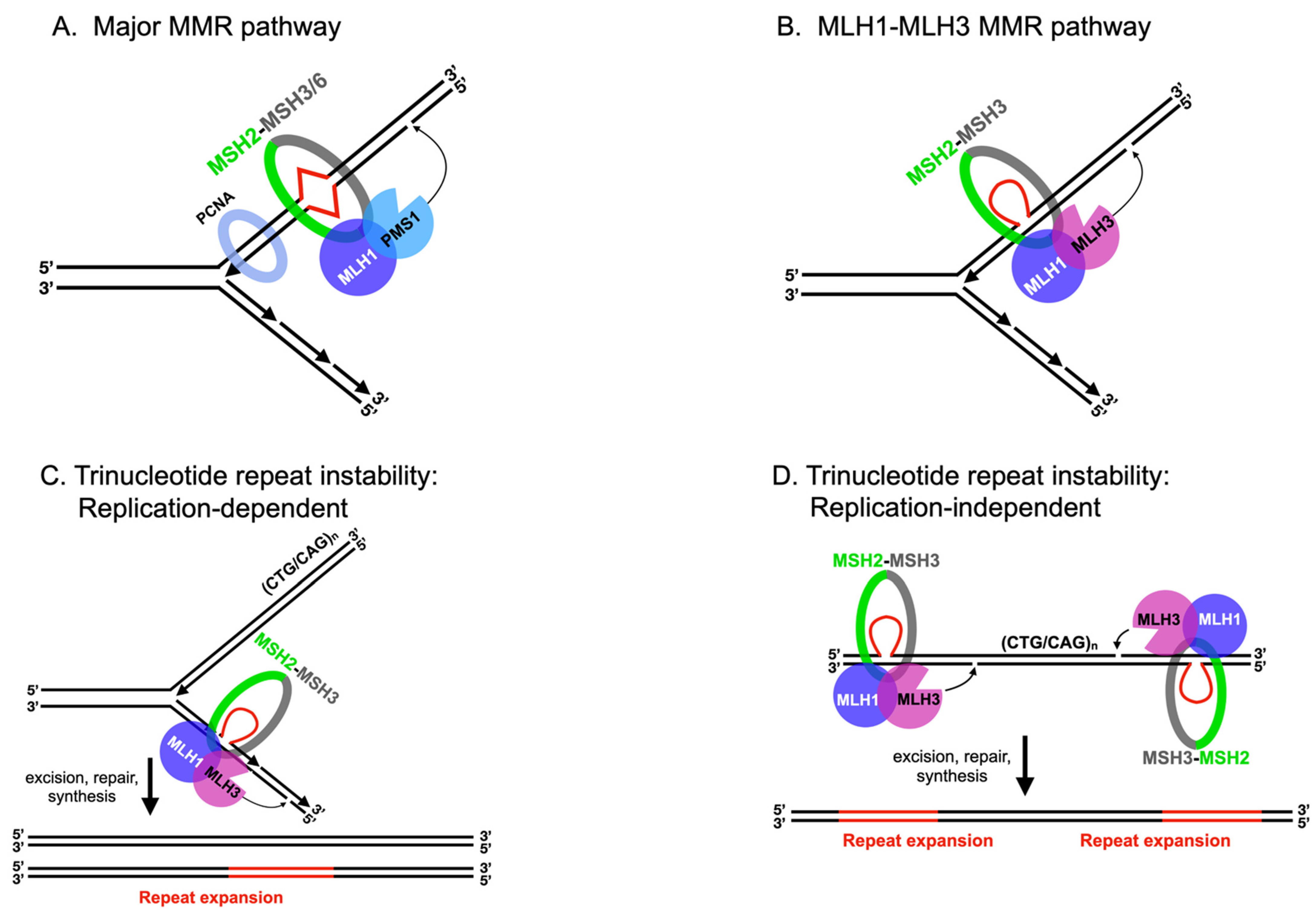
2.1. MLH Family Proteins Function Together in Post-Replicative DNA-Mismatch Repair
2.2. MLH1-PMS1 and MLH1-MLH3 Promote Trinucleotide Repeat Expansion in Mammals
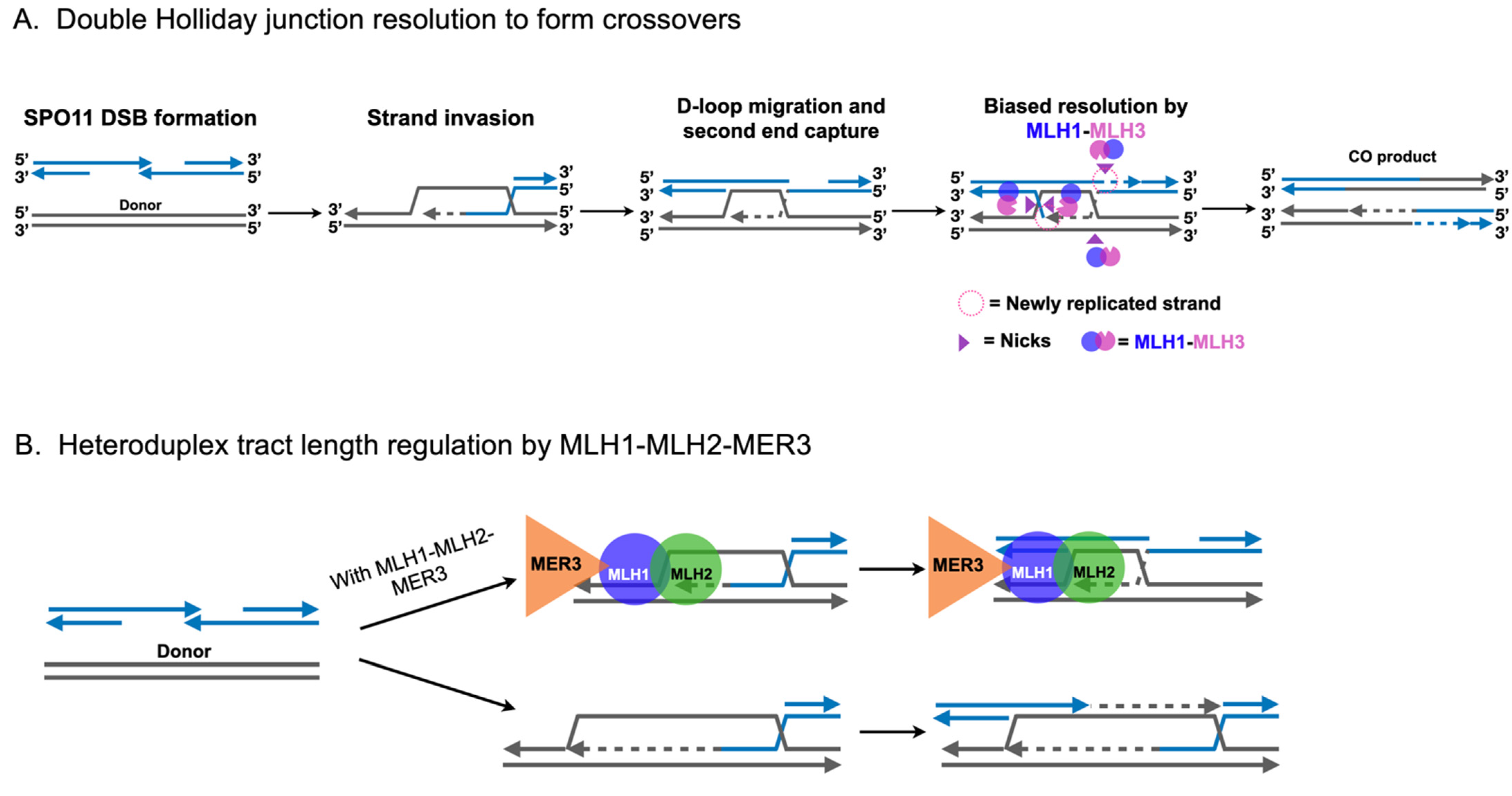
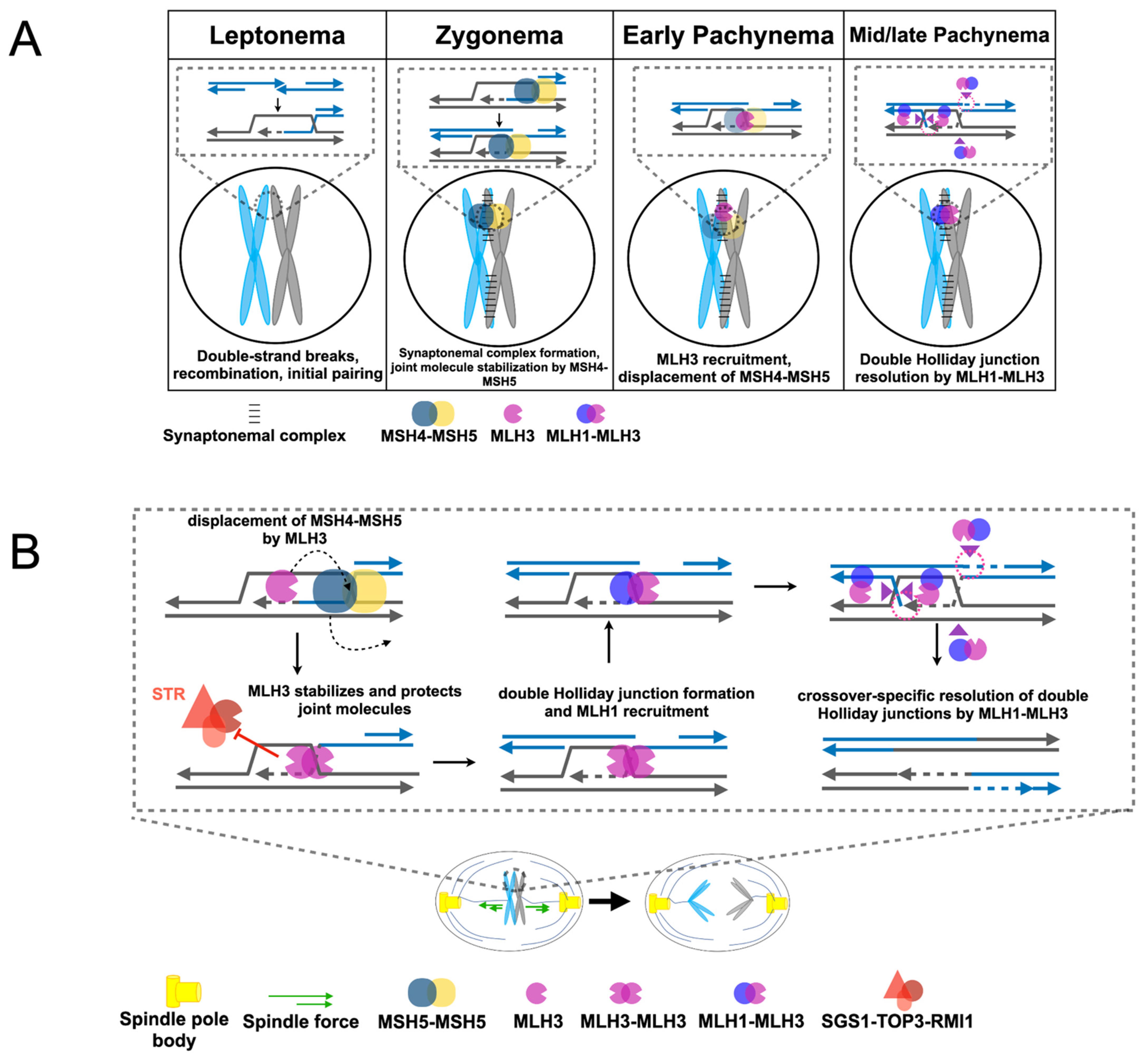
2.3. Roles for MLH1-MLH3 and MLH1-MLH2 in Meiotic Recombination
3. Independent Roles for MLH Proteins in Homologous Recombination and Meiosis
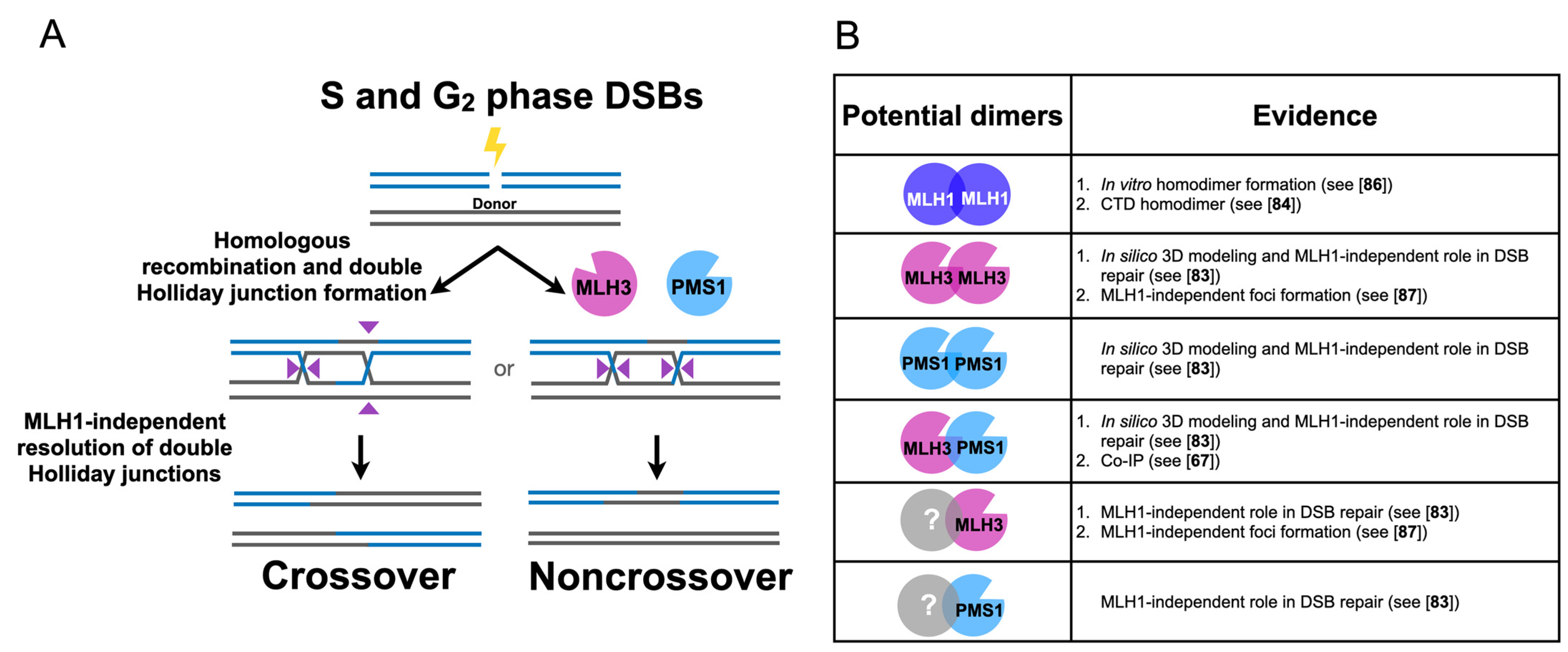
3.1. Human MLH3 and PMS2 (Homolog of Baker’s Yeast PMS1) Function Separately from MLH1 in Somatic Homologous Recombination
3.2. Differential Timing and Location of Mouse MLH1 and MLH3 Foci Formation in Meiotic Recombination
3.3. Differential Crossing Over and Spore Viability Phenotypes of MSH and MLH Mutants in Yeast Suggest CO-Independent Mechanisms of Chromosome Segregation
3.4. Contrasting Meiotic Phenotypes for MLH1 and MLH3 in Yeast Hint at an Alternative Role for MLH3 in Meiosis
4. MLH3 may Promote Meiotic Homolog Disjunction through a Crossover-Independent Mechanism
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef]
- Hunter, N.; Borts, R.H. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev. 1997, 11, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.F.F.; Hoffmann, E.R.; Cotton, V.E.; Borts, R.H. A role for the MutL homologue MLH2 in controlling heteroduplex formation and in regulating between two different crossover pathways in budding yeast. Cytogenet. Genome Res. 2004, 107, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Duroc, Y.; Kumar, R.; Ranjha, L.; Adam, C.; Guérois, R.; Muntaz, K.M.; Marsolier-Kergoat, M.C.; Dingli, F.; Laureau, R.; Loew, D.; et al. Concerted action of the MutLβ heterodimer and Mer3 helicase regulates the global extent of meiotic gene conversion. Elife 2017, 6, e21900. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.; Shields, D. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef]
- Vakirlis, N.; Sarilar, V.; Drillon, G.; Fleiss, A.; Agier, N.; Meyniel, J.P.; Blanpain, L.; Carbone, A.; Devillers, H.; Dubois, K.; et al. Reconstruction of ancestral chromosome architecture and gene repertoire reveals principles of genome evolution in a model yeast genus. Genome Res. 2016, 26, 918–932. [Google Scholar] [CrossRef]
- Campbell, C.S.; Hombauer, H.; Srivatsan, A.; Bowen, N.; Gries, K.; Desai, A.; Putnam, C.D.; Kolodner, R.D. Mlh2 Is an Accessory Factor for DNA Mismatch Repair in Saccharomyces cerevisiae. PLoS Genet. 2014, 10, e1004327. [Google Scholar] [CrossRef]
- Furman, C.M.; Elbashir, R.; Pannafino, G.P.; Clark, N.L.; Alani, E. Experimental exchange of paralogous domains in the MLH family provides evidence of sub-functionalization after gene duplication. G3. in press. [CrossRef]
- Liu, J.; Hanne, J.; Britton, B.M.; Bennett, J.; Kim, D.; Lee, J.B.; Fishel, R. Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 2016, 539, 583–587. [Google Scholar] [CrossRef]
- Burdett, V.; Baitinger, C.; Viswanathan, M.; Lovett, S.T.; Modrich, P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc. Natl. Acad. Sci. USA 2001, 98, 6765–6770. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in E. coli and Human Mismatch Repair (Nobel Lecture). Angew. Chemie Int. Ed. 2016, 55, 8490–8501. [Google Scholar] [CrossRef]
- Gradia, S.; Acharya, S.; Fishel, R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell 1997, 91, 995–1005. [Google Scholar] [CrossRef]
- Acharya, S.; Foster, P.L.; Brooks, P.; Fishel, R. The Coordinated Functions of the E. coli MutS and MutL Proteins in Mismatch Repair of hereditary nonpolyposis colorectal cancer (HNPCC) families. Mol. Cell 2003, 12, 233–246. [Google Scholar] [CrossRef]
- Jensen, L.E.; Jauert, P.A.; Kirkpatrick, D.T. The large loop repair and mismatch repair pathways of Saccharomyces cerevisiae act on distinct substrates during meiosis. Genetics 2005, 170, 1033–1043. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLα in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Kawasoe, Y.; Tsurimoto, T.; Nakagawa, T.; Masukata, H.; Takahashi, T.S. MutSα maintains the mismatch repair capability by inhibiting PCNA unloading. Elife 2016, 5, e15155. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D. A personal historical view of DNA mismatch repair with an emphasis on eukaryotic DNA mismatch repair. DNA Repair 2016, 38, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Harfe, B.D.; Minesinger, B.K.; Jinks-Robertson, S. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 2000, 10, 145–148. [Google Scholar] [CrossRef]
- Flores-Rozas, H.; Kolodner, R.D. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA 1998, 95, 12404–12409. [Google Scholar] [CrossRef]
- Romanova, N.V.; Crouse, G.F. Different roles of eukaryotic MutS and MutL complexes in repair of small insertion and deletion loops in yeast. PLoS Genet. 2013, 9, e1003920. [Google Scholar] [CrossRef]
- Rogacheva, M.V.; Manhart, C.M.; Chen, G.; Guarne, A.; Surtees, J.; Alani, E. Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J. Biol. Chem. 2014, 289, 5664–5673. [Google Scholar] [CrossRef]
- Ban, C.; Yang, W. Crystal structure and ATPase activity of MutL: Implications for DNA repair and mutagenesis. Cell 1998, 95, 541–552. [Google Scholar] [CrossRef]
- Sacho, E.J.; Kadyrov, F.A.; Modrich, P.; Kunkel, T.A.; Erie, D.A. Direct visualization of asymmetric adenine nucleotide-induced conformational changes in MutLα. Mol. Cell 2008, 29, 112–121. [Google Scholar] [CrossRef]
- Hall, M.C.; Shcherbakova, P.V.; Kunkel, T.A. Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and Pms1 proteins. J. Biol. Chem. 2002, 277, 3673–3679. [Google Scholar] [CrossRef]
- Tran, P.T.; Liskay, R.M. Functional Studies on the Candidate ATPase Domains of Saccharomyces cerevisiae MutLα. Mol. Cell. Biol. 2000, 20, 6390–6398. [Google Scholar] [CrossRef]
- Claeys Bouuaert, C.; Keeney, S. Distinct DNA-binding surfaces in the ATPase and linker domains of MutLγ determine its substrate specificities and exert separable functions in meiotic recombination and mismatch repair. PLoS Genet. 2017, 13, e1006722. [Google Scholar] [CrossRef]
- Reyes, G.X.; Zhao, B.; Schmidt, T.T.; Gries, K.; Kloor, M.; Hombauer, H. Identification of MLH2/hPMS1 dominant mutations that prevent DNA mismatch repair function. Commun. Biol. 2020, 3, 751. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Holmes, S.F.; Arana, M.E.; Lukianova, O.A.; O’Donnell, M.; Kunkel, T.A.; Modrich, P. Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. J. Biol. Chem. 2007, 282, 37181–37190. [Google Scholar] [CrossRef]
- Ranjha, L.; Anand, R.; Cejka, P. The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to holliday junctions. J. Biol. Chem. 2014, 289, 5674–5686. [Google Scholar] [CrossRef]
- Manhart, C.M.; Ni, X.; White, M.A.; Ortega, J.; Surtees, J.A.; Alani, E. The mismatch repair and meiotic recombination endonuclease Mlh1-Mlh3 is activated by polymer formation and can cleave DNA substrates in trans. PLoS Biol. 2017, 15, e2001164. [Google Scholar] [CrossRef]
- Lenhart, J.S.; Pillon, M.C.; Guarné, A.; Biteen, J.S.; Simmons, L.A. Mismatch repair in Gram-positive bacteria. Res. Microbiol. 2016, 167, 4–12. [Google Scholar] [CrossRef]
- Genschel, J.; Kadyrova, L.Y.; Iyer, R.R.; Dahal, B.K.; Kadyrov, F.A.; Modrich, P. Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLα activation and function in mismatch repair. Proc. Natl. Acad. Sci. USA 2017, 114, 4930–4935. [Google Scholar] [CrossRef]
- Schmidt, M.H.M.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef]
- Pinto, R.M.; Dragileva, E.; Kirby, A.; Lloret, A.; Lopez, E.; Claire, J.S.; Panigrahi, G.B.; Hou, C.; Holloway, K.; Gillis, T.; et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. 2013, 9, e1003930. [Google Scholar] [CrossRef]
- Lee, J.M.; Chao, M.J.; Harold, D.; Elneel, K.A.; Gillis, T.; Holmans, P.; Jones, L.; Orth, M.; Myers, R.H.; Kwak, S.; et al. A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet. 2017, 26, 3859–3867. [Google Scholar] [CrossRef]
- Halabi, A.; Fuselier, K.T.B.; Grabczyk, E. GAA•TTC repeat expansion in human cells is mediated by mismatch repair complex MutL and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res. 2018, 46, 4022–4032. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Gujar, V.; Burdett, V.; Modrich, P.L.; Kadyrov, F.A. Human MutLγ, the MLH1–MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Pereira, M.; Fortune, M.T.; Ingram, L.; McAbney, J.P.; Monckton, D.G. Pms2 is a genetic enhancer of trinucleotide CAG-CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet. 2004, 13, 1815–1825. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wilkins, K.; Edelmann, W.; Usdin, K. MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 2018, 14, e1007719. [Google Scholar] [CrossRef]
- Lee, J.M.; Correia, K.; Loupe, J.; Kim, K.H.; Barker, D.; Hong, E.P.; Chao, M.J.; Long, J.D.; Lucente, D.; Vonsattel, J.P.G.; et al. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef]
- Ciosi, M.; Maxwell, A.; Cumming, S.A.; Moss, D.J.H.; Alshammari, A.M.; Flower, M.D.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Holmans, P.; et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 2019, 48, 568–580. [Google Scholar] [CrossRef]
- Lee, J.M.; Wheeler, V.C.; Chao, M.J.; Vonsattel, J.P.G.; Pinto, R.M.; Lucente, D.; Abu-Elneel, K.; Ramos, E.M.; Mysore, J.S.; Gillis, T.; et al. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef]
- Moss, D.J.H.; Tabrizi, S.J.; Mead, S.; Lo, K.; Pardiñas, A.F.; Holmans, P.; Jones, L.; Langbehn, D.; Coleman, A.; Santos, R.D.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Su, X.A.; Freudenreich, C.H. Cytosine deamination and base excision repair cause R-loop–induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E8392–E8401. [Google Scholar] [CrossRef]
- Pluciennik, A.; Burdett, V.; Baitinger, C.; Iyer, R.R.; Shi, K.; Modrich, P. Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLa endonuclease activation. Proc. Natl. Acad. Sci. USA 2013, 110, 12277–12282. [Google Scholar] [CrossRef]
- Kantartzis, A.; Williams, G.A.; Balakrishnan, L.; Roberts, R.L.; Surtees, J.A.; Bambara, R.A. Msh2-Msh3 interferes with Okazaki fragment processing to promote trinucleotide repeat expansions. Cell Rep. 2012, 2, 216–222. [Google Scholar] [CrossRef]
- Marston, A.L.; Amon, A. Meiosis: Cell-cycle controls shuffle and deal. Nat. Rev. Mol. Cell Biol. 2004, 5, 983–997. [Google Scholar] [CrossRef]
- Lamb, N.E.; Sherman, S.L.; Hassold, T.J. Effect of meiotic recombination on the production of aneuploid gametes in humans. Cytogenet. Genome Res. 2005, 111, 250–255. [Google Scholar] [CrossRef]
- Hunter, N. Meiotic recombination: The essence of heredity. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–35. [Google Scholar] [CrossRef]
- Zickler, D.; Kleckner, N. Recombination, pairing, and synapsis of homologs during meiosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a016626. [Google Scholar] [CrossRef]
- Soler, A.; Morales, C.; Mademont-Soler, I.; Margarit, E.; Borrell, A.; Borobio, V.; Munõz, M.; Sánchez, A. Overview of chromosome abnormalities in first trimester miscarriages: A series of 1011 consecutive chorionic Villi sample karyotypes. Cytogenet. Genome Res. 2017, 152, 81–89. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef]
- Cao, L.; Alani, E.; Kleckner, N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 1990, 61, 1089–1101. [Google Scholar] [CrossRef]
- Robine, N.; Uematsu, N.; Amiot, F.; Gidrol, X.; Barillot, E.; Nicolas, A.; Borde, V. Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, L.A.; Stahl, F.A. A test of the double-strand break repair model for meiotic recombination in Saccharomyces cerevisiae. Genetics 1996, 144, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Tsubouchi, T.; Rockmill, B.; Sandler, J.S.; Richards, D.R.; Vader, G.; Hochwagen, A.; Roeder, G.S.; Fung, J.C. Global analysis of the meiotic crossover landscape. Dev. Cell 2008, 15, 401–415. [Google Scholar] [CrossRef]
- Allers, T.; Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 2001, 106, 47–57. [Google Scholar] [CrossRef]
- Hunter, N.; Kleckner, N. The single-end invasion: An asymmetric intermediate at the double-strand break to double-Holliday Junction transition of meiotic recombination. Cell 2004, 106, 59–70. [Google Scholar] [CrossRef]
- Furman, C.M.; Elbashir, R.; Alani, E. Expanded roles for the MutL family of DNA mismatch repair proteins. Yeast 2020, 38, 39–53. [Google Scholar] [CrossRef]
- Pedrazzi, G.; Perrera, C.; Blaser, H.; Kuster, P.; Marra, G.; Davies, S.L.; Ryu, G.H.; Freire, R.; Hickson, I.D.; Jiricny, J.; et al. Direct association of Bloom’s syndrome gene product with the human mismatch repair protein MLH1. Nucleic Acids Res. 2001, 29, 4378–4386. [Google Scholar] [CrossRef]
- Langland, G.; Kordich, J.; Creaney, J.; Heppner Goss, K.; Lillard-Wetherell, K.; Bebenek, K.; Kunkel, T.A.; Groden, J. The BLM helicase interacts with hMLH1 but is not required for DNA mismatch repair. J. Biol. Chem. 2001, 276, 30031–30035. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.F.; Kung, W.M. Supercomplex formation between Mlh1-Mlh3 and Sgs1-Top3 heterocomplexes in meiotic yeast cells. Biochem. Biophys. Res. Commun. 2002, 296, 949–953. [Google Scholar] [CrossRef]
- Pyatnitskaya, A.; Borde, V.; De Muyt, A. Crossing and zipping: Molecular duties of the ZMM proteins in meiosis. Chromosoma 2019, 128, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Zakharyevich, K.; Tang, S.; Ma, Y.; Hunter, N. Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 2012, 149, 334–347. [Google Scholar] [CrossRef]
- Cannavo, E.; Sanchez, A.; Anand, R.; Ranjha, L.; Hugener, J.; Adam, C.; Acharya, A.; Weyland, N.; Aran-Guiu, X.; Charbonnier, J.B.; et al. Regulation of the MLH1–MLH3 endonuclease in meiosis. Nature 2020, 586, 618–622. [Google Scholar] [CrossRef]
- Kulkarni, D.S.; Owens, S.N.; Honda, M.; Ito, M.; Yang, Y.; Corrigan, M.W.; Chen, L.; Quan, A.L.; Hunter, N. PCNA activates the MutLγ endonuclease to promote meiotic crossing over. Nature 2020, 586, 623–627. [Google Scholar] [CrossRef]
- Sanchez, A.; Adam, C.; Rauh, F.; Duroc, Y.; Ranjha, L.; Lombard, B.; Mu, X.; Wintrebert, M.; Loew, D.; Guarné, A.; et al. Exo1 recruits Cdc5 polo kinase to MutLγ to ensure efficient meiotic crossover formation. Proc. Natl. Acad. Sci. USA 2020, 117, 30577–30588. [Google Scholar] [CrossRef]
- Matos, J.; Blanco, M.G.; Maslen, S.; Skehel, J.M.; West, S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011, 147, 158–172. [Google Scholar] [CrossRef]
- De los Santos, T.; Hunter, N.; Lee, C.; Larkin, B.; Loidl, J.; Hollingsworth, N.M. The MUS81/MMS4 endonuclease acts independently of double-holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 2003, 164, 81–94. [Google Scholar]
- Holloway, J.K.; Booth, J.; Edelmann, W.; McGowan, C.H.; Cohen, P.E. MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet. 2008, 4, e1000186. [Google Scholar] [CrossRef]
- De Muyt, A.; Jessop, L.; Kolar, E.; Sourirajan, A.; Chen, J.; Davani, Y.; Lichten, M. BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Mol. Cell 2012, 46, 43–53. [Google Scholar] [CrossRef]
- Lipkin, S.M.; Moens, P.B.; Wang, V.; Lenzi, M.; Shanmugarajah, D.; Gilgeous, A.; Thomas, J.; Cheng, J.; Touchman, J.W.; Green, E.D.; et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat. Genet. 2002, 31, 385–390. [Google Scholar] [CrossRef]
- Toledo, M.; Sun, X.; Brieño-Enríquez, M.A.; Raghavan, V.; Gray, S.; Pea, J.; Milano, C.R.; Venkatesh, A.; Patel, L.; Borst, P.L.; et al. A mutation in the endonuclease domain of mouse MLH3 reveals novel roles for mutlγ during crossover formation in meiotic prophase I. PLoS Genet. 2019, 15, e1008177. [Google Scholar] [CrossRef]
- Edelmann, W.; Cohen, P.E.; Kane, M.; Lau, K.; Morrow, B.; Bennett, S.; Umar, A.; Kunkel, T.; Cattoretti, G.; Chaganti, R.; et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell 1996, 85, 1125–1134. [Google Scholar] [CrossRef]
- Wang, T.F.; Kleckner, N.; Hunter, N. Functional specificity of MutL homologs in yeast: Evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc. Natl. Acad. Sci. USA 1999, 96, 13914–13919. [Google Scholar] [CrossRef] [PubMed]
- Argueso, J.L.; Kijas, A.W.; Sarin, S.; Heck, J.; Waase, M.; Alani, E. Systematic Mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol. Cell. Biol. 2003, 23, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Argueso, J.L.; Wanat, J.; Gemici, Z.; Alani, E. Competing crossover pathways act during meiosis in Saccharomyces cerevisiae. Genetics 2004, 168, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Nishant, K.T.; Plys, A.J.; Alani, E. A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 2008, 179, 747–755. [Google Scholar] [CrossRef]
- Schwartz, E.K.; Heyer, W.D. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma 2011, 120, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Susperregui, A.; Piazza, I.; Dörig, C.; Oke, A.; Arter, M.; Yamaguchi, M.; Hilditch, A.T.; Vuina, K.; Chan, K.C.; et al. Network rewiring of homologous recombination enzymes during mitotic proliferation and meiosis. Mol. Cell 2019, 75, 859–874.e4. [Google Scholar] [CrossRef] [PubMed]
- Marsolier-Kergoat, M.C.; Khan, M.M.; Schott, J.; Zhu, X.; Llorente, B. Mechanistic view and genetic control of DNA recombination during meiosis. Mol. Cell 2018, 70, 9–20.e6. [Google Scholar] [CrossRef]
- Peterson, S.E.; Keeney, S.; Jasin, M. Mechanistic Insight into Crossing over during Mouse Meiosis. Mol. Cell 2020, 78, 1252–1263.e3. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Mohiuddin, M.; Keka, I.S.; Yamada, K.; Tsuda, M.; Sasanuma, H.; Andreani, J.; Guerois, R.; Borde, V.; Charbonnier, J.B.; et al. Genetic evidence for the involvement of mismatch repair proteins, PMS2 and MLH3, in a late step of homologous recombination. J. Biol. Chem. 2020, 295, 17460–17475. [Google Scholar] [CrossRef] [PubMed]
- Gueneau, E.; Dherin, C.; Legrand, P.; Tellier-Lebegue, C.; Gilquin, B.; Bonnesoeur, P.; Londino, F.; Quemener, C.; Le Du, M.H.; Márquez, J.A.; et al. Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat. Struct. Mol. Biol. 2013, 20, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Al-Sweel, N.; Raghavan, V.; Dutta, A.; Ajith, V.P.; Di Vietro, L.; Khondakar, N.; Manhart, C.M.; Surtees, J.A.; Nishant, K.T.; Alani, E. mlh3 mutations in baker’s yeast alter meiotic recombination outcomes by increasing noncrossover events genome-wide. PLoS Genet. 2017, 13, e1006974. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, P.V.; Hall, M.C.; Lewis, M.S.; Bennett, S.E.; Martin, K.J.; Bushel, P.R.; Afshari, C.A.; Kunkel, T.A. Inactivation of DNA Mismatch Repair by Increased Expression of Yeast MLH1. Mol. Cell. Biol. 2001, 21, 940–951. [Google Scholar] [CrossRef]
- Kolas, N.K.; Svetlanov, A.; Lenzi, M.L.; Macaluso, F.P.; Lipkin, S.M.; Liskay, R.M.; Greally, J.; Edelmann, W.; Cohen, P.E. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J. Cell Biol. 2005, 171, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Nishant, K.T.; Chen, C.; Shinohara, M.; Shinohara, A.; Alani, E. Genetic analysis of baker’s yeast Msh4-Msh5 reveals a threshold crossover level for meiotic viability. PLoS Genet. 2010, 6, e1001083. [Google Scholar] [CrossRef]
- Brown, M.S.; Lim, E.; Chen, C.; Nishant, K.T.; Alani, E. Genetic analysis of mlh3 mutations reveals interactions between crossover promoting factors during meiosis in baker’s yeast. G3 2013, 3, 9–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zakharyevich, K.; Ma, Y.; Tang, S.; Hwang, P.Y.; Boiteux, S.; Hunter, N. Temporally and biochemically distinct activities of Exo1 during meiosis: Double-strand-break resection and resolution of double-Holliday Junctions. Mol. Cell 2010, 40, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Kaback, D.B.; Steensma, H.Y.; de Jonge, P. Enhanced meiotic recombination on the smallest chromosome of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1989, 86, 3694–3698. [Google Scholar] [CrossRef]
- Kaback, D.B.; Barber, D.; Mahon, J.; Lamb, J.; You, J. Chromosome size-dependent control of meiotic reciprocal recombination in Saccharomyces cerevisiae: The role of crossover interference. Genetics 1999, 152, 1475–1486. [Google Scholar] [CrossRef]
- Gerton, J.L.; DeRisi, J.; Shroff, R.; Lichten, M.; Brown, P.O.; Petes, T.D. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 11383–11390. [Google Scholar] [CrossRef]
- Murakami, H.; Lam, I.; Huang, P.C.; Song, J.; van Overbeek, M.; Keeney, S. Multilayered mechanisms ensure that short chromosomes recombine in meiosis. Nature 2020, 582, 124–128. [Google Scholar] [CrossRef]
- Storlazzi, A.; Gargano, S.; Ruprich-Robert, G.; Falque, M.; David, M.; Kleckner, N.; Zickler, D. Recombination proteins mediate meiotic spatial chromosome organization and pairing. Cell 2010, 141, 94–106. [Google Scholar] [CrossRef]
- Santucci-Darmanin, S.; Walpita, D.; Lespinasse, F.; Desnuelle, C.; Ashley, T.; Paquis-Flucklinger, V. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 2000, 14, 1539–1547. [Google Scholar] [CrossRef]
- Hall, M.C.; Wang, H.; Erie, D.A.; Kunkel, T.A. High affinity cooperative DNA binding by the yeast Mlh1-Pms1 heterodimer. J. Mol. Biol. 2001, 312, 637–647. [Google Scholar] [CrossRef]
- Surtees, J.A.; Alani, E. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J. Mol. Biol. 2006, 360, 523–536. [Google Scholar] [CrossRef]
- Holloway, J.K.; Morelli, M.A.; Borst, P.L.; Cohen, P.E. Mammalian BLM helicase is critical for integrating multiple pathways of meiotic recombination. J. Cell Biol. 2010, 188, 779–789. [Google Scholar] [CrossRef]
- Arter, M.; Hurtado-Nieves, V.; Oke, A.; Zhuge, T.; Wettstein, R.; Fung, J.C.; Blanco, M.G.; Matos, J. Regulated crossing-over requires inactivation of Yen1/GEN1 resolvase during meiotic prophase I. Dev. Cell 2018, 45, 785–800.e6. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pannafino, G.; Alani, E. Coordinated and Independent Roles for MLH Subunits in DNA Repair. Cells 2021, 10, 948. https://doi.org/10.3390/cells10040948
Pannafino G, Alani E. Coordinated and Independent Roles for MLH Subunits in DNA Repair. Cells. 2021; 10(4):948. https://doi.org/10.3390/cells10040948
Chicago/Turabian StylePannafino, Gianno, and Eric Alani. 2021. "Coordinated and Independent Roles for MLH Subunits in DNA Repair" Cells 10, no. 4: 948. https://doi.org/10.3390/cells10040948
APA StylePannafino, G., & Alani, E. (2021). Coordinated and Independent Roles for MLH Subunits in DNA Repair. Cells, 10(4), 948. https://doi.org/10.3390/cells10040948