
The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Genome-Wide Replication Timing Analysis
2.3. DNA Combing
2.4. Immunofluorescence Staining
2.5. Visualization of Replication Sites in the Cell Nucleus
2.6. Immuno-DNA FISH
2.7. Microarray Analysis
2.8. Methylation Analysis by Southern Blotting
2.9. Imaging System and Measurements
3. Results
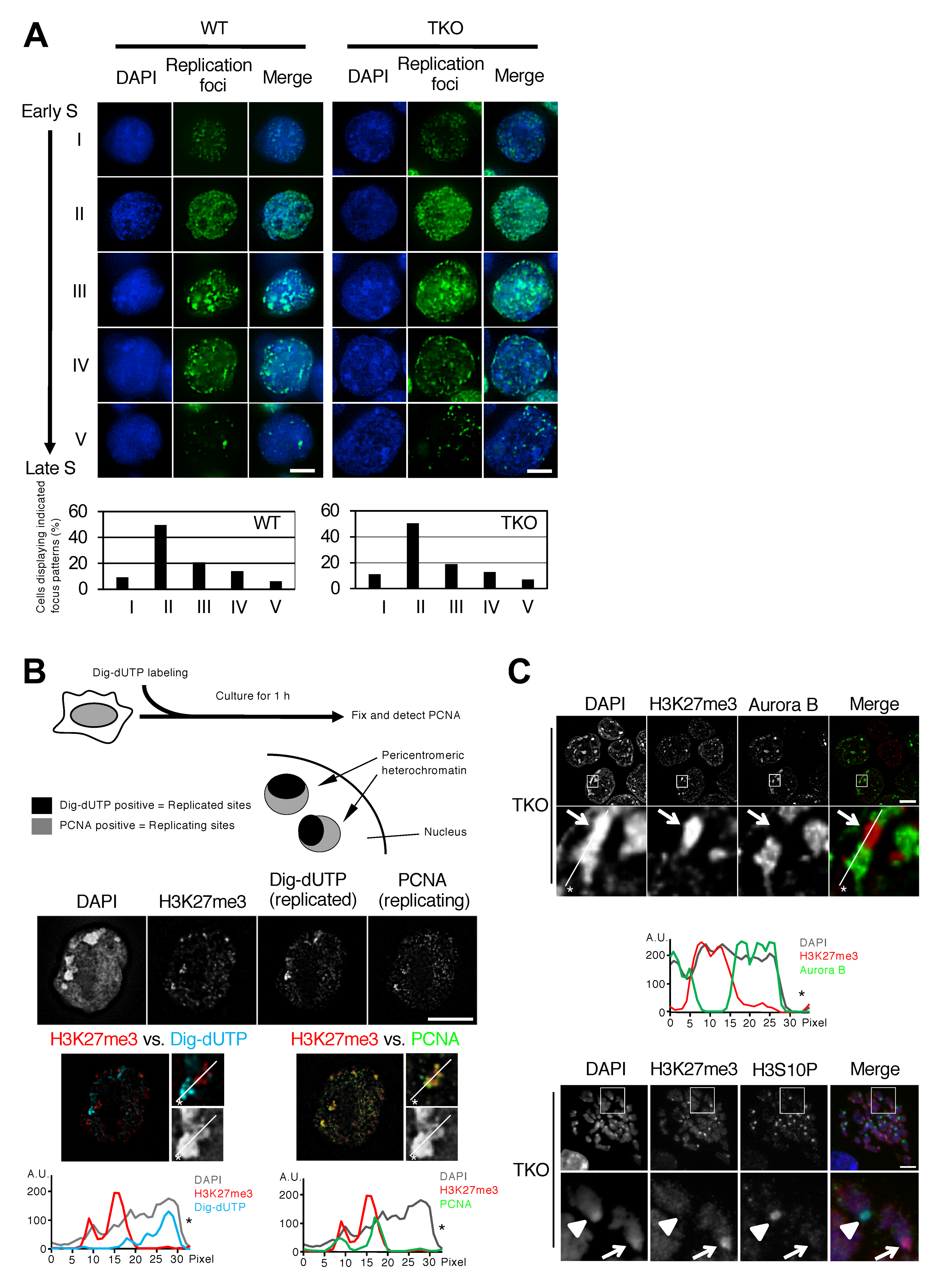
3.1. H3K27me3 Foci Formed in Mouse Embryonic Stem Cells with Severely Hypomethylated DNA Coincide with Later Replication of Pericentromeric Heterochromatin
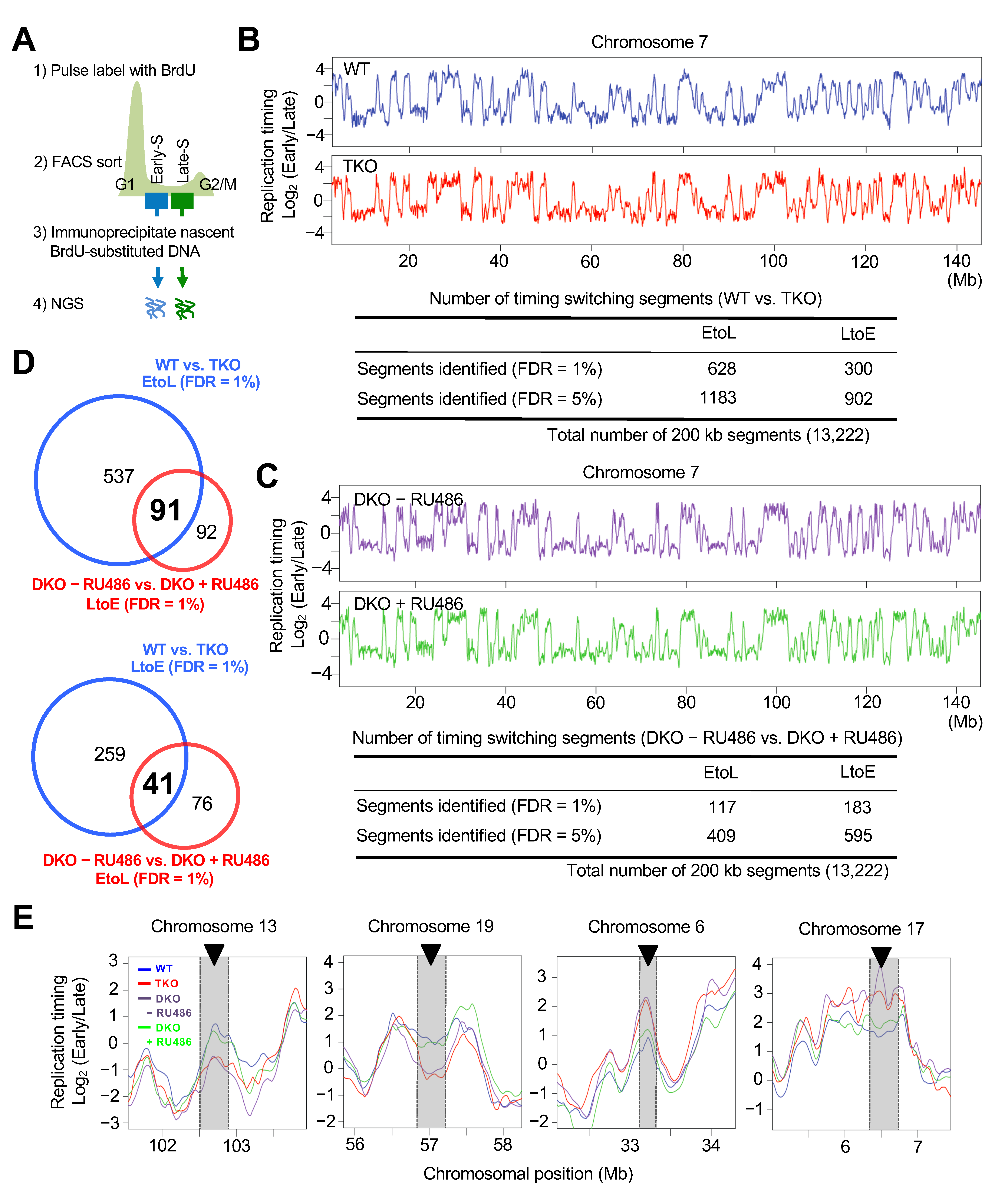
3.2. Genome-Wide Replication Timing Analysis Identified a Subset of Chromosomal Regions Sensitive to Dnmt Loss
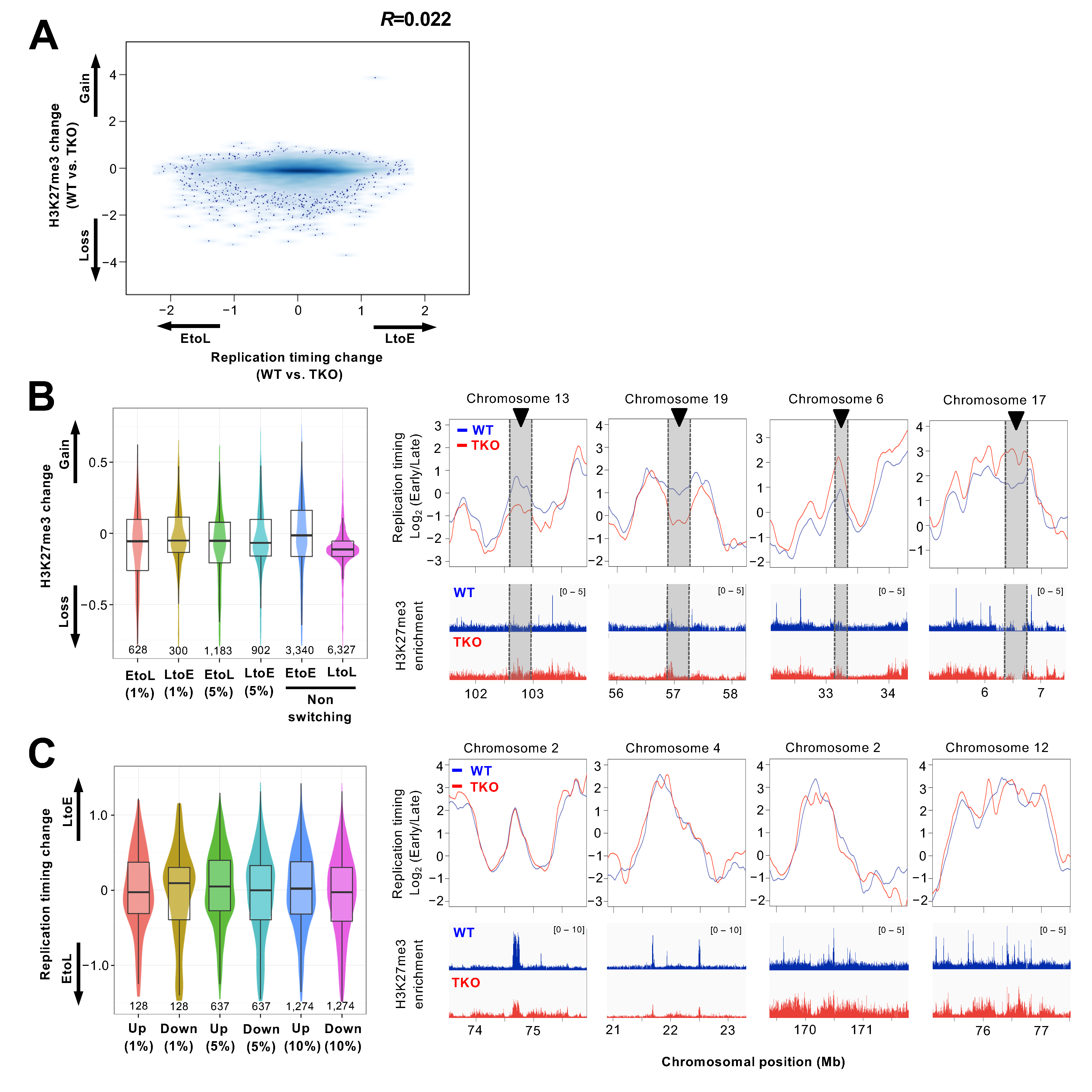
3.3. Redistribution of the H3K27me3 Mark Is not Associated with Replication Timing Changes in the Chromosomal Arm Regions
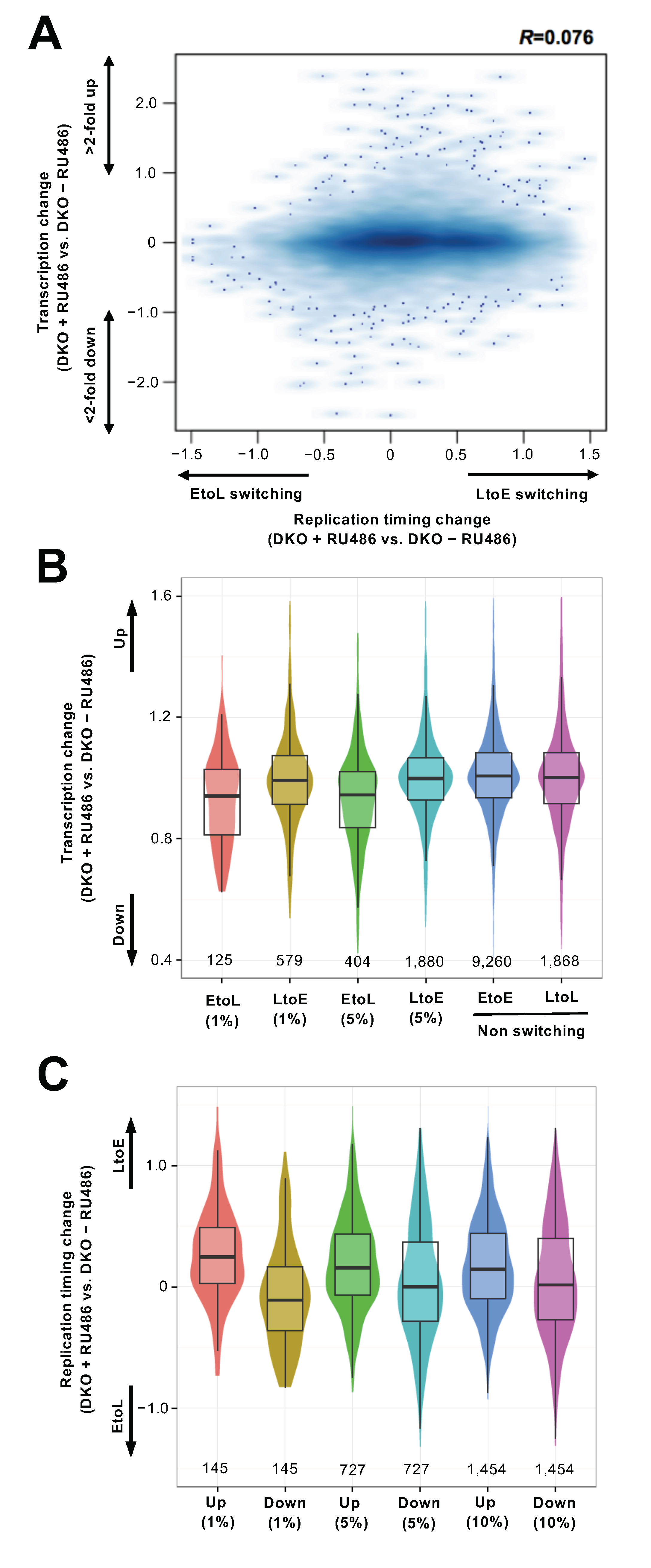
3.4. Replication Timing Changes Frequently Coincide with Transcriptional Changes in the Chromosomal Arm Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef]
- Pope, B.D.; Gilbert, D.M. The replication domain model: Regulating replicon firing in the context of large-scale chromosome architecture. J. Mol. Biol. 2013, 425, 4690–4695. [Google Scholar] [CrossRef]
- Takebayashi, S.-I.; Ogata, M.; Okumura, K. Anatomy of mammalian replication domains. Genes 2017, 8, 110. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Rathjen, J.; Kulik, M.; Papp, B.; Fussner, E.; Bazett-Jones, D.P.; Plath, K.; Dalton, S.; et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2009, 20, 155–169. [Google Scholar] [CrossRef]
- Ryba, T.; Battaglia, D.; Chang, B.H.; Shirley, J.W.; Buckley, Q.; Pope, B.D.; Devidas, M.; Druker, B.J.; Gilbert, D.M. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012, 22, 1833–1844. [Google Scholar] [CrossRef]
- Dileep, V.; Rivera-Mulia, J.C.; Sima, J.; Gilbert, D.M. Large-scale chromatin structure–function relationships during the cell cycle and development: Insights from replication timing. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, H.; Page, A.W.; Weier, H.-U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef]
- Bachman, K.E.; Rountree, M.R.; Baylin, S.B. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 2001, 276, 32282–32287. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Jackson-Grusby, L.; Beard, C.; Possemato, R.; Tudor, M.; Fambrough, D.; Csankovszki, G.; Dausman, J.; Lee, P.; Wilson, C.; Lander, E.; et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001, 27, 31–39. [Google Scholar] [CrossRef]
- Oda, M.; Yamagiwa, A.; Yamamoto, S.; Nakayama, T.; Tsumura, A.; Sasaki, H.; Nakao, K.; Li, E.; Okano, M. DNA methylation regulates long-range gene silencing of an X-linked homeobox gene cluster in a lineage-specific manner. Genes Dev. 2006, 20, 3382–3394. [Google Scholar] [CrossRef][Green Version]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta Bioenerg. 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Tuck-Muller, C.M.; Narayan, A.; Tsien, F.; Smeets, D.F.; Sawyer, J.; Fiala, E.S.; Sohn, O.S.; Ehrlich, M. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell Genet. 2000, 89, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Okano, M.; Dick, F.; Tsujimoto, N.; Chen, T.; Wang, S.; Ueda, Y.; Dyson, N.; Li, E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005, 280, 17986–17991. [Google Scholar] [CrossRef]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.-A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capellà, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef]
- Suzuki, M.; Oda, M.; Ramos, M.-P.; Pascual, M.; Lau, K.; Stasiek, E.; Agyiri, F.; Thompson, R.F.; Glass, J.L.; Jing, Q.; et al. Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome. Genome Res. 2011, 21, 1833–1840. [Google Scholar] [CrossRef]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.-I.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 2006, 11, 805–814. [Google Scholar] [CrossRef]
- Wan, Y.; Coxe, K.K.; Thackray, V.G.; Housley, P.R.; Nordeen, S.K. Separable features of the ligand-binding domain determine the differential subcellular localization and ligand-binding specificity of glucocorticoid receptor and progesterone receptor. Mol. Endocrinol. 2001, 15, 17–31. [Google Scholar] [CrossRef][Green Version]
- Vegeto, E.; Allan, G.F.; Schrader, W.T.; Tsai, M.-J.; McDonnell, D.P.; O’Malley, B.W. The mechanism of RU486 antagonism is dependent on the conformation of the carboxy-terminal tail of the human progesterone receptor. Cell 1992, 69, 703–713. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Pierson, T.; O’Malley, B.W.; Tsai, S.Y. Positive and negative regulation of gene expression in eukaryotic cells with an inducible transcriptional regulator. Gene Ther. 1997, 4, 432–441. [Google Scholar] [CrossRef][Green Version]
- Ryba, T.; Battaglia, D.; Pope, B.D.; Hiratani, I.; Gilbert, D.M. Genome-scale analysis of replication timing: From bench to bioinformatics. Nat. Protoc. 2011, 6, 870–895. [Google Scholar] [CrossRef]
- Takebayashi, S.-I.; Lei, I.; Ryba, T.; Sasaki, T.; Dileep, V.; Battaglia, D.; Gao, X.; Fang, P.; Fan, Y.; Esteban, M.A.; et al. Murine esBAF chromatin remodeling complex subunits BAF250a and Brg1 are necessary to maintain and reprogram pluripotency-specific replication timing of select replication domains. Epigenet. Chromatin 2013, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.-I.; Ogata, S.; Ogata, M.; Okumura, K. Mapping mammalian replication domains using the ion torrent semiconductor sequencing platform. Biosci. Biotechnol. Biochem. 2018, 82, 2098–2100. [Google Scholar] [CrossRef]
- Ryba, T.; Hiratani, I.; Sasaki, T.; Battaglia, D.; Kulik, M.; Zhang, J.; Dalton, S.; Gilbert, D.M. Replication timing: A fingerprint for cell identity and pluripotency. PLoS Comput. Biol. 2011, 7, e1002225. [Google Scholar] [CrossRef]
- Kuriya, K.; Higashiyama, E.; Avşar-Ban, E.; Tamaru, Y.; Ogata, S.; Takebayashi, S.-I.; Ogata, M.; Okumura, K. Direct visualization of dna replication dynamics in zebrafish cells. Zebrafish 2015, 12, 432–439. [Google Scholar] [CrossRef]
- Michalet, X.; Ekong, R.; Fougerousse, F.; Rousseaux, S.; Schurra, C.; Hornigold, N.; van Slegtenhorst, M.; Wolfe, J.; Povey, S.; Beckmann, J.S.; et al. Dynamic molecular combing: Stretching the whole human genome for high-resolution studies. Science 1997, 277, 1518–1523. [Google Scholar] [CrossRef]
- Koberna, K.; Staněk, D.; Malínský, J.; Eltsov, M.; Pliss, A.; Čtrnáctá, V.; Cermanová, Š.; Raška, I. Nuclear organization studied with the help of a hypotonic shift: Its use permits hydrophilic molecules to enter into living cells. Chromosoma 1999, 108, 325–335. [Google Scholar] [CrossRef]
- Hino, S.; Sakamoto, A.; Nagaoka, K.; Anan, K.; Wang, Y.; Mimasu, S.; Umehara, T.; Yokoyama, S.; Kosai, K.-I.; Nakao, M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Takebayashi, S.-I.; Tamura, T.; Matsuoka, C.; Okano, M. Major and essential role for the DNA methylation mark in mouse embryogenesis and stable association of DNMT1 with newly replicated regions. Mol. Cell. Biol. 2007, 27, 8243–8258. [Google Scholar] [CrossRef]
- Panning, M.M.; Gilbert, D.M. Spatio-temporal organization of DNA replication in murine embryonic stem, primary, and immortalized cells. J. Cell. Biochem. 2005, 95, 74–82. [Google Scholar] [CrossRef]
- Takebayashi, S.-I.; Sugimura, K.; Saito, T.; Sato, C.; Fukushima, Y.; Taguchi, H.; Okumura, K. Regulation of replication at the R/G chromosomal band boundary and pericentromeric heterochromatin of mammalian cells. Exp. Cell Res. 2005, 304, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Dienstbier, M.; Hassan, R.; Schermelleh, L.; Sharif, J.; Blackledge, N.P.; De Marco, V.; Elderkin, S.; Koseki, H.; Klose, R.; et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014, 7, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.H.; Smith, L.; Huang, J.; Thayer, M. Chromosomes with delayed replication timing lead to checkpoint activation, delayed recruitment of Aurora B and chromosome instability. Oncogene 2006, 26, 1852–1861. [Google Scholar] [CrossRef]
- Cornacchia, D.; Dileep, V.; Quivy, J.-P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B.C. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.A.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendergast, J.G.D.; Mjoseng, H.K.; et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013, 14, R25. [Google Scholar] [CrossRef]
- King, A.D.; Huang, K.; Rubbi, L.; Liu, S.; Wang, C.-Y.; Wang, Y.; Pellegrini, M.; Fan, G. Reversible Regulation of promoter and enhancer histone landscape by DNA methylation in mouse embryonic stem cells. Cell Rep. 2016, 17, 289–302. [Google Scholar] [CrossRef]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA Methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. elife 2012, 1, e00205. [Google Scholar] [CrossRef]
- Wu, X.; Johansen, J.V.; Helin, K. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 2013, 49, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Brustel, J.; Kirstein, N.; Izard, F.; Grimaud, C.; Prorok, P.; Cayrou, C.; Schotta, G.; Abdelsamie, A.F.; Déjardin, J.; Méchali, M.; et al. Histone H4K20 tri-methylation at late-firing origins ensures timely heterochromatin replication. EMBO J. 2017, 36, 2726–2741. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.F.; Azuara, V.; Amoils, S.; Spivakov, M.; Terry, A.; Nesterova, T.B.; Cobb, B.S.; Ramsahoye, B.; Merkenschlager, M.; Fisher, A.G. The impact of chromatin modifiers on the timing of locus replication in mouse embryonic stem cells. Genome Biol. 2007, 8, R169. [Google Scholar] [CrossRef]
- Peters, A.H.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 2003, 12, 1577–1589. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late S phase. Nature 2002, 420, 198–202. [Google Scholar] [CrossRef]
- Hassan-Zadeh, V.; Rugg-Gunn, P.; Bazett-Jones, D.P. DNA methylation is dispensable for changes in global chromatin architecture but required for chromocentre formation in early stem cell differentiation. Chromosoma 2017, 126, 605–614. [Google Scholar] [CrossRef]
- Nothjunge, S.; Nührenberg, T.G.; Grüning, B.A.; Doppler, S.A.; Preissl, S.; Schwaderer, M.; Rommel, C.; Krane, M.; Hein, L.; Gilsbach, R. DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takebayashi, S.-i.; Ryba, T.; Wimbish, K.; Hayakawa, T.; Sakaue, M.; Kuriya, K.; Takahashi, S.; Ogata, S.; Hiratani, I.; Okumura, K.; et al. The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases. Cells 2021, 10, 266. https://doi.org/10.3390/cells10020266
Takebayashi S-i, Ryba T, Wimbish K, Hayakawa T, Sakaue M, Kuriya K, Takahashi S, Ogata S, Hiratani I, Okumura K, et al. The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases. Cells. 2021; 10(2):266. https://doi.org/10.3390/cells10020266
Chicago/Turabian StyleTakebayashi, Shin-ichiro, Tyrone Ryba, Kelsey Wimbish, Takuya Hayakawa, Morito Sakaue, Kenji Kuriya, Saori Takahashi, Shin Ogata, Ichiro Hiratani, Katsuzumi Okumura, and et al. 2021. "The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases" Cells 10, no. 2: 266. https://doi.org/10.3390/cells10020266
APA StyleTakebayashi, S.-i., Ryba, T., Wimbish, K., Hayakawa, T., Sakaue, M., Kuriya, K., Takahashi, S., Ogata, S., Hiratani, I., Okumura, K., Okano, M., & Ogata, M. (2021). The Temporal Order of DNA Replication Shaped by Mammalian DNA Methyltransferases. Cells, 10(2), 266. https://doi.org/10.3390/cells10020266