
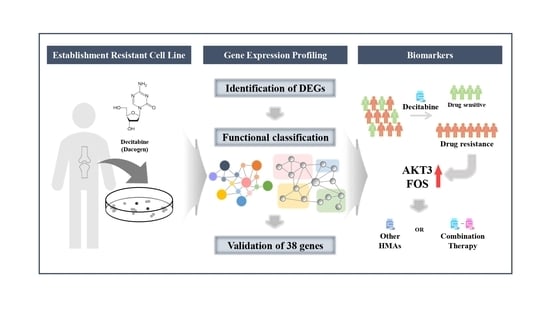
Gene Expression Profiles Identify Biomarkers of Resistance to Decitabine in Myelodysplastic Syndromes


Abstract
:
1. Introduction
2. Materials and Methods
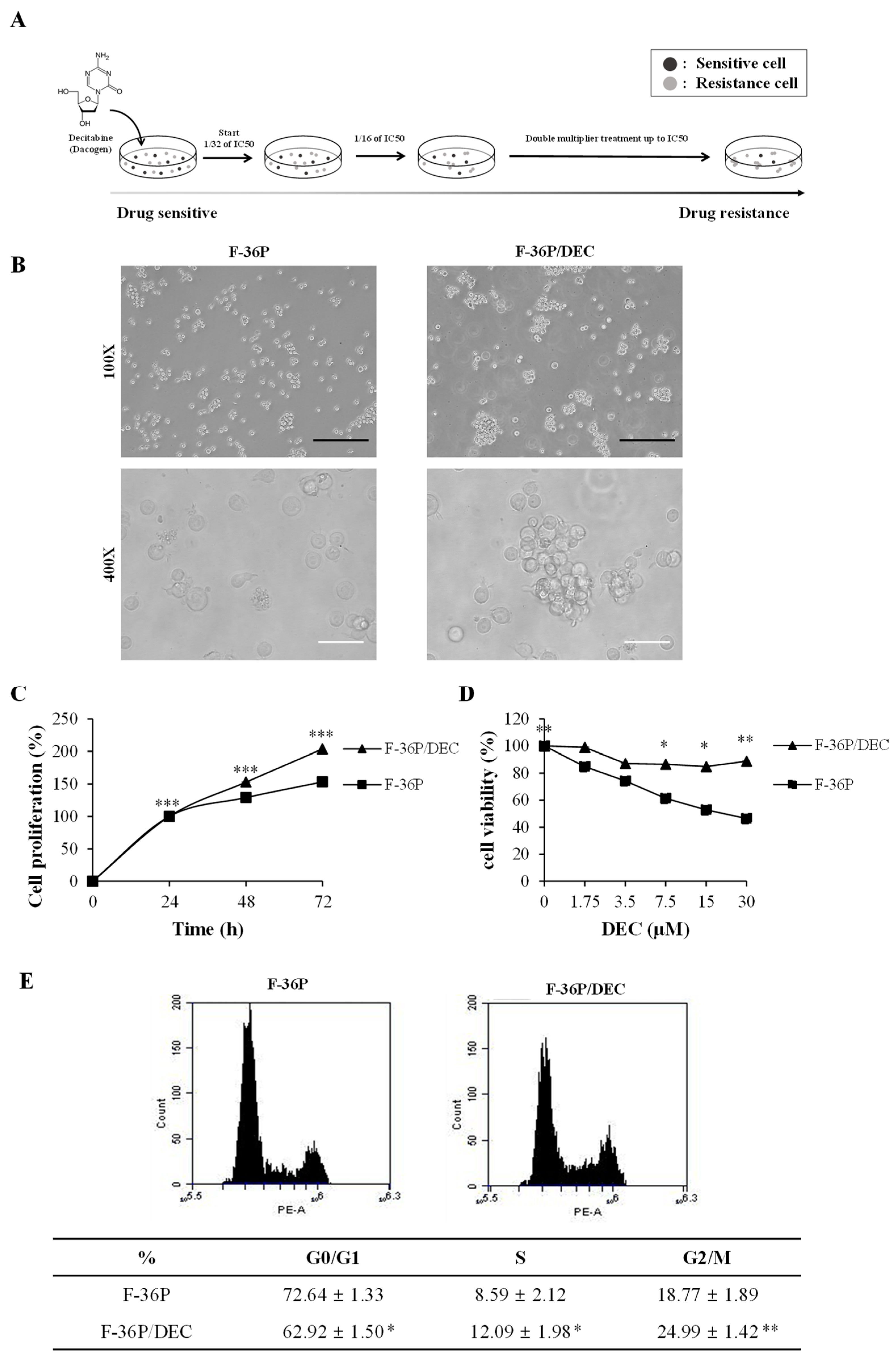
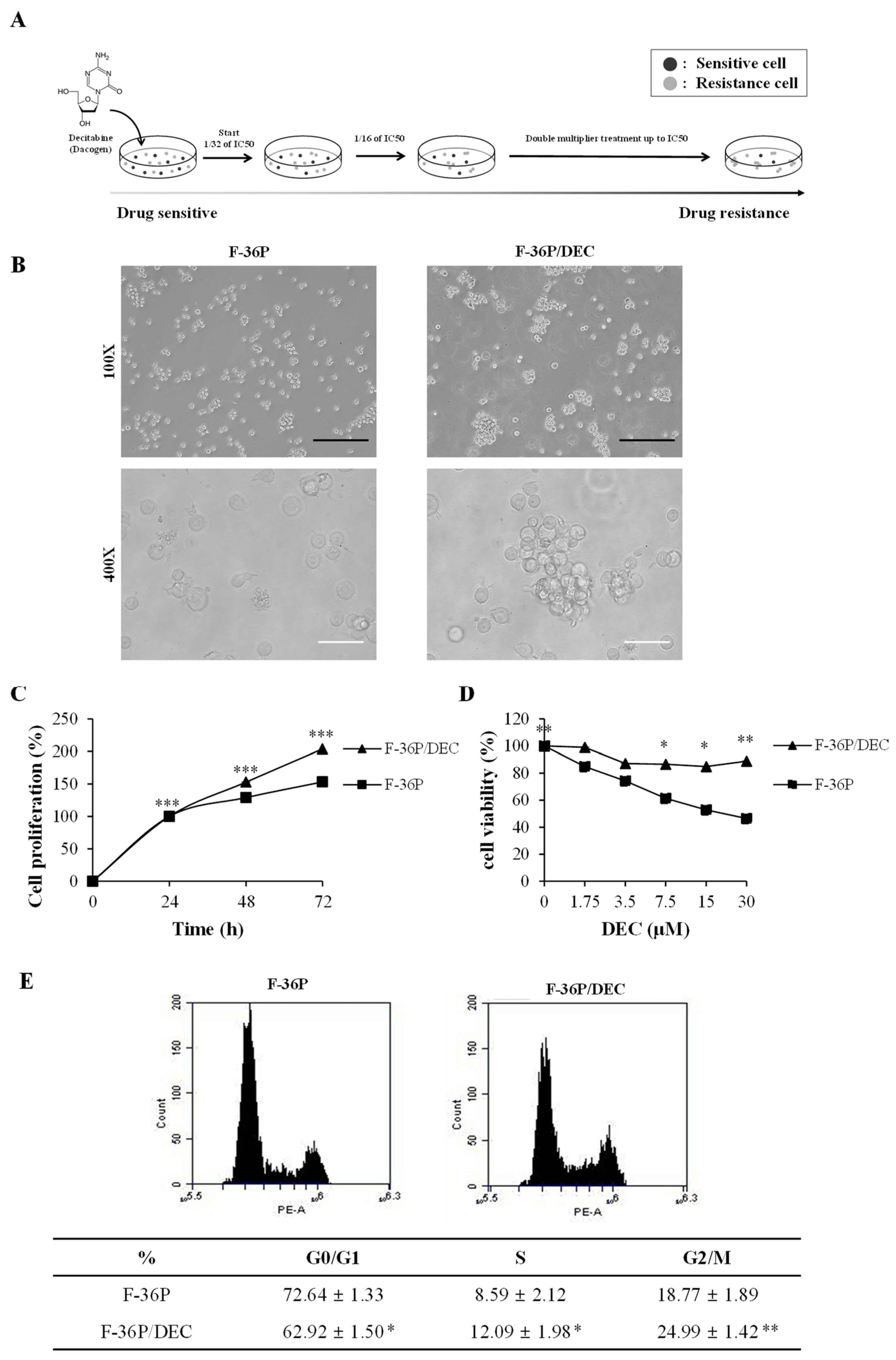
2.1. DEC-Resistant Cell Selection and Culture
2.2. Cell Morphology and Measurement of Drug Sensitivity
2.3. Fluorescence-Activated Cell Sorting (FACS) Analysis
2.4. RNA Isolation
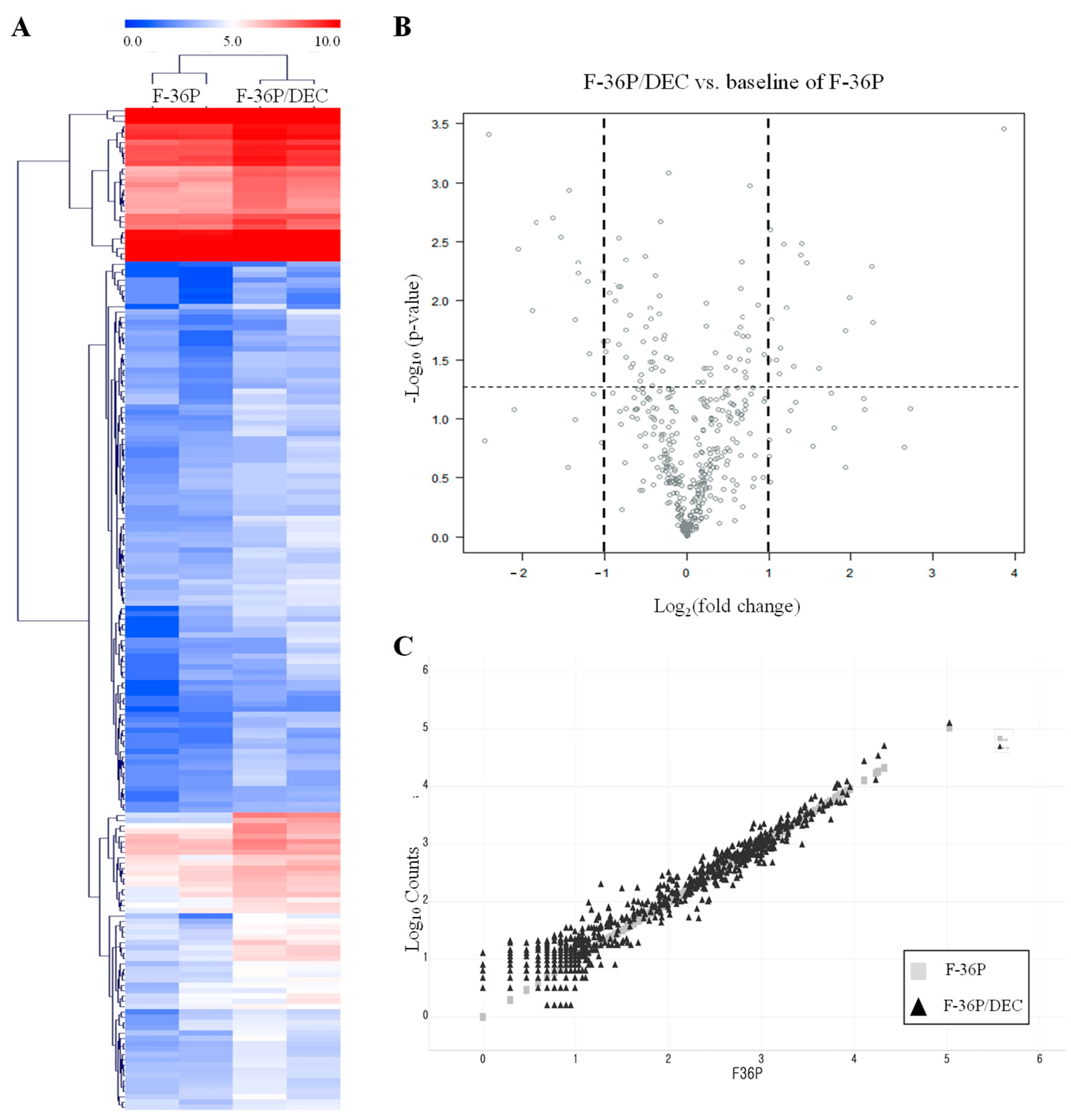
2.5. NanoString Targeted Gene Expression
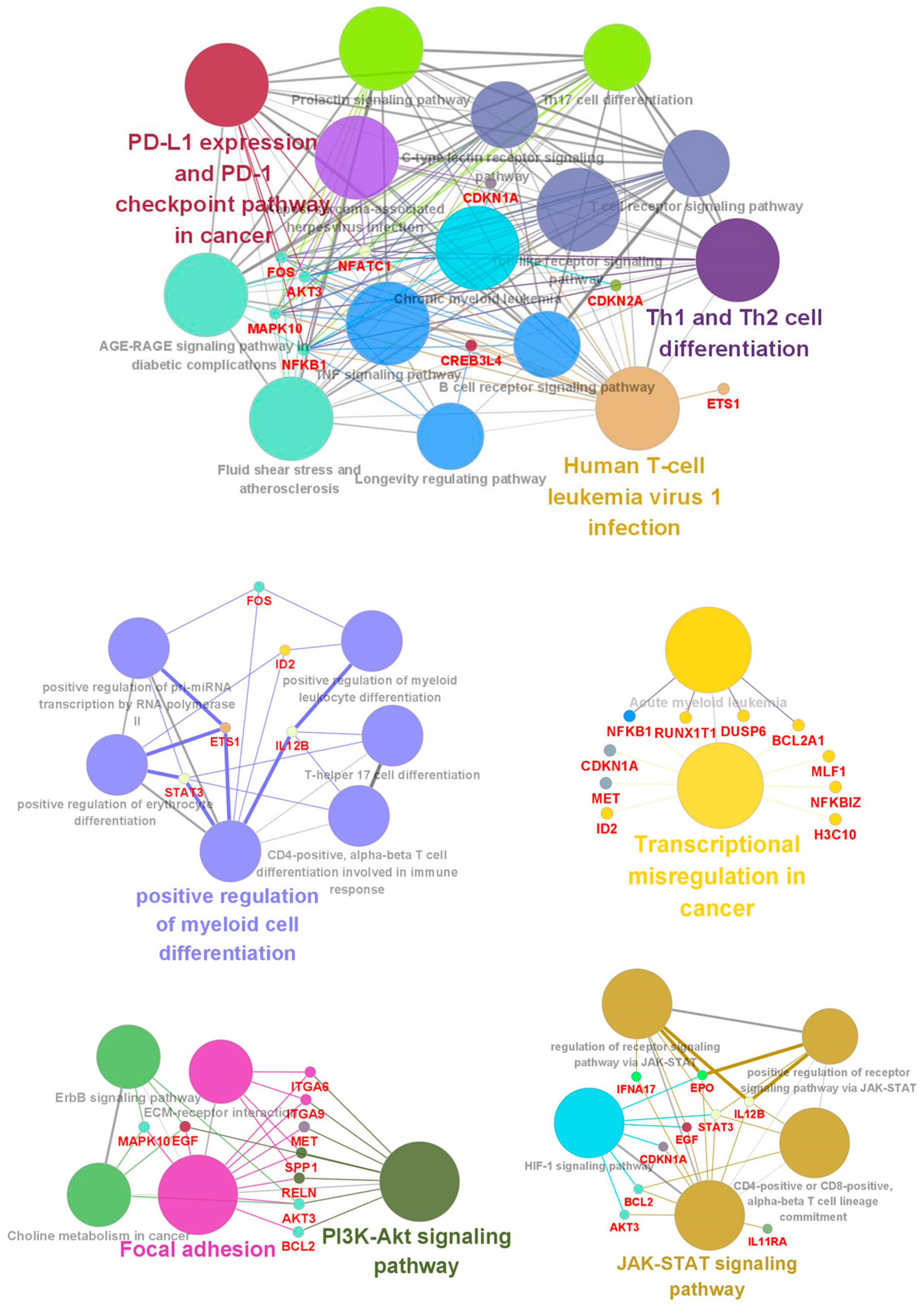
2.6. Gene and Pathway Enrichment Analyses of Differentially Expressed Genes (DEGs)
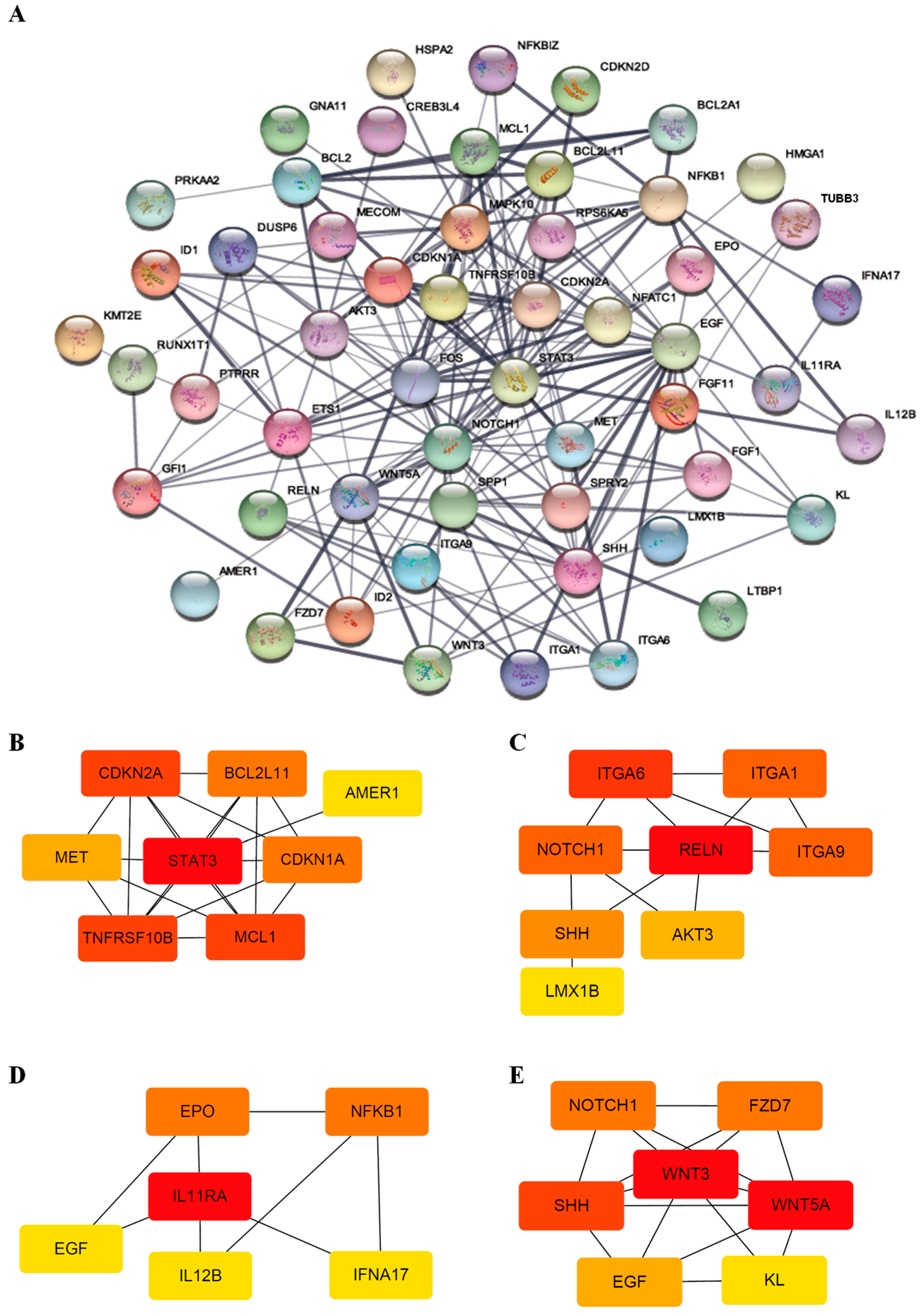
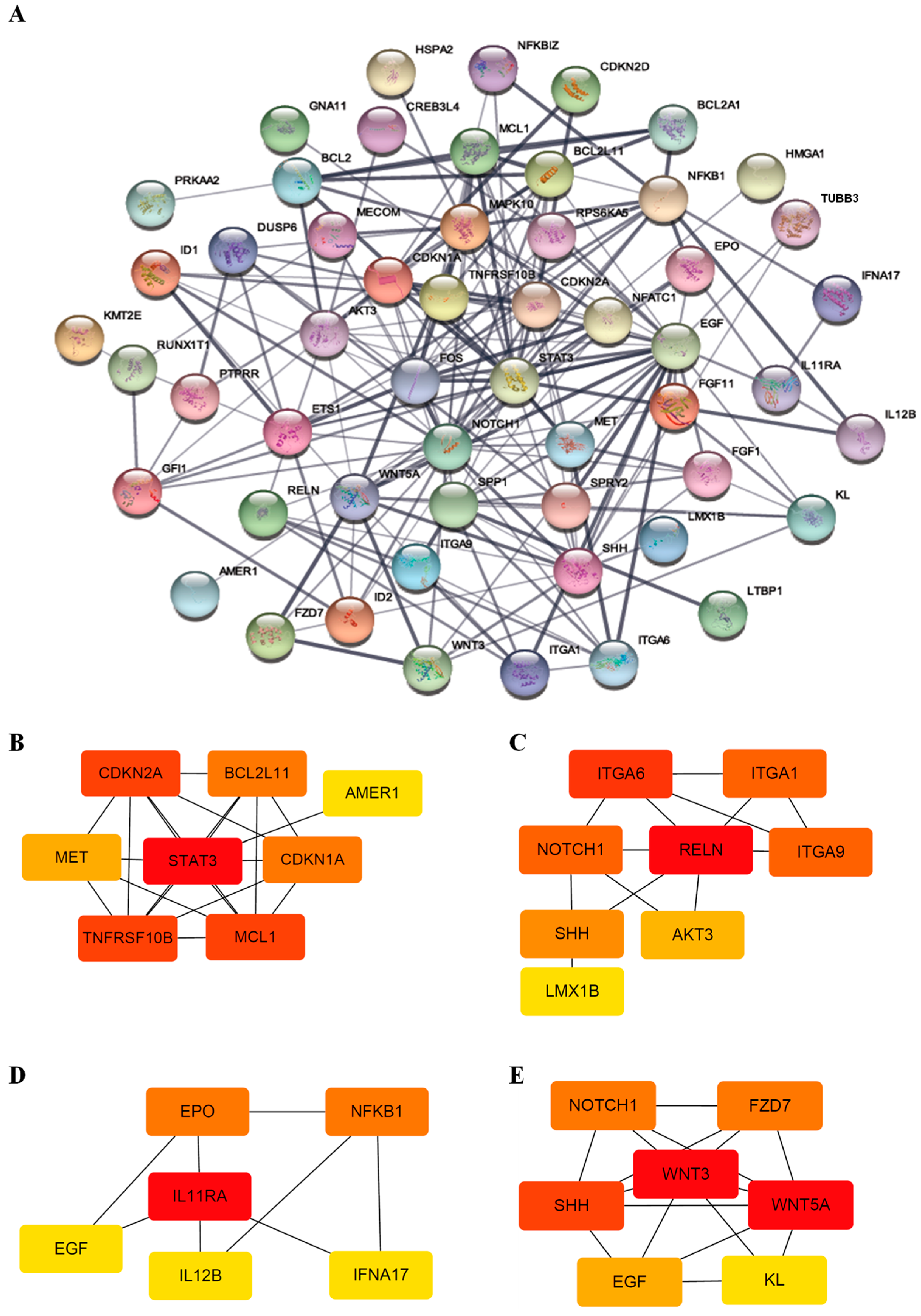
2.7. Protein–Protein Network and Module Analysis
2.8. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
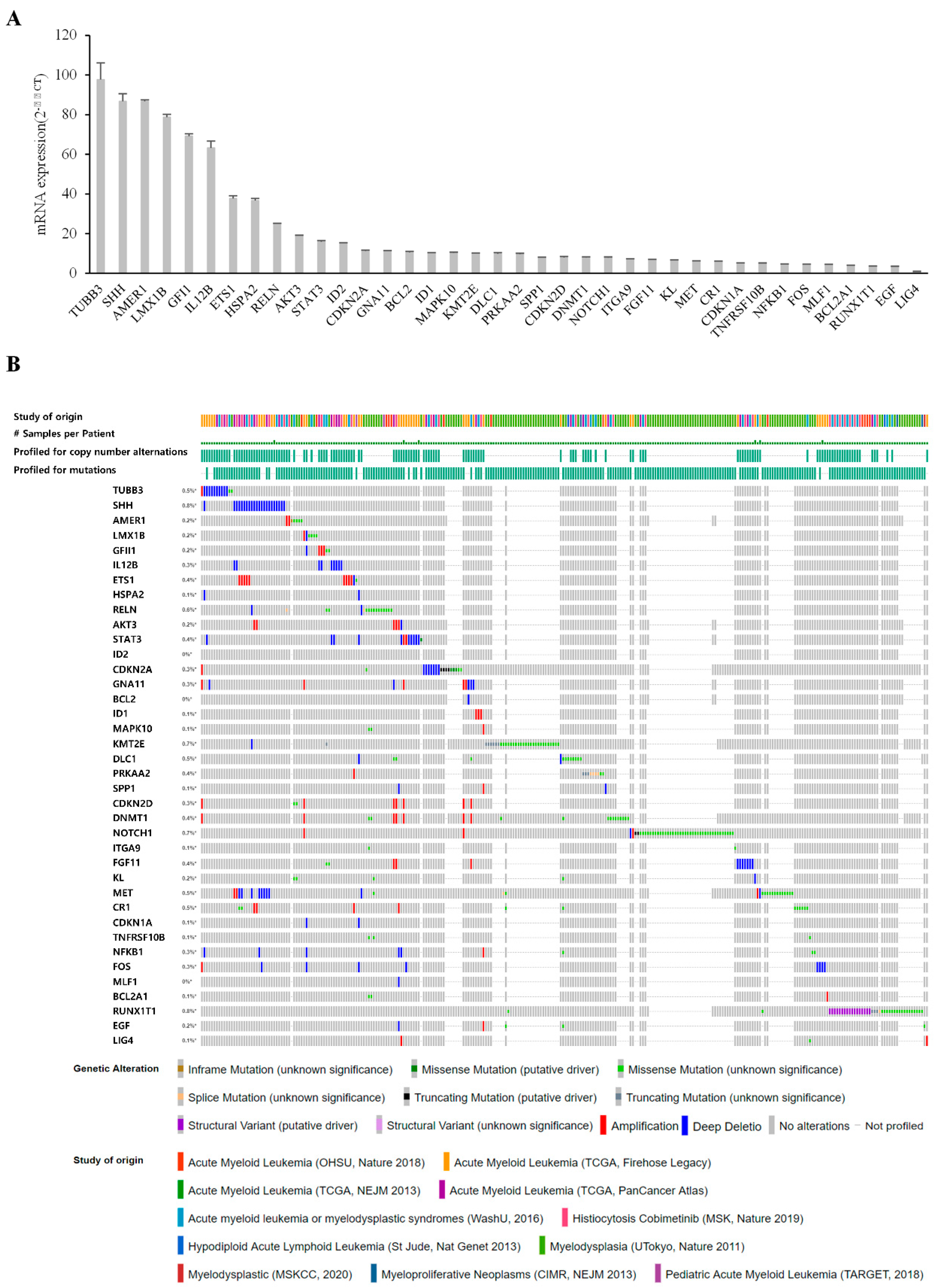
2.9. Validation of Genetic Alterations in Candidate Genes
2.10. Patient Enrollment and Treatment–Bone Marrow Samples
2.11. Statistical Analysis
3. Results
3.1. Establishment of the DEC-Resistant Cell Line, F-36P/DEC
3.2. Identification of Genes That Are Differentially Expressed between F-36P/DEC and F-36P Cells
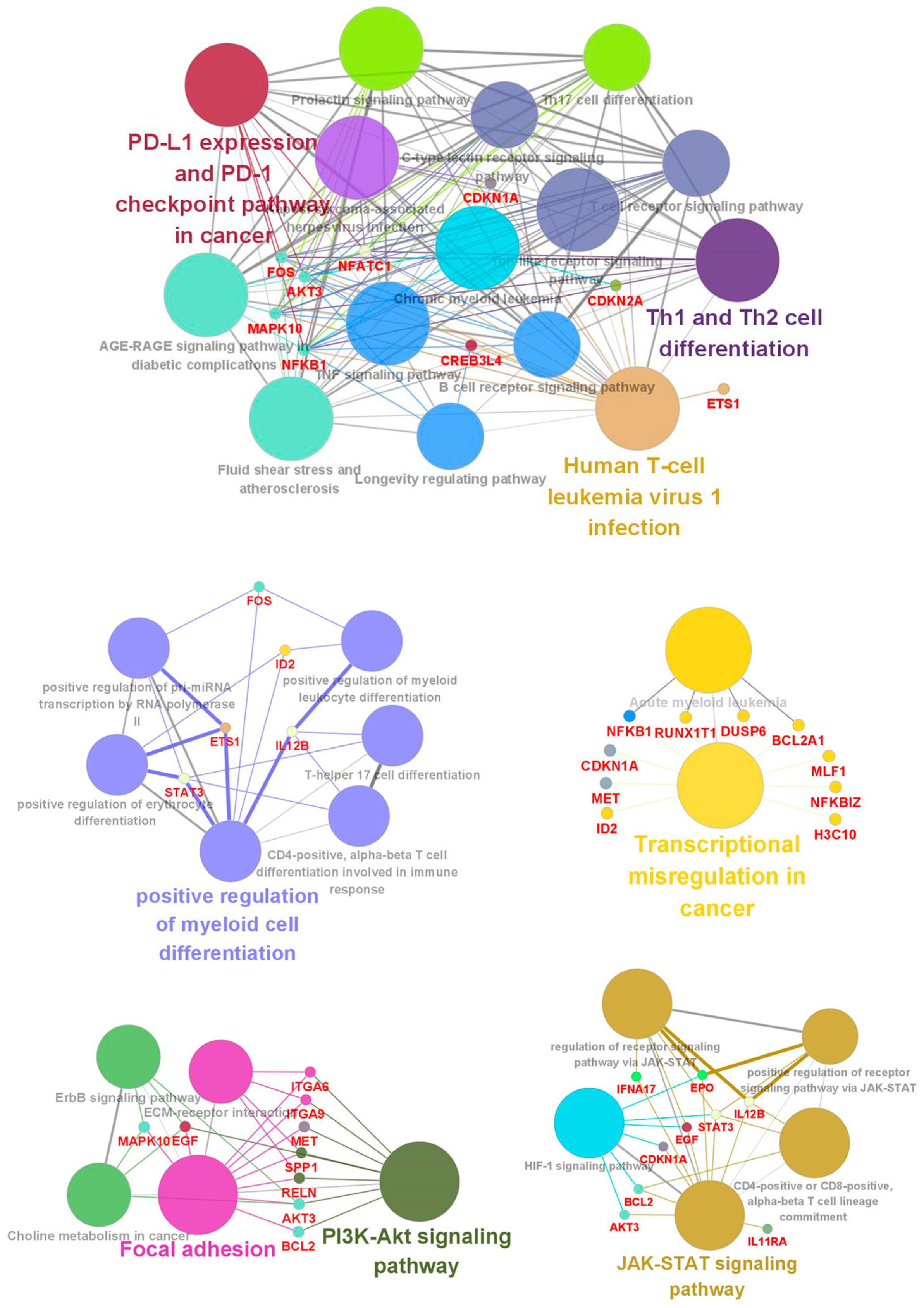
3.3. Functional Classification of DEGs Associated with DEC Resistance of the Cell Line, F-36P/DEC
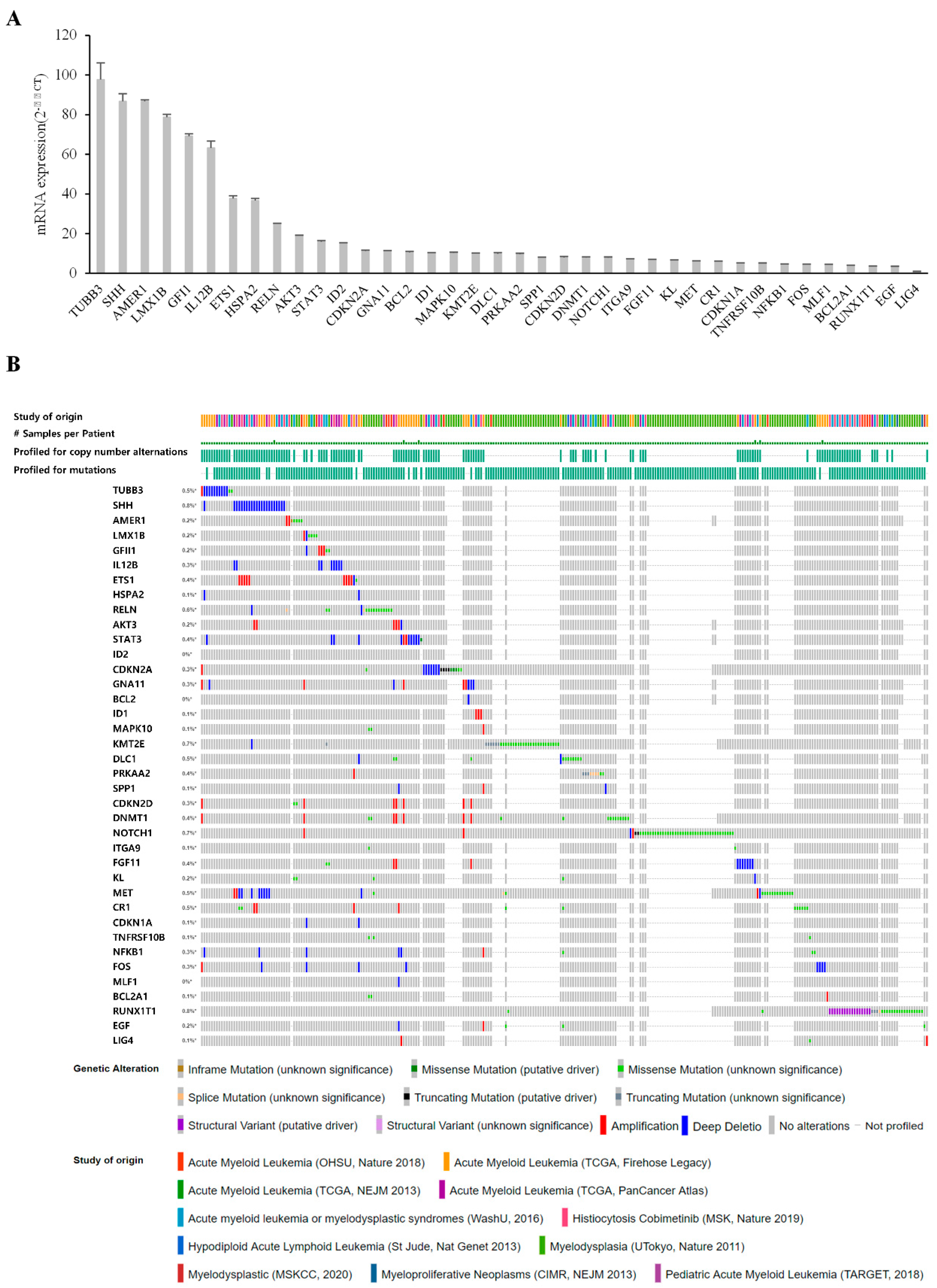
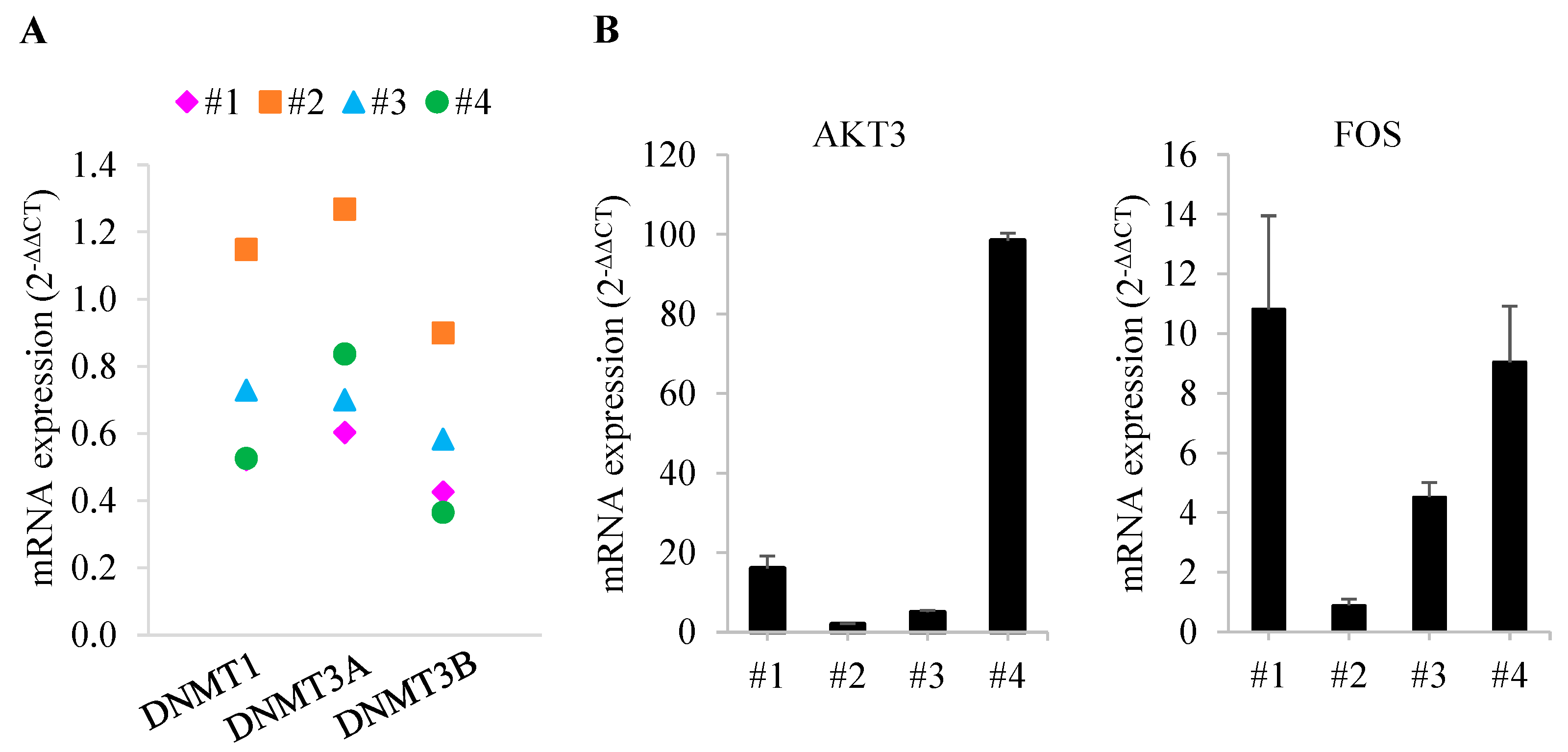
3.4. Validation of Candidates Identified by RNA-Seq Analysis
3.5. Comparison of Gene Expression in Bone Marrow from Patients with MDS
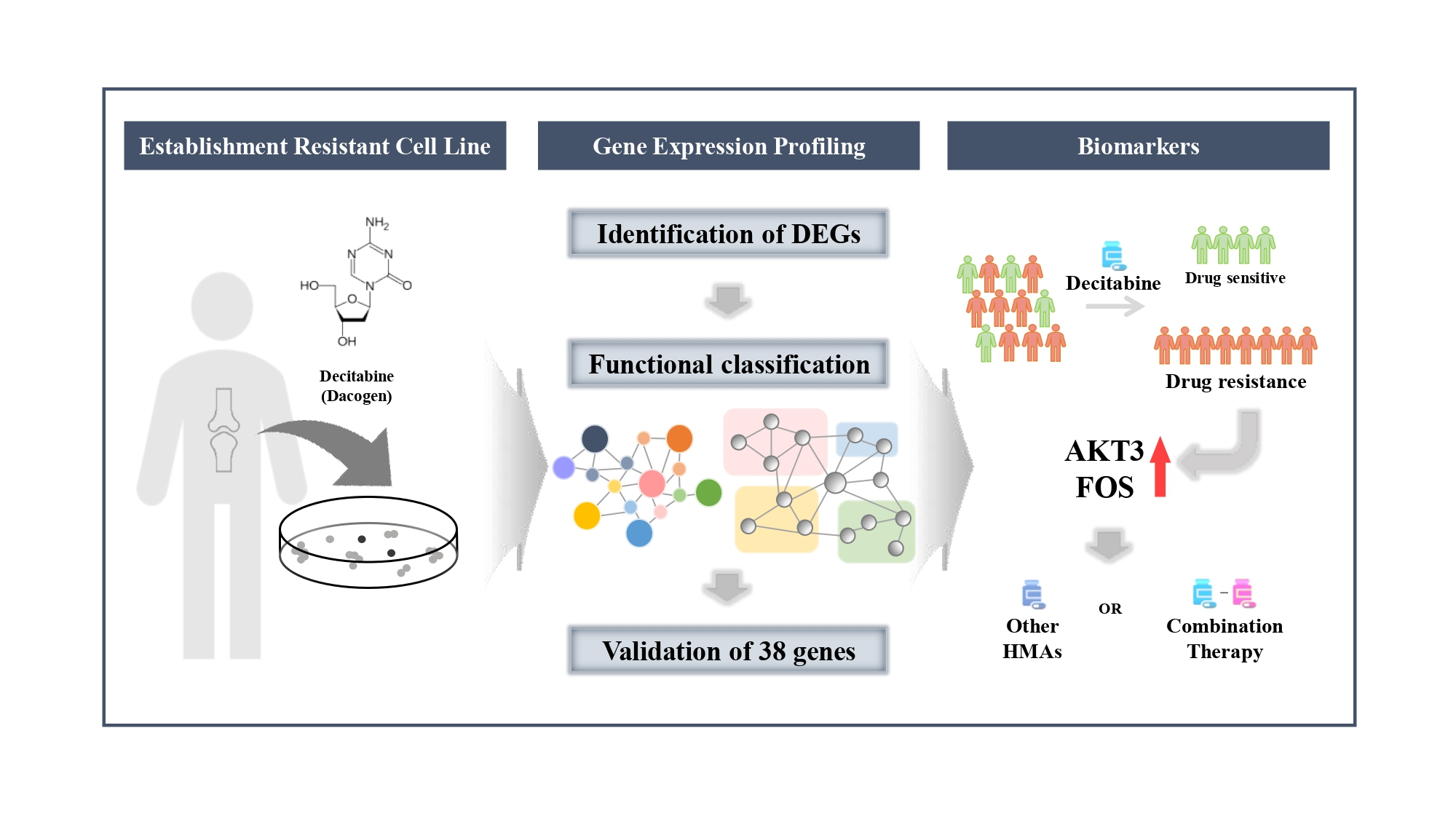
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Le, Y. Screening and identification of key candidate genes and pathways in myelodysplastic syndrome by bioinformatic analysis. PeerJ 2019, 7, e8162. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F. Revised international prognostic scoring system for myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Scalzulli, E.; Pepe, S.; Colafigli, G.; Breccia, M. Therapeutic strategies in low and high-risk MDS: What does the future have to offer? Blood Rev. 2020, 45, 100689. [Google Scholar] [CrossRef]
- Prebet, T.; Sun, Z.; Figueroa, M.E.; Ketterling, R.; Melnick, A.; Greenberg, P.L.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: Results of the US Leukemia Intergroup trial E1905. J. Clin. Oncol. 2014, 32, 1242. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R. Low-dose decitabine versus best supportive care in elderly patients with intermediate-or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lübbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Kantarjian, H.; Issa, J.P.J.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; De Castro, C.; Ravandi, F. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 106, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.R.; McKenzie, D.R.; Peterson, B.L.; Holland, J.F.; Backstrom, J.T.; Beach, C.; Larson, R.A. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: Studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin. Oncol. 2006, 24, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P.; Baer, M.R.; Slack, J.L.; Buckstein, R.; Godley, L.A.; Garcia-Manero, G.; Albitar, M.; Larsen, J.S.; Arora, S.; Cullen, M.T. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: The alternative dosing for outpatient treatment (ADOPT) trial. J. Clin. Oncol. 2009, 27, 3842. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P. Mechanisms of resistance to 5-aza-2’-deoxycytidine in human cancer cell lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, E.-H.; Jung, S.-H.; Goo, B.-K.; Moon, J.; Choi, Y.; Choi, D.R.; Chung, Y.-J.; Lee, J.-H. Establishment and characterization of hypomethylating agent-resistant cell lines, MOLM/AZA-1 and MOLM/DEC-5. Oncotarget 2017, 8, 11748. [Google Scholar] [CrossRef] [Green Version]
- Alva, A.S.; Hahn, N.M.; Aparicio, A.M.; Singal, R.; Yellapragada, S.; Sonpavde, G. Hypomethylating agents for urologic cancers. Future Oncol. 2011, 7, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Sorrentino, V.G.; Thota, S.; Gonzalez, E.A.; Rameshwar, P.; Chang, V.T.; Etchegaray, J.-P. Hypomethylating Chemotherapeutic Agents as Therapy for Myelodysplastic Syndromes and Prevention of Acute Myeloid Leukemia. Pharmaceuticals 2021, 14, 641. [Google Scholar] [CrossRef]
- Chang, C.K.; Zhao, Y.S.; Xu, F.; Guo, J.; Zhang, Z.; He, Q.; Wu, D.; Wu, L.Y.; Su, J.Y.; Song, L.X. TP53 mutations predict decitabine-induced complete responses in patients with myelodysplastic syndromes. Br. J. Haematol. 2017, 176, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Garcia-Manero, G.; Batty, N.; Shan, J.; O’Brien, S.; Cortes, J.; Ravandi, F.; Issa, J.P.; Kantarjian, H. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer 2010, 116, 3830–3834. [Google Scholar] [CrossRef] [Green Version]
- Kadia, T.M.; Jabbour, E.; Kantarjian, H. Failure of hypomethylating agent–based therapy in myelodysplastic syndromes. Semin Oncol. 2011, 38, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P. Genomic biomarkers to predict resistance to hypomethylating agents in patients with myelodysplastic syndromes using artificial intelligence. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Lv, N.; Li, Y.; Wang, L.; Yu, L. Predictors of clinical responses to hypomethylating agents in acute myeloid leukemia or myelodysplastic syndromes. Ann. Hematol. 2018, 97, 2025–2038. [Google Scholar] [CrossRef]
- Stomper, J.; Lübbert, M. Can we predict responsiveness to hypomethylating agents in AML? Semin Hematol. 2019, 56, 118–124. [Google Scholar] [CrossRef]
- Chiba, S.; Takaku, F.; Tange, T.; Shibuya, K.; Misawa, C.; Sasaki, K.; Miyagawa, K.; Yazaki, Y.; Hirai, H. Establishment and erythroid differentiation of a cytokine-dependent human leukemic cell line F-36: A parental line requiring granulocyte-macrophage colony-stimulating factor or interleukin-3, and a subline requiring erythropoietin. Blood. 1991, 78, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood J. Am. Soc. Hematol. 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011, 94. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.O. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Fontanillo, C. Protein–protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterman, T.J.; Terry, M.; Miller, R.S. Improving Cancer Data Interoperability: The Promise of the Minimal Common Oncology Data Elements (mCODE) Initiative. JCO Clin. Cancer Inform. 2020, 4, 993–1001. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Galon, J.; Bindea, G. Comprehensive functional analysis of large lists of genes and proteins. J. Proteom. 2018, 171, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Choi, Y.; Kim, S.D.; Kim, D.Y.; Lee, J.H.; Lee, K.H.; Lee, S.M.; Lee, W.S.; Joo, Y.D. Clinical outcome after failure of hypomethylating therapy for myelodysplastic syndrome. Eur. J. Haematol. 2015, 94, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Kharfan-Dabaja, M.A.; Komrokji, R.S. Beyond hypomethylating agents failure in patients with myelodysplastic syndromes. Curr. Opin. Hematol. 2014, 21, 123. [Google Scholar] [CrossRef]
- Coley, H.M. Development of drug-resistant models. Methods Mol. Med. 2004, 88, 267–273. [Google Scholar]
- Wen, X.M.; Zhang, T.J.; Ma, J.C.; Zhou, J.D.; Xu, Z.J.; Zhu, X.W.; Yuan, Q.; Ji, R.B.; Chen, Q.; Deng, Z.Q. Establishment and molecular characterization of decitabine-resistant K562 cells. J. Cell. Mol. Med. 2019, 23, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.X.; Cai, H.; Mao, Y.Y.; Wu, Q.; Zhang, L.; Zhou, D.B.; Li, J. Next-generation sequencing-based genetic landscape and its clinical implications for Chinese acute myeloid leukemia patients. Cancer Cell Int. 2018, 18, 215. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Li, Y.; Li, T.; Li, Y.; Xing, H.; Sun, H.; Sun, L.; Wan, D.; Liu, Y.; Xie, X. Gene mutational analysis by NGS and its clinical significance in patients with myelodysplastic syndrome and acute myeloid leukemia. Exp. Hematol. Oncol. 2020, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 transcription factors as regulators of immune responses in cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [Green Version]
- Follo, M.Y.; Pellagatti, A.; Armstrong, R.N.; Ratti, S.; Mongiorgi, S.; De Fanti, S.; Bochicchio, M.T.; Russo, D.; Gobbi, M.; Miglino, M. Response of high-risk MDS to azacitidine and lenalidomide is impacted by baseline and acquired mutations in a cluster of three inositide-specific genes. Leukemia 2019, 33, 2276–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mias, G.I.; Chen, R.; Zhang, Y.; Sridhar, K.; Sharon, D.; Xiao, L.; Im, H.; Snyder, M.P.; Greenberg, P.L. Specific plasma autoantibody reactivity in myelodysplastic syndromes. Sci. Rep. 2013, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Description | Count | p-Value |
|---|---|---|---|---|
| BP | GO:0045893 | Regulation of transcription, DNA-templated | 15 | 5.89 × 10−7 |
| GO:0045944 | Regulation of transcription from RNA polymerase II promoter | 18 | 1.01 × 10−5 | |
| GO:0008285 | Negative regulation of cell proliferation | 9 | 3.59 × 10−5 | |
| GO:0043066 | Negative regulation of apoptotic process | 9 | 9.52 × 10−5 | |
| GO:0007165 | Signal transduction | 12 | 1.00 × 10−3 | |
| GO:0007411 | Axon guidance | 5 | 0.0017 | |
| GO:0007050 | Cell cycle arrest | 4 | 0.0105 | |
| GO:0007568 | Aging | 4 | 0.0160 | |
| GO:0016055 | Wnt signaling pathway | 4 | 0.0223 | |
| GO:0071425 | Hematopoietic stem cell proliferation | 2 | 0.0441 | |
| CC | GO:0005654 | Nucleoplasm | 25 | 0.0000 |
| GO:0005634 | Nucleus | 29 | 0.0005 | |
| GO:0008305 | Integrin complex | 3 | 0.0029 | |
| GO:0009925 | Basal plasma membrane | 3 | 0.0036 | |
| GO:0009986 | Cell surface | 7 | 0.0052 | |
| GO:0005829 | Cytosol | 19 | 0.0055 | |
| GO:0005576 | Extracellular region | 12 | 0.0068 | |
| GO:0005886 | Plasma membrane | 20 | 0.0241 | |
| GO:0005925 | Focal adhesion | 5 | 0.0285 | |
| GO:0005737 | Cytoplasm | 23 | 0.0377 | |
| MF | GO:0005515 | Protein binding | 46 | 0.0000 |
| GO:0003700 | Transcription factor activity, sequence-specific DNA binding | 13 | 0.0000 | |
| GO:0008134 | Transcription factor binding | 6 | 0.0021 | |
| GO:0001205 | Transcriptional activator activity, RNA polymerase II distal enhancer sequence-specific binding | 3 | 0.0029 | |
| GO:0003682 | Chromatin binding | 6 | 0.0081 | |
| GO:0003677 | DNA binding | 12 | 0.0155 | |
| GO:0046982 | Protein heterodimerization activity | 6 | 0.0162 | |
| GO:0019903 | Protein phosphatase binding | 3 | 0.0173 | |
| GO:0004672 | Protein kinase activity | 5 | 0.0276 | |
| GO:0019901 | Protein kinase binding | 5 | 0.0320 |
| Sample No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Sex | Female | Male | Male | Female |
| Age (years) | 46 | 73 | 74 | 61 |
| Weight (kg) | 60 | 66 | 76 | 55 |
| Height (m) | 1.56 | 1.6 | 1.63 | 1.47 |
| BMI (kg/m2) | 24.7 | 25.8 | 28.6 | 25.5 |
| Underlying disease | History of allogeneic HSCT due to aplastic anemia, paroxysmal nocturnal hemoglobinuria | Diabetes mellitus | Diabetes mellitus | None |
| Baseline clinical characteristics | ||||
| WBC (×106/L) | 3370 | 6700 | 2970 | 3490 |
| ANC (×106/L) | 135 | 1100 | 535 | 733 |
| Hb (g/dL) | 9.7 | 8.7 | 7 | 10.4 |
| Platelets (×109/L) | 11 | 37 | 32 | 200 |
| BM blasts (%) | 10.3 | 20 | 6 | 12.5 |
| Cytogenetic abnormalities | Complex karyotype 1 | None | Complex karyotype 2 | del (20q) |
| IPSS | 3.0 | 2.0 | 2.0 | 1.5 |
| IPSS risk category | High | Int-2 | Int-2 | Int-2 |
| IPSS-R | 9.5 | 6.0 | 9.0 | 4.5 |
| IPSS-R risk category | Very high | High | Very high | Int |
| MDS subtypes (WHO) | MDS-EB2 | MDS-EB2 | MDS-EB1 | MDS-EB2 |
| Treatment cycle of DEC | 2 | 16 | 11 | 5 |
| Best response | CR | PR | CR | CR |
| Progression (Leukemic transformation) | Yes | Yes | Yes | Yes |
| PFS (month) | 9 | 46 | 11 | 24 |
| Allogeneic HSCT | Yes | No | No | Yes |
| Time to HSCT (month) | 4 | None | None | 7 |
| F/U period (month) | 11 | 52 | 11 | 24 |
| F/U result | Dead | Dead | Dead | Dead |
| Cause of death | Leukemia | Leukemia | Leukemia | Pneumonia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Shin, D.-Y.; Kim, D.; Oh, S.; Hong, J.; Kim, I.; Kim, E. Gene Expression Profiles Identify Biomarkers of Resistance to Decitabine in Myelodysplastic Syndromes. Cells 2021, 10, 3494. https://doi.org/10.3390/cells10123494
Kim S, Shin D-Y, Kim D, Oh S, Hong J, Kim I, Kim E. Gene Expression Profiles Identify Biomarkers of Resistance to Decitabine in Myelodysplastic Syndromes. Cells. 2021; 10(12):3494. https://doi.org/10.3390/cells10123494
Chicago/Turabian StyleKim, Seungyoun, Dong-Yeop Shin, Dayeon Kim, Somi Oh, Junshik Hong, Inho Kim, and Eunju Kim. 2021. "Gene Expression Profiles Identify Biomarkers of Resistance to Decitabine in Myelodysplastic Syndromes" Cells 10, no. 12: 3494. https://doi.org/10.3390/cells10123494
APA StyleKim, S., Shin, D.-Y., Kim, D., Oh, S., Hong, J., Kim, I., & Kim, E. (2021). Gene Expression Profiles Identify Biomarkers of Resistance to Decitabine in Myelodysplastic Syndromes. Cells, 10(12), 3494. https://doi.org/10.3390/cells10123494