
Protein Aggregation in the ER: Calm behind the Storm


Abstract
1. Introduction
2. ER Protein Folding and Quality Control Systems
2.1. ER Chaperones
| Keypoints: ER Chaperones |
|
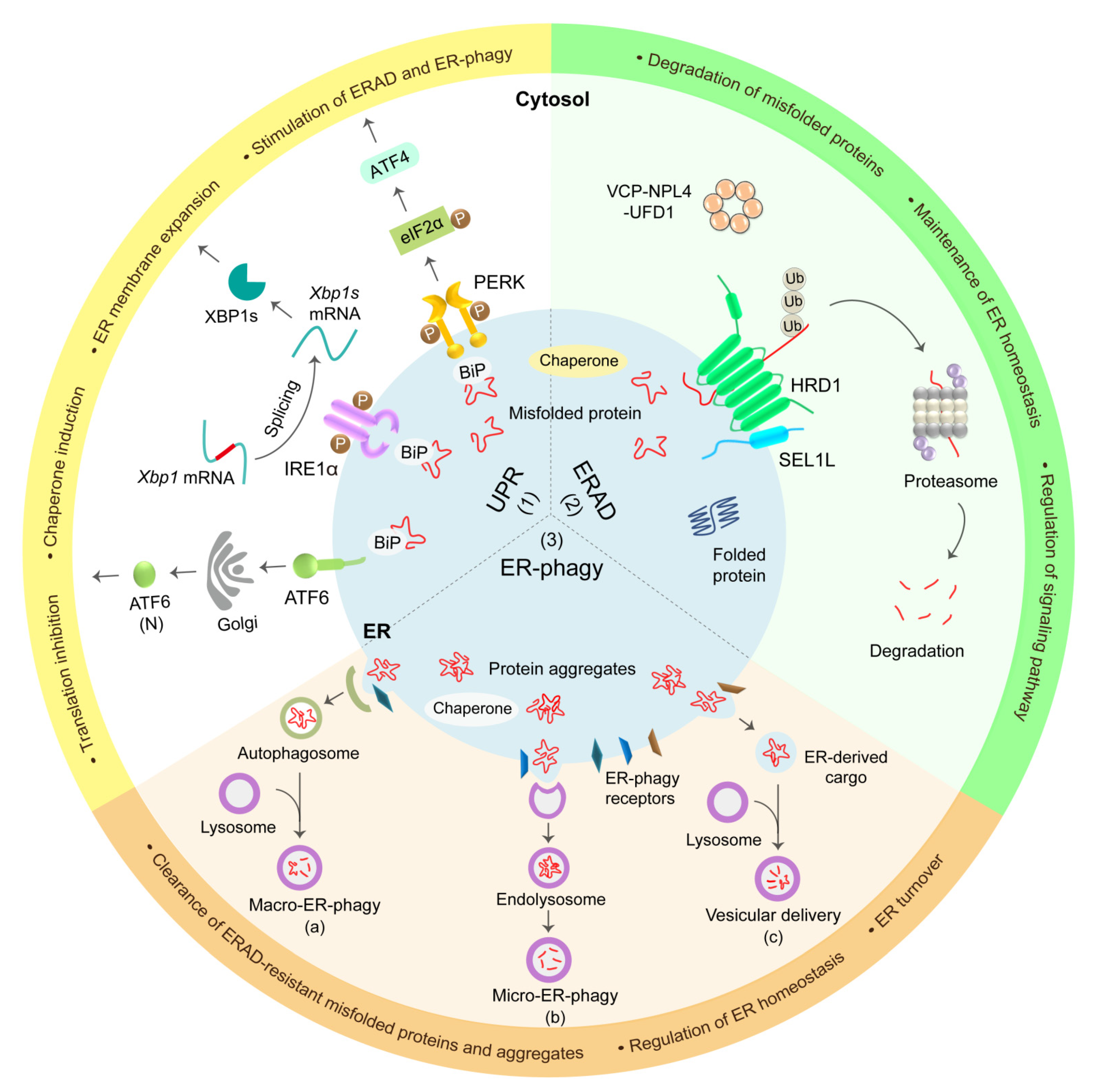
2.2. UPR
| Keypoints: UPR |
|
2.3. ERAD
| Keypoints: ERAD |
|
2.4. ER-Phagy
| Keypoints: ER-phagy |
|
2.5. Crosstalk among the Quality Control Pathways
3. ER Storage Diseases
3.1. α1-Antitrypsin Deficiency (AATD)
3.2. Familial Neurohypophyseal Diabetes Insipidus (FNDI)
3.3. Mutant INS-Gene-Induced Diabetes of Youth (MIDY)
3.4. Hepatic Fibrinogen Storage Disease (HFSD)
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- English, A.R.; Zurek, N.; Voeltz, G.K. Peripheral ER structure and function. Curr. Opin. Cell Biol. 2009, 21, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough sheets and smooth tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef]
- De Matteis, M.A.; Rega, L.R. Endoplasmic reticulum-Golgi complex membrane contact sites. Curr. Opin. Cell Biol. 2015, 35, 43–50. [Google Scholar] [CrossRef]
- Choudhary, V.; Schneiter, R. A Unique Junctional Interface at Contact Sites Between the Endoplasmic Reticulum and Lipid Droplets. Front. Cell Dev. Biol. 2021, 9, 650186. [Google Scholar] [CrossRef]
- Vincenz-Donnelly, L.; Holthusen, H.; Körner, R.; Hansen, E.C.; Presto, J.; Johansson, J.; Sawarkar, R.; Hartl, F.U.; Hipp, M.S. High capacity of the endoplasmic reticulum to prevent secretion and aggregation of amyloidogenic proteins. EMBO J. 2018, 37, 337–350. [Google Scholar] [CrossRef]
- Shao, S.; Hegde, R.S. Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. Cell Dev. Biol. 2011, 27, 25–56. [Google Scholar] [CrossRef]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef]
- Vincenz-Donnelly, L.; Hipp, M.S. The endoplasmic reticulum: A hub of protein quality control in health and disease. Free Radic. Biol. Med. 2017, 108, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Aspects Med. 2015, 42, 105–118. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein Quality Control in the Endoplasmic Reticulum and Cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage diseases. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef]
- Callea, F.; Desmet, V. The Discovery of Endoplasmic Reticulum Storage Disease. The Connection between an H&E Slide and the Brain. Int. J. Mol. Sci. 2021, 22, 2899. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Arvan, P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: Disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev. 1998, 19, 173–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rutishauser, J.; Spiess, M. Endoplasmic reticulum storage diseases. Swiss Med. Wkly. 2002, 132, 211–222. [Google Scholar]
- Hegde, R.S.; Bernstein, H.D. The surprising complexity of signal sequences. Trends Biochem. Sci. 2006, 31, 563–571. [Google Scholar] [CrossRef]
- Nicchitta, C.V.; Murphy, E.C., III; Haynes, R.; Shelness, G.S. Stage- and ribosome-specific alterations in nascent chain-Sec61p interactions accompany translocation across the ER membrane. J. Cell Biol. 1995, 129, 957–970. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638. [Google Scholar] [CrossRef]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.H.; Inaba, K.; Okumura, M. The Protein Disulfide Isomerase Family: From proteostasis to pathogenesis. Biochim. Biophys. Acta-Gen. Subj. 2020, 1864, 129338. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Gehring, K. Calnexin cycle—Structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340. [Google Scholar] [CrossRef]
- Zuiderweg, E.R.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.P.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 2013, 328, 99–153. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Andréasson, C.; Rampelt, H.; Fiaux, J.; Druffel-Augustin, S.; Bukau, B. The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J. Biol. Chem. 2010, 285, 12445–12453. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef]
- Preissler, S.; Rohland, L.; Yan, Y.; Chen, R.; Read, R.J.; Ron, D. AMPylation targets the rate-limiting step of BiP’s ATPase cycle for its functional inactivation. eLife 2017, 6, e29428. [Google Scholar] [CrossRef]
- Preissler, S.; Rato, C.; Chen, R.; Antrobus, R.; Ding, S.; Fearnley, I.M.; Ron, D. AMPylation matches BiP activity to client protein load in the endoplasmic reticulum. eLife 2015, 4, e12621. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Chen, A.J.; Nakayasu, E.S.; Lazar, C.S.; Zbornik, E.A.; Worby, C.A.; Koller, A.; Mattoo, S. A novel link between Fic (filamentation induced by cAMP)-mediated adenylylation/AMPylation and the unfolded protein response. J. Biol. Chem. 2015, 290, 8482–8499. [Google Scholar] [CrossRef]
- Preissler, S.; Chambers, J.E.; Crespillo-Casado, A.; Avezov, E.; Miranda, E.; Perez, J.; Hendershot, L.M.; Harding, H.P.; Ron, D. Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. eLife 2015, 4, e08961. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pareja, K.A.; Kaiser, C.A.; Sevier, C.S. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. eLife 2014, 3, e03496. [Google Scholar] [CrossRef]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Parodi, A.J. Getting in and out from calnexin/calreticulin cycles. J. Biol. Chem. 2008, 283, 10221–10225. [Google Scholar] [CrossRef]
- Deprez, P.; Gautschi, M.; Helenius, A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell 2005, 19, 183–195. [Google Scholar] [CrossRef]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Xu, Y.; Brenner, M.B. Retention of unassembled components of integral membrane proteins by calnexin. Science 1994, 263, 387–390. [Google Scholar] [CrossRef]
- D’Alessio, C.; Caramelo, J.J.; Parodi, A.J. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell Dev. Biol. 2010, 21, 491–499. [Google Scholar] [CrossRef]
- Soldà, T.; Galli, C.; Kaufman, R.J.; Molinari, M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol. Cell 2007, 27, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Tannous, A.; Patel, N.; Tamura, T.; Hebert, D.N. Reglucosylation by UDP-glucose:glycoprotein glucosyltransferase 1 delays glycoprotein secretion but not degradation. Mol. Biol. Cell 2015, 26, 390–405. [Google Scholar] [CrossRef]
- Kim, G.H.; Shi, G.; Somlo, D.R.; Haataja, L.; Song, S.; Long, Q.; Nillni, E.A.; Low, M.J.; Arvan, P.; Myers, M.G., Jr.; et al. Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Investig. 2018, 128, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Somlo, D.R.M.; Kim, G.H.; Prescianotto-Baschong, C.; Sun, S.; Beuret, N.; Long, Q.; Rutishauser, J.; Arvan, P.; Spiess, M.; et al. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Investig. 2017, 127, 3897–3912. [Google Scholar] [CrossRef]
- Liu, M.; Hodish, I.; Haataja, L.; Lara-Lemus, R.; Rajpal, G.; Wright, J.; Arvan, P. Proinsulin misfolding and diabetes: Mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 2010, 21, 652–659. [Google Scholar] [CrossRef]
- Bulleid, N.J. Disulfide bond formation in the mammalian endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2012, 4, a013219. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef]
- Goldberger, R.F.; Epstein, C.J.; Anfinsen, C.B. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J. Biol. Chem. 1963, 238, 628–635. [Google Scholar] [CrossRef]
- Klappa, P.; Freedman, R.B.; Zimmermann, R. Protein disulphide isomerase and a lumenal cyclophilin-type peptidyl prolyl cis-trans isomerase are in transient contact with secretory proteins during late stages of translocation. Eur. J. Biochem. 1995, 232, 755–764. [Google Scholar] [CrossRef]
- Frickel, E.M.; Frei, P.; Bouvier, M.; Stafford, W.F.; Helenius, A.; Glockshuber, R.; Ellgaard, L. ERp57 is a multifunctional thiol-disulfide oxidoreductase. J. Biol. Chem. 2004, 279, 18277–18287. [Google Scholar] [CrossRef]
- Zapun, A.; Darby, N.J.; Tessier, D.C.; Michalak, M.; Bergeron, J.J.; Thomas, D.Y. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 1998, 273, 6009–6012. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Bridges, J.P.; Apsley, K.; Xu, Y.; Weaver, T.E. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 2008, 19, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Pincus, D.; Chevalier, M.W.; Aragón, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Ellis, R.E.; Lee, K.; Liu, C.Y.; Yang, K.; Solomon, A.; Yoshida, H.; Morimoto, R.; Kurnit, D.M.; Mori, K.; et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., III; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Werner, E.D.; Dubas, M.E.; Goeckeler, J.L.; Kruse, K.B.; McCracken, A.A. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 1999, 274, 3453–3460. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Böhmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895. [Google Scholar] [CrossRef]
- Olivari, S.; Galli, C.; Alanen, H.; Ruddock, L.; Molinari, M. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 2005, 280, 2424–2428. [Google Scholar] [CrossRef]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef]
- Tremblay, L.O.; Herscovics, A. Cloning and expression of a specific human alpha 1,2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology 1999, 9, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef]
- Shenkman, M.; Groisman, B.; Ron, E.; Avezov, E.; Hendershot, L.M.; Lederkremer, G.Z. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J. Biol. Chem. 2013, 288, 2167–2178. [Google Scholar] [CrossRef]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef]
- He, K.; Cunningham, C.N.; Manickam, N.; Liu, M.; Arvan, P.; Tsai, B. PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol. Biol. Cell 2015, 26, 3413–3423. [Google Scholar] [CrossRef]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]
- Pye, V.E.; Beuron, F.; Keetch, C.A.; McKeown, C.; Robinson, C.V.; Meyer, H.H.; Zhang, X.; Freemont, P.S. Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. USA 2007, 104, 467–472. [Google Scholar] [CrossRef]
- Li, G.; Zhao, G.; Zhou, X.; Schindelin, H.; Lennarz, W.J. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8348–8353. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [PubMed]
- Hassink, G.; Kikkert, M.; van Voorden, S.; Lee, S.J.; Spaapen, R.; van Laar, T.; Coleman, C.S.; Bartee, E.; Früh, K.; Chau, V.; et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Iwase, I.; Yamasaki, Y.; Takai, T.; Wu, Y.; Kanemoto, S.; Matsuhisa, K.; Asada, R.; Okuma, Y.; Watanabe, T.; et al. Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci. Rep. 2016, 6, 30955. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Qi, L. ER-associated degradation in health and disease - from substrate to organism. J. Cell Sci. 2019, 132, jcs232850. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef]
- Shrestha, N.; Reinert, R.B.; Qi, L. Endoplasmic Reticulum Protein Quality Control in β Cells. Semin. Cell Dev. Biol. 2020, 103, 59–67. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Shen, Y.; Sun, A.; Zhu, X.; Fang, S.; Shen, Y. The ubiquitin ligase Hrd1 promotes degradation of the Z variant alpha 1-antitrypsin and increases its solubility. Mol. Cell. Biochem. 2011, 346, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gnann, A.; Riordan, J.R.; Wolf, D.H. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol. Biol. Cell 2004, 15, 4125–4135. [Google Scholar] [CrossRef]
- Sato, T.; Sako, Y.; Sho, M.; Momohara, M.; Suico, M.A.; Shuto, T.; Nishitoh, H.; Okiyoneda, T.; Kokame, K.; Kaneko, M.; et al. STT3B-dependent posttranslational N-glycosylation as a surveillance system for secretory protein. Mol. Cell 2012, 47, 99–110. [Google Scholar] [CrossRef]
- Sha, H.; Sun, S.; Francisco, A.B.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.D.; Panhwar, M.S.; et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef]
- Tyler, R.E.; Pearce, M.M.; Shaler, T.A.; Olzmann, J.A.; Greenblatt, E.J.; Kopito, R.R. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol. Biol. Cell 2012, 23, 4668–4678. [Google Scholar] [CrossRef]
- Wei, J.; Chen, L.; Li, F.; Yuan, Y.; Wang, Y.; Xia, W.; Zhang, Y.; Xu, Y.; Yang, Z.; Gao, B.; et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. 2018, 37, e98942. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Sun, S.; Wang, H.; Liu, M.; Long, Q.; Yin, L.; Kersten, S.; Zhang, K.; Qi, L. Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 2018, 37, e99277. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Kim, H.; Yang, L.; Sha, H.; Roman, C.A.; Long, Q.; Qi, L. The Sel1L-Hrd1 Endoplasmic Reticulum-Associated Degradation Complex Manages a Key Checkpoint in B Cell Development. Cell Rep. 2016, 16, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kong, S.; Zhang, Y.; Melo-Cardenas, J.; Gao, B.; Zhang, Y.; Zhang, D.D.; Zhang, B.; Song, J.; Thorp, E.; et al. The endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in B cell development in mice. J. Biol. Chem. 2018, 293, 12934–12944. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qiu, Q.; Gao, B.; Kong, S.; Lin, Z.; Fang, D. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J. Exp. Med. 2014, 211, 2467–2479. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Löhr, F.; Wakatsuki, S.; Dötsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 2019, 29, 846–855. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Hölper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Müller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M. ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev. Cell 2021, 56, 949–966. [Google Scholar] [CrossRef]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef]
- Cunningham, C.N.; Williams, J.M.; Knupp, J.; Arunagiri, A.; Arvan, P.; Tsai, B. Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol. Cell 2019, 75, 442–456. [Google Scholar] [CrossRef]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef]
- Omari, S.; Makareeva, E.; Roberts-Pilgrim, A.; Mirigian, L.; Jarnik, M.; Ott, C.; Lippincott-Schwartz, J.; Leikin, S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E10099–E10108. [Google Scholar] [CrossRef]
- Fregno, I.; Fasana, E.; Bergmann, T.J.; Raimondi, A.; Loi, M.; Soldà, T.; Galli, C.; D’Antuono, R.; Morone, D.; Danieli, A.; et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018, 37, e99847. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Zielke, S.; Kardo, S.; Zein, L.; Mari, M.; Covarrubias-Pinto, A.; Kinzler, M.N.; Meyer, N.; Stolz, A.; Fulda, S.; Reggiori, F.; et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Horimoto, S.; Ninagawa, S.; Okada, T.; Koba, H.; Sugimoto, T.; Kamiya, Y.; Kato, K.; Takeda, S.; Mori, K. The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J. Biol. Chem. 2013, 288, 31517–31527. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Loi, M.; Molinari, M. Mechanistic insights in recov-ER-phagy: Micro-ER-phagy to recover from stress. Autophagy 2020, 16, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.L.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018, 9, 3671. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha(1)-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Alpha1-antitrypsin deficiency--a model for conformational diseases. N. Engl. J. Med. 2002, 346, 45–53. [Google Scholar] [CrossRef]
- Schaeffer, C.; Merella, S.; Pasqualetto, E.; Lazarevic, D.; Rampoldi, L. Mutant uromodulin expression leads to altered homeostasis of the endoplasmic reticulum and activates the unfolded protein response. PLoS ONE 2017, 12, e0175970. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Lucia, J.P.; Galgano, S.J.; Markowitz, G.S.; D’Agati, V.D. Uromodulin storage disease. Kidney Int. 2008, 73, 971–976. [Google Scholar] [CrossRef][Green Version]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Creemers, J.W.; Lee, Y.S.; Oliver, R.L.; Bahceci, M.; Tuzcu, A.; Gokalp, D.; Keogh, J.; Herber, S.; White, A.; O’Rahilly, S.; et al. Mutations in the amino-terminal region of proopiomelanocortin (POMC) in patients with early-onset obesity impair POMC sorting to the regulated secretory pathway. J. Clin. Endocrinol. Metab. 2008, 93, 4494–4499. [Google Scholar] [CrossRef]
- Ito, M.; Jameson, J.L.; Ito, M. Molecular basis of autosomal dominant neurohypophyseal diabetes insipidus. Cellular toxicity caused by the accumulation of mutant vasopressin precursors within the endoplasmic reticulum. J. Clin. Investig. 1997, 99, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yu, R.N.; Jameson, J.L. Mutant vasopressin precursors that cause autosomal dominant neurohypophyseal diabetes insipidus retain dimerization and impair the secretion of wild-type proteins. J. Biol. Chem. 1999, 274, 9029–9037. [Google Scholar] [CrossRef]
- Medeiros-Neto, G.A.; Knobel, M.; Cavaliere, H.; Simonetti, J.; Mattar, E. Hereditary congenital goitre with thyroglobulin deficiency causing hypothyroidism. Clin. Endocrinol. 1984, 20, 631–642. [Google Scholar] [CrossRef]
- Kim, P.S.; Kwon, O.Y.; Arvan, P. An endoplasmic reticulum storage disease causing congenital goiter with hypothyroidism. J. Cell Biol. 1996, 133, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xiong, Y.; Li, X.; Haataja, L.; Chen, W.; Mir, S.A.; Lv, L.; Madley, R.; Larkin, D.; Anjum, A.; et al. Role of Proinsulin Self-Association in Mutant INS Gene-Induced Diabetes of Youth. Diabetes 2020, 69, 954–964. [Google Scholar] [CrossRef]
- Wang, H.; Saint-Martin, C.; Xu, J.; Ding, L.; Wang, R.; Feng, W.; Liu, M.; Shu, H.; Fan, Z.; Haataja, L.; et al. Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations. Mol. Cell. Endocrinol. 2020, 518, 111025. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Duga, S. Hereditary Hypofibrinogenemia with Hepatic Storage. Int. J. Mol. Sci. 2020, 21, 7830. [Google Scholar] [CrossRef]
- Brennan, S.O.; Maghzal, G.; Shneider, B.L.; Gordon, R.; Magid, M.S.; George, P.M. Novel fibrinogen gamma375 Arg-->Trp mutation (fibrinogen aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology 2002, 36, 652–658. [Google Scholar] [CrossRef]
- Sorensen, S.; Ranheim, T.; Bakken, K.S.; Leren, T.P.; Kulseth, M.A. Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J. Biol. Chem 2006, 281, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kizhakkedath, P.; John, A.; Al-Sawafi, B.K.; Al-Gazali, L.; Ali, B.R. Endoplasmic reticulum quality control of LDLR variants associated with familial hypercholesterolemia. FEBS Open Biol. 2019, 9, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Kuivaniemi, H.; Tromp, G.; Prockop, D.J. Mutations in collagen genes: Causes of rare and some common diseases in humans. FASEB J. 1991, 5, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; San Antonio, J.D.; Milgrom, S.; Hyland, J.C.; Korkko, J.; Prockop, D.J.; De Paepe, A.; et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Kolářová, H.; Tesařová, M.; Švecová, Š.; Stránecký, V.; Přistoupilová, A.; Zima, T.; Uhrová, J.; Volgina, S.Y.; Zeman, J.; Honzík, T. Lipoprotein lipase deficiency: Clinical, biochemical and molecular characteristics in three patients with novel mutations in the LPL gene. Folia Biol. 2014, 60, 235–243. [Google Scholar]
- Rahalkar, A.R.; Giffen, F.; Har, B.; Ho, J.; Morrison, K.M.; Hill, J.; Wang, J.; Hegele, R.A.; Joy, T. Novel LPL mutations associated with lipoprotein lipase deficiency: Two case reports and a literature review. Can. J. Physiol. Pharmacol. 2009, 87, 151–160. [Google Scholar] [CrossRef]
- Karatas, E.; Bouchecareilh, M. Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int. J. Mol. Sci. 2020, 21, 1493. [Google Scholar] [CrossRef]
- Miyata, T.; Hagiwara, D.; Hodai, Y.; Miwata, T.; Kawaguchi, Y.; Kurimoto, J.; Ozaki, H.; Mitsumoto, K.; Takagi, H.; Suga, H.; et al. Degradation of Mutant Protein Aggregates within the Endoplasmic Reticulum of Vasopressin Neurons. iScience 2020, 23, 101648. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Allard, C.; Alvarez-Mercado, A.I.; Fuselier, T.; Kim, J.H.; Coons, L.A.; Hewitt, S.C.; Urano, F.; Korach, K.S.; Levin, E.R.; et al. Estrogens Promote Misfolded Proinsulin Degradation to Protect Insulin Production and Delay Diabetes. Cell Rep. 2018, 24, 181–196. [Google Scholar] [CrossRef]
- Asselta, R.; Robusto, M.; Braidotti, P.; Peyvandi, F.; Nastasio, S.; D’Antiga, L.; Perisic, V.N.; Maggiore, G.; Caccia, S.; Duga, S. Hepatic fibrinogen storage disease: Identification of two novel mutations (p.Asp316Asn, fibrinogen Pisa and p.Gly366Ser, fibrinogen Beograd) impacting on the fibrinogen γ-module. J. Thromb. Haemost. 2015, 13, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Venick, R.; Bhuta, S.; Li, X.; Wang, H.L. Hepatic Fibrinogen Storage Disease in a Patient with Hypofibrinogenemia: Report of a Case with a Missense Mutation of the FGA Gene. Semin. Liver Dis. 2015, 35, 439–443. [Google Scholar] [CrossRef]
- Sveger, T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N. Engl. J. Med. 1976, 294, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Ogushi, F.; Fells, G.A.; Hubbard, R.C.; Straus, S.D.; Crystal, R.G. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. J. Clin. Investig. 1987, 80, 1366–1374. [Google Scholar] [CrossRef]
- Tafaleng, E.N.; Chakraborty, S.; Han, B.; Hale, P.; Wu, W.; Soto-Gutierrez, A.; Feghali-Bostwick, C.A.; Wilson, A.A.; Kotton, D.N.; Nagaya, M.; et al. Induced pluripotent stem cells model personalized variations in liver disease resulting from α1-antitrypsin deficiency. Hepatology 2015, 62, 147–157. [Google Scholar] [CrossRef]
- Le, A.; Ferrell, G.A.; Dishon, D.S.; Le, Q.Q.; Sifers, R.N. Soluble aggregates of the human PiZ alpha 1-antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesis. J. Biol. Chem. 1992, 267, 1072–1080. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Miranda, E.; Perez, J.; Ekeowa, U.I.; Hadzic, N.; Kalsheker, N.; Gooptu, B.; Portmann, B.; Belorgey, D.; Hill, M.; Chambers, S.; et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology 2010, 52, 1078–1088. [Google Scholar] [CrossRef]
- Qu, D.; Teckman, J.H.; Omura, S.; Perlmutter, D.H. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem. 1996, 271, 22791–22795. [Google Scholar] [CrossRef] [PubMed]
- Teckman, J.H.; Burrows, J.; Hidvegi, T.; Schmidt, B.; Hale, P.D.; Perlmutter, D.H. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J. Biol. Chem. 2001, 276, 44865–44872. [Google Scholar] [CrossRef] [PubMed]
- Granell, S.; Baldini, G.; Mohammad, S.; Nicolin, V.; Narducci, P.; Storrie, B.; Baldini, G. Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function. Mol. Biol. Cell 2008, 19, 572–586. [Google Scholar] [CrossRef]
- Granell, S.; Baldini, G. Inclusion bodies and autophagosomes: Are ER-derived protective organelles different than classical autophagosomes? Autophagy 2008, 4, 375–377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmidt, B.Z.; Perlmutter, D.H. Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G444–G455. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell 2006, 17, 203–212. [Google Scholar] [CrossRef]
- Teckman, J.H.; Perlmutter, D.H. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G961–G974. [Google Scholar] [CrossRef]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef]
- Leon, C.; Bouchecareilh, M. The Autophagy Pathway: A Critical Route in the Disposal of Alpha 1-Antitrypsin Aggregates That Holds Many Mysteries. Int. J. Mol. Sci. 2021, 22, 1875. [Google Scholar] [CrossRef]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. 2010, 235, 700–709. [Google Scholar] [CrossRef]
- Tang, Y.; Fickert, P.; Trauner, M.; Marcus, N.; Blomenkamp, K.; Teckman, J. Autophagy induced by exogenous bile acids is therapeutic in a model of α-1-AT deficiency liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G156–G165. [Google Scholar] [CrossRef]
- Pastore, N.; Blomenkamp, K.; Annunziata, F.; Piccolo, P.; Mithbaokar, P.; Maria Sepe, R.; Vetrini, F.; Palmer, D.; Ng, P.; Polishchuk, E.; et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 2013, 5, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Burrows, J.A.; Willis, L.K.; Perlmutter, D.H. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc. Natl. Acad. Sci. USA 2000, 97, 1796–1801. [Google Scholar] [CrossRef] [PubMed]
- Castino, R.; Davies, J.; Beaucourt, S.; Isidoro, C.; Murphy, D. Autophagy is a prosurvival mechanism in cells expressing an autosomal dominant familial neurohypophyseal diabetes insipidus mutant vasopressin transgene. FASEB J. 2005, 19, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Murphy, D. Autophagy in hypothalamic neurones of rats expressing a familial neurohypophysial diabetes insipidus transgene. J. Neuroendocrinol. 2002, 14, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, D.; Arima, H.; Morishita, Y.; Wenjun, L.; Azuma, Y.; Ito, Y.; Suga, H.; Goto, M.; Banno, R.; Sugimura, Y.; et al. Arginine vasopressin neuronal loss results from autophagy-associated cell death in a mouse model for familial neurohypophysial diabetes insipidus. Cell Death Dis. 2014, 5, e1148. [Google Scholar] [CrossRef]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Ariav, Y.; Tirosh, B.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 2013, 62, 1227–1237. [Google Scholar] [CrossRef]
- Puls, F.; Goldschmidt, I.; Bantel, H.; Agne, C.; Bröcker, V.; Dämmrich, M.; Lehmann, U.; Berrang, J.; Pfister, E.D.; Kreipe, H.H.; et al. Autophagy-enhancing drug carbamazepine diminishes hepatocellular death in fibrinogen storage disease. J. Hepatol. 2013, 59, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.M.; Bichet, D.G. Molecular biology of hereditary diabetes insipidus. J. Am. Soc. Nephrol. 2005, 16, 2836–2846. [Google Scholar] [CrossRef]
- Spiess, M.; Friberg, M.; Beuret, N.; Prescianotto-Baschong, C.; Rutishauser, J. Role of protein aggregation and degradation in autosomal dominant neurohypophyseal diabetes insipidus. Mol. Cell. Endocrinol. 2020, 501, 110653. [Google Scholar] [CrossRef] [PubMed]
- Rittig, S.; Robertson, G.L.; Siggaard, C.; Kovács, L.; Gregersen, N.; Nyborg, J.; Pedersen, E.B. Identification of 13 new mutations in the vasopressin-neurophysin II gene in 17 kindreds with familial autosomal dominant neurohypophyseal diabetes insipidus. Am. J. Hum. Genet. 1996, 58, 107–117. [Google Scholar]
- Birk, J.; Friberg, M.A.; Prescianotto-Baschong, C.; Spiess, M.; Rutishauser, J. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J. Cell Sci. 2009, 122, 3994–4002. [Google Scholar] [CrossRef] [PubMed]
- Friberg, M.A.; Spiess, M.; Rutishauser, J. Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J. Biol. Chem. 2004, 279, 19441–19447. [Google Scholar] [CrossRef]
- Kimura, T.; Matsui, K.; Sato, T.; Yoshinaga, K. Mechanism of carbamazepine (Tegretol)-induced antidiuresis: Evidence for release of antidiuretic hormone and impaired excretion of a water load. J. Clin. Endocrinol. Metab. 1974, 38, 356–362. [Google Scholar] [CrossRef]
- Meinders, A.E.; Cejka, V.; Robertson, G.L. The antidiuretic action of carbamazepine in man. Clin. Sci. Mol. Med. 1974, 47, 289–299. [Google Scholar] [CrossRef]
- Wales, J.K. Treatment of diabetes insipidus with carbamazepine. Lancet 1975, 2, 948–951. [Google Scholar] [CrossRef]
- Castino, R.; Thepparit, C.; Bellio, N.; Murphy, D.; Isidoro, C. Akt induces apoptosis in neuroblastoma cells expressing a C98X vasopressin mutant following autophagy suppression. J. Neuroendocrinol. 2008, 20, 1165–1175. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Hagiwara, D.; Miyata, T.; Hodai, Y.; Kurimoto, J.; Takagi, H.; Suga, H.; Kobayashi, T.; Sugiyama, M.; Onoue, T.; et al. Endoplasmic reticulum chaperone BiP/GRP78 knockdown leads to autophagy and cell death of arginine vasopressin neurons in mice. Sci. Rep. 2020, 10, 19730. [Google Scholar] [CrossRef]
- Hagiwara, D.; Grinevich, V.; Arima, H. A novel mechanism of autophagy-associated cell death of vasopressin neurons in familial neurohypophysial diabetes insipidus. Cell Tissue Res. 2019, 375, 259–266. [Google Scholar] [CrossRef]
- Dodson, G.; Steiner, D. The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. [Google Scholar] [CrossRef]
- Cunningham, C.N.; He, K.; Arunagiri, A.; Paton, A.W.; Paton, J.C.; Arvan, P.; Tsai, B. Chaperone-Driven Degradation of a Misfolded Proinsulin Mutant in Parallel With Restoration of Wild-Type Insulin Secretion. Diabetes 2017, 66, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Haataja, L.; Wright, J.; Wickramasinghe, N.P.; Hua, Q.X.; Phillips, N.F.; Barbetti, F.; Weiss, M.A.; Arvan, P. Mutant INS-gene induced diabetes of youth: Proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE 2010, 5, e13333. [Google Scholar] [CrossRef]
- Hodish, I.; Liu, M.; Rajpal, G.; Larkin, D.; Holz, R.W.; Adams, A.; Liu, L.; Arvan, P. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J. Biol. Chem. 2010, 285, 685–694. [Google Scholar] [CrossRef]
- Yoshioka, M.; Kayo, T.; Ikeda, T.; Koizumi, A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997, 46, 887–894. [Google Scholar] [CrossRef]
- Wang, J.; Takeuchi, T.; Tanaka, S.; Kubo, S.K.; Kayo, T.; Lu, D.; Takata, K.; Koizumi, A.; Izumi, T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Investig. 1999, 103, 27–37. [Google Scholar] [CrossRef]
- Renner, S.; Braun-Reichhart, C.; Blutke, A.; Herbach, N.; Emrich, D.; Streckel, E.; Wünsch, A.; Kessler, B.; Kurome, M.; Bähr, A.; et al. Permanent neonatal diabetes in INS(C94Y) transgenic pigs. Diabetes 2013, 62, 1505–1511. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Allen, J.R.; Nguyen, L.X.; Sargent, K.E.; Lipson, K.L.; Hackett, A.; Urano, F. High ER stress in beta-cells stimulates intracellular degradation of misfolded insulin. Biochem. Biophys. Res. Commun. 2004, 324, 166–170. [Google Scholar] [CrossRef]
- Riahi, Y.; Israeli, T.; Yeroslaviz, R.; Chimenez, S.; Avrahami, D.; Stolovich-Rain, M.; Alter, I.; Sebag, M.; Polin, N.; Bernal-Mizrachi, E.; et al. Inhibition of mTORC1 by ER stress impairs neonatal β-cell expansion and predisposes to diabetes in the Akita mouse. eLife 2018, 7, e38472. [Google Scholar] [CrossRef]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 2012, 61, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Gorasia, D.G.; Dudek, N.L.; Safavi-Hemami, H.; Perez, R.A.; Schittenhelm, R.B.; Saunders, P.M.; Wee, S.; Mangum, J.E.; Hubbard, M.J.; Purcell, A.W. A prominent role of PDIA6 in processing of misfolded proinsulin. Biochim. Biophys. Acta 2016, 1864, 715–723. [Google Scholar] [CrossRef]
- Oyadomari, S.; Yun, C.; Fisher, E.A.; Kreglinger, N.; Kreibich, G.; Oyadomari, M.; Harding, H.P.; Goodman, A.G.; Harant, H.; Garrison, J.L.; et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 2006, 126, 727–739. [Google Scholar] [CrossRef]
- Ladiges, W.C.; Knoblaugh, S.E.; Morton, J.F.; Korth, M.J.; Sopher, B.L.; Baskin, C.R.; MacAuley, A.; Goodman, A.G.; LeBoeuf, R.C.; Katze, M.G. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005, 54, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, B.; Liu, L.; Gan, Q.; Liu, X.; Chen, L.; Chen, L. Hepatic fibrinogen storage disease and hypofibrinogenemia caused by fibrinogen Aguadilla mutation: A case report. J. Int. Med. Res. 2020, 48, 300060519898033. [Google Scholar] [CrossRef] [PubMed]
- Rubbia-Brandt, L.; Neerman-Arbez, M.; Rougemont, A.L.; Malé, P.J.; Spahr, L. Fibrinogen gamma375 arg-->trp mutation (fibrinogen aguadilla) causes hereditary hypofibrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am. J. Surg. Pathol. 2006, 30, 906–911. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Schoenecker, J.G.; Flick, M.J. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019, 133, 511–520. [Google Scholar] [CrossRef]
- Liu, X.; Shi, B. Progress in research on the role of fibrinogen in lung cancer. Open Life Sci. 2020, 15, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Neerman-Arbez, M.; de Moerloose, P. Heterogeneity of congenital afibrinogenemia, from epidemiology to clinical consequences and management. Blood Rev. 2020, 100793. [Google Scholar] [CrossRef]
- Laurens, N.; Koolwijk, P.; de Maat, M.P. Fibrin structure and wound healing. J. Thromb. Haemost. 2006, 4, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Arai, S.; Nagaya, H.; Mizuguchi, J.; Wada, I. Stepwise assembly of fibrinogen is assisted by the endoplasmic reticulum lectin-chaperone system in HepG2 cells. PLoS ONE 2013, 8, e74580. [Google Scholar] [CrossRef]
- Roy, S.; Sun, A.; Redman, C. In vitro assembly of the component chains of fibrinogen requires endoplasmic reticulum factors. J. Biol. Chem. 1996, 271, 24544–24550. [Google Scholar] [CrossRef]
- Lord, S.T. Fibrinogen and fibrin: Scaffold proteins in hemostasis. Curr. Opin. Hematol. 2007, 14, 236–241. [Google Scholar] [CrossRef]
- de Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital fibrinogen disorders: An update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef]
- Brennan, S.O.; Wyatt, J.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma284 Gly-->Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Brennan, S.O.; Davis, R.L.; Conard, K.; Savo, A.; Furuya, K.N. Novel fibrinogen mutation γ314Thr→Pro (fibrinogen AI duPont) associated with hepatic fibrinogen storage disease and hypofibrinogenaemia. Liver Int. 2010, 30, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Giovannoni, I.; Sari, S.; Aksu, A.U.; Esendagly, G.; Dalgic, B.; Boldrini, R.; Akyol, G.; Francalanci, P.; Bellacchio, E. A novel fibrinogen gamma chain mutation (c.1096C>G; p.His340Asp), fibrinogen Ankara, causing hypofibrinogenaemia and hepatic storage. Pathology 2017, 49, 534–537. [Google Scholar] [CrossRef]
- Dib, N.; Quelin, F.; Ternisien, C.; Hanss, M.; Michalak, S.; De Mazancourt, P.; Rousselet, M.C.; Calès, P. Fibrinogen angers with a new deletion (gamma GVYYQ 346-350) causes hypofibrinogenemia with hepatic storage. J. Thromb. Haemost. 2007, 5, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Ogiwara, N.; Mukai, S.; Takezawa, Y.; Sugano, M.; Honda, T.; Okumura, N. The fibrous form of intracellular inclusion bodies in recombinant variant fibrinogen-producing cells is specific to the hepatic fibrinogen storage disease-inducible variant fibrinogen. Int. J. Hematol. 2017, 105, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Suh, T.T.; Holmbäck, K.; Jensen, N.J.; Daugherty, C.C.; Small, K.; Simon, D.I.; Potter, S.; Degen, J.L. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995, 9, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Le Fourn, V.; Park, S.; Jang, I.; Gaplovska-Kysela, K.; Guhl, B.; Lee, Y.; Cho, J.W.; Zuber, C.; Roth, J. Large protein complexes retained in the ER are dislocated by non-COPII vesicles and degraded by selective autophagy. Cell. Mol. Life Sci. 2013, 70, 1985–2002. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Redman, C. The degradation of nascent fibrinogen chains is mediated by the ubiquitin proteasome pathway. Biochem. Biophys. Res. Commun. 1999, 261, 590–597. [Google Scholar] [CrossRef]
- Kruse, K.B.; Dear, A.; Kaltenbrun, E.R.; Crum, B.E.; George, P.M.; Brennan, S.O.; McCracken, A.A. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: An explanation for liver disease. Am. J. Pathol. 2006, 168, 1299–1308. [Google Scholar] [CrossRef]
- Verdile, V.; De Paola, E.; Paronetto, M.P. Aberrant Phase Transitions: Side Effects and Novel Therapeutic Strategies in Human Disease. Front. Genet. 2019, 10, 173. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Alberti, S. Protein Phase Separation as a Stress Survival Strategy. Cold Spring Harb. Perspect. Biol. 2019, 11, a034058. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020, 30, 672–675. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disorder | Protein | Genetic | Pathagenic Variants | Tissue Mutation | Symptoms (Retention) | Ref. |
|---|---|---|---|---|---|---|
| α1-antitrypsin deficiency (AATD) | α1-antitrypsin | Over 100 | Recessive | Liver | Hepatic fibrosis Pulmonary emphysema Plasma AAT shortage | [151,171] |
| Tubulointerstitial kidney disease (ADTKD) | Uromodulin | Over 100 | Dominant | Kidney | Gout Hyperuricaemia Low uromodulin | [153,154] |
| Obesity due to proopiomelanocortin deficiency | Proopiomelanocortin | 8 * | Recessive | Hypothalamus | Hyperphagia Insulin resistance Low plasma α-MSH | [155,156] |
| Congenital hypothyroid goiter with thyroglobulin deficiency | Thyroglobulin | Over 40 | Recessive | Thyroid | Deficient thyroid hormone | [159,160] |
| Mutant INS-gene-induced diabetes of youth (MIDY) | Proinsulin | 30 * | Dominant | Pancreas | Hyperglycemia Insulin deficiency | [161,162] |
| Familial hypercholesterolemia (FH) | Low-density lipoprotein receptor | Over 2000 | Dominant | Liver # | Tendon xanthomas Hypercholesterolemia Premature coronary heart disease | [165,166] |
| Osteogenesis imperfecta (OI) | Procollagen | Over 1500 | Dominant | Bone | Osteopenia Multiple fracture Skeletal malformation Short stature | [167,168] |
| Familial lipoprotein lipase decifiency (LPLD) | Lipoprotein lipase | Over 100 | Recessive | Adipose Muscle | Eruptive xanthomas Hypertriglyceridaemia Hepatosplenomegaly Recurrent abdominal pain | [169,170] |
| Familial neurohypophyseal diabetes insipidus (FNDI) | Vasopressin prohormone | Over 70 | Dominant | Hypothalamus | Polyuria Polydipsia Serum AVP deficiency | [172,173] |
| Hepatic fibrinogen storage disease (HFSD) | Fibrinogen | 8 | Dominant | Liver | Hemorrhage Hypofibrinogenemia | [174,175] |
| DisorderTreatment | Model | Mechanisms | Effects | Reference | |
| AATD | Carbamazepine | Z-AAT mice | Promotes Z-AAT aggregate degradation Raises autophagy activity | Liver fibrosis ↓ | [192] |
| Rapamycin | Z-AAT mice | Decreases hepatic Z-AAT accumulation Enhances autophagy activity | Liver fibrosis ↓ | [193] | |
| Norursodeoxycholic acid | Z-AAT mice | Reduces intrahepatic Z-AAT aggregates Boosts autophagic activation | Liver injury ↓ Hepatocellular death ↓ | [194] | |
| TFEB overexpression | Z-AAT mice | Enhances Z-AAT polymer degradation Increases autophagy flux Inhibits Z-AAT expression | Liver fibrosis ↓ Hepatic apoptosis ↓ | [195] | |
| 4-phenylbutyric acid | Z-AAT mice | Augments Z-AAT secretion Facilitates protein folding | Blood AAT level ↑ | [196] | |
| FNDI | Rapamycin | FNDI mice | Reduces mutant pro-AVP aggregates Increases autophagy flux | Unclear | [172] |
| Carbamazepine | Patients with FNDI | Unclear | Urine volume ↓ Urine osmolality ↑ | [197,198,199] | |
| MIDY | Estrogen | Akita mice | Increases Akita proinsulin degradation Stabilizes SEL1L-HRD1 ERAD | Hyperglycemia ↓ Insulin secretion ↑ | [173] |
| Rapamycin | Akita mice | Decreases ER stress Boosts autophagy flux | Hyperglycemia ↓ Insulin production ↑ β-cell apoptosis ↓ | [200] | |
| HFSD | Carbamazepine | Patients with HFSD | Enhances autophagy | Liver damages ↓ ALT and AST levels ↓ | [201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Sun, S. Protein Aggregation in the ER: Calm behind the Storm. Cells 2021, 10, 3337. https://doi.org/10.3390/cells10123337
Li H, Sun S. Protein Aggregation in the ER: Calm behind the Storm. Cells. 2021; 10(12):3337. https://doi.org/10.3390/cells10123337
Chicago/Turabian StyleLi, Haisen, and Shengyi Sun. 2021. "Protein Aggregation in the ER: Calm behind the Storm" Cells 10, no. 12: 3337. https://doi.org/10.3390/cells10123337
APA StyleLi, H., & Sun, S. (2021). Protein Aggregation in the ER: Calm behind the Storm. Cells, 10(12), 3337. https://doi.org/10.3390/cells10123337