
Tissue-Nonspecific Alkaline Phosphatase, a Possible Mediator of Cell Maturation: Towards a New Paradigm

 ,
,  , , ,
, , ,
Abstract
1. General Property of Alkaline Phosphatase (ALP)
2. ALP Isoforms and Their Detailed Properties
2.1. PLALP
2.2. IALP
2.3. GCALP
2.4. EAP (in Mice)
2.5. TNSALP
2.5.1. Creation of Akp2 KI or KO Mice
2.5.2. Creation of Conditional Akp2 KO Mice
2.5.3. Creation of Akp2 Mutant Mice through N-Ethyl-N-Nitrosourea (ENU) Mutagenesis
2.5.4. Creation of Tg Mice Overexpressing TNSALP
3. Various Biological Roles of TNSALP
3.1. Osteogenic System
3.2. Lipid and Energy Metabolism of Fat Cells
3.3. Neuronal System
3.4. Immune System
3.5. Vascular Calcification
3.6. Role of TNSALP in Renal Cell Carcinoma (RCC)
3.7. Role of TNSALP Fibroblastic-Like Cell Lines
3.8. Role of TNSALP in the Hair-Inductive Capacity of Human Dermal Papilla Spheres
3.9. TNSALP May Be Involved in Premature Bone Aging
3.10. Mitochondrial TNSALP Controls Thermogenesis
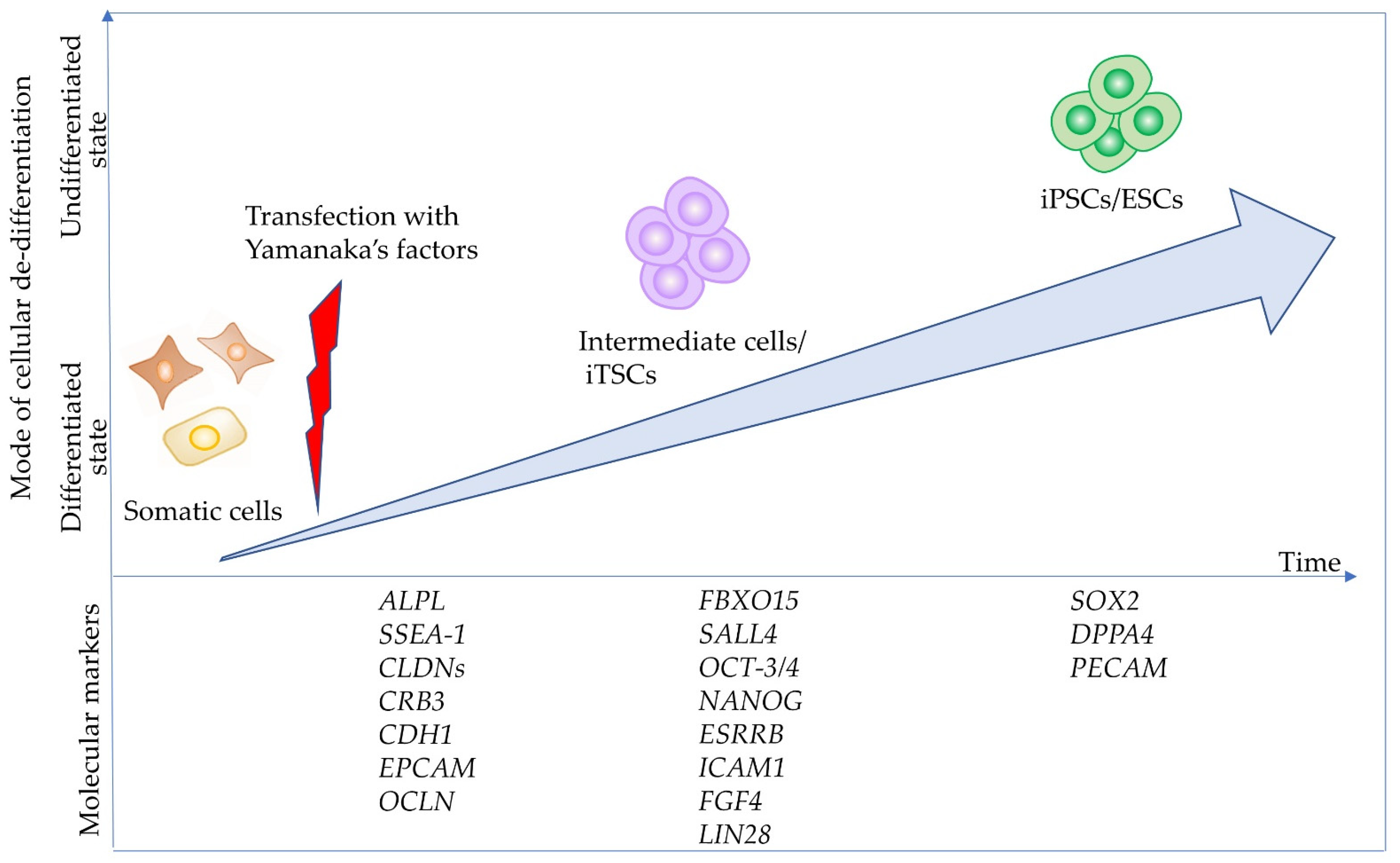
4. Possible Involvement of TNSALP in Differentiation and Maintenance in Juvenile State
4.1. TNSALP as an Early Marker Expressed in Intermediate Cells towards iPSCs
4.2. Possible Involvement of Wnt/β-Catenin Signaling Pathway in Generation of iPSCs
4.3. Possible Involvement of BMP-2 Signaling Pathway in the Generation of iPSCs
5. TNSALP as a Signal Regulator
5.1. TNSALP May Be Involved in Lineage Switching
5.2. Overexpression or Suppression of ALPL May Affect the Expression of Some Genes
5.3. Expression of ALPL May Be Affected by Some Genes
6. Therapeutic Aspect of TNSALP
7. ALP as Possible New Tools for Cell Research
7.1. Usefulness of Live Staining Kit for Isolating Live ALP-Expressing Cells without Fixation
7.2. Usefulness of Ecto-Alkaline Phosphatase-Mediated System to Eliminate ALP-Positive Cells
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Millán, J.L. Alkaline Phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Fedde, K.N.; Lane, C.C.; Whyte, M.P. Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch. Biochem. Biophys. 1988, 264, 400–409. [Google Scholar] [CrossRef]
- Say, J.C.; Ciuffi, K.; Furriel, R.P.; Ciancaglini, P.; Leone, F.A. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta 1991, 1074, 256–262. [Google Scholar] [CrossRef]
- Buchet, R.; Millán, J.L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol. Biol. 2013, 1053, 27–51. [Google Scholar] [CrossRef]
- Mulnard, J.; Huygens, R. Ultrastructural localization of non-specific alkaline phosphatase during cleavage and blastocyst formation in the mouse. J. Embryol. Exp. Morphol. 1978, 44, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, S.; Hofmann, M.C.; Ziomek, C.A.; Millán, J.L. Embryonic alkaline phosphatase is expressed at M-phase in the spermatogenic lineage of the mouse. Development 1992, 116, 159–165. [Google Scholar] [CrossRef]
- Pease, S.; Braghetta, P.; Gearing, D.; Grail, D.; Williams, R.L. Isolation of embryonic stem (ES) cells in media supplemented with recombinant leukemia inhibitory factor (LIF). Dev. Biol. 1990, 141, 344–352. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Heath, J.K. Characterization of a xenogeneic antiserum raised against the fetal germ cells of the mouse: Cross-reactivity with embryonal carcinoma cells. Cell 1978, 15, 299–306. [Google Scholar] [CrossRef]
- Wang, P.; Suo, L.-J.; Shang, H.; Li, Y.; Li, G.-X.; Li, Q.-W.; Hu, J.-H. Differentiation of spermatogonial stem cell-like cells from murine testicular tissue into haploid male germ cells in vitro. Cytotechnology 2014, 66, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Braun, N.; Shukla, V.; Füllgrabe, M.; Schomerus, C.; Korf, H.W.; Gachet, C.; Ikehara, Y.; Sévigny, J.; Robson, S.C.; et al. Extracellular nucleotide signaling in adult neural stem cells: Synergism with growth factor-mediated cellular proliferation. Development 2006, 133, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef]
- Rico, L.G.; Juncà, J.; Ward, M.D.; Bradford, J.; Petriz, J. Is alkaline phosphatase the smoking gun for highly refractory primitive leukemic cells? Oncotarget 2016, 7, 72057–72066. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Salles, J.P. Hypophosphatasia: Biological and clinical aspects, avenues for therapy. Clin. Biochem. Rev. 2020, 41, 13–27. [Google Scholar] [CrossRef]
- Villa-Suárez, J.M.; García-Fontana, C.; Andújar-Vera, F.; González-Salvatierra, S.; de Haro-Muñoz, T.; Contreras-Bolívar, V.; García-Fontana, B.; Muñoz-Torres, M. Hypophosphatasia: A unique disorder of bone mineralization. Int. J. Mol. Sci. 2021, 22, 4303. [Google Scholar] [CrossRef]
- Sharma, U.; Pal, D.; Prasad, R. Alkaline phosphatase: An overview. Indian J. Clin. Biochem. 2014, 29, 269–278. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 5313–5329. [Google Scholar]
- Linder, C.H.; Englund, U.H.; Narisawa, S.; Millán, J.L.; Magnusson, P. Isozyme profile and tissue-origin of alkaline phosphatases in mouse serum. Bone 2013, 53, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Skynner, M.J.; Drage, D.J.; Dean, W.L.; Turner, S.; Watt, D.J.; Allen, N.D. Transgenic mice ubiquitously expressing human placental alkaline phosphatase (PLAP): An additional reporter gene for use in tandem with beta-galactosidase (lacZ). Int. J. Dev. Biol. 1999, 43, 85–90. [Google Scholar]
- DePrimo, S.E.; Stambrook, P.J.; Stringer, J.R. Human placental alkaline phosphatase as a histochemical marker of gene expression in transgenic mice. Transgenic Res. 1996, 5, 459–466. [Google Scholar] [CrossRef]
- Narisawa, S.; Huang, L.; Iwasaki, A.; Hasegawa, H.; Alpers, D.H.; Millán, J.L. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol. Cell Biol. 2003, 23, 7525–7530. [Google Scholar] [CrossRef] [PubMed]
- Brun, L.R.; Lombarte, M.; Roma, S.; Perez, F.; Millán, J.L.; Rigalli, A. Increased calcium uptake and improved trabecular bone properties in intestinal alkaline phosphatase knockout mice. J. Bone Min. Metab. 2018, 36, 661–667. [Google Scholar] [CrossRef]
- Narisawa, S.; Smans, K.A.; Avis, J.; Hoylaerts, M.F.; Millán, J.L. Transgenic mice expressing the tumor marker germ cell alkaline phosphatase: An in vivo tumor model for human cancer antigens. Proc. Natl. Acad. Sci. USA 1993, 90, 5081–5085. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, S.; Fröhlander, N.; Millán, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Dehghani, H.; Narisawa, S.; Millán, J.L.; Hahnel, A.C. Effects of disruption of the embryonic alkaline phosphatase gene on preimplantation development of the mouse. Dev. Dyn. 2000, 217, 440–448. [Google Scholar] [CrossRef]
- MacGregor, G.R.; Zambrowicz, B.P.; Soriano, P. Tissue non-specific alkaline phosphatase is expressed in both embryonic and extraembryonic lineages during mouse embryogenesis but is not required for migration of primordial germ cells. Development 1995, 121, 1487–1496. [Google Scholar] [CrossRef]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; Coburn, S.P. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Gámez-Belmonte, R.; Ocón, B.; Martínez-Moya, P.; Wirtz, S.; Sánchez de Medina, F.; Martínez-Augustin, O. Tissue non-specific alkaline phosphatase expression is needed for the full stimulation of T cells and T cell-dependent colitis. J. Crohns Colitis 2017, 11, 857–870. [Google Scholar] [CrossRef][Green Version]
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millán, J.L.; MacGregor, G.R.; et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026. [Google Scholar] [CrossRef]
- Hanics, J.; Barna, J.; Xiao, J.; Millán, J.L.; Fonta, C.; Négyessy, L. Ablation of TNAP function compromises myelination and synaptogenesis in the mouse brain. Cell Tissue Res. 2012, 349, 459–471. [Google Scholar] [CrossRef]
- Lomelí, H.; Ramos-Mejía, V.; Gertsenstein, M.; Lobe, C.G.; Nagy, A. Targeted insertion of Cre recombinase into the TNAP gene: Excision in primordial germ cells. Genesis 2000, 26, 116–117. [Google Scholar] [CrossRef]
- Narisawa, S. Genetically modified mice for studying TNAP function. Subcell. Biochem. 2015, 76, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millán, J.L.; Savinova, O.V. Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J. Am. Heart Assoc. 2015, 4, e002499. [Google Scholar] [CrossRef]
- Hahnel, A.C.; Rappolee, D.A.; Millán, J.L.; Manes, T.; Ziomek, C.A.; Theodosiou, N.G.; Werb, Z.; Pedersen, R.A.; Schultz, G.A. Two alkaline phosphatase genes are expressed during early development in the mouse embryo. Development 1990, 110, 555–564. [Google Scholar] [CrossRef]
- Manes, T.; Glade, K.; Ziomek, C.A.; Millán, J.L. Genomic structure and comparison of mouse tissue-specific alkaline phosphatase genes. Genomics 1990, 8, 541–554. [Google Scholar] [CrossRef]
- Lynes, M.; Narisawa, S.; Millán, J.L.; Widmaier, E.P. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1738–R1747. [Google Scholar] [CrossRef]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Weiss, M.J.; Henthorn, P.S.; Lafferty, M.A.; Slaughter, C.; Raducha, M.; Harris, H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 7182–7186. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Tashiro, K.; Hattori, M.; Sakaki, Y.; Ikehara, Y. Primary structure of rat liver alkaline phosphatase deduced from its cDNA. Biochem. J. 1988, 249, 661–668. [Google Scholar] [CrossRef]
- Abdul, R.B.; Shafquat, A.; Mohammad, T. Differentially expressed three non-coding alternate exons at 5’ UTR of regulatory type I beta subunit gene of mouse. Mol. Biol. Rep. 2012, 3375–3383. [Google Scholar] [CrossRef]
- Matsuura, S.; Kishi, F.; Kajii, T. Characterization of a 5’-flanking region of the human liver/bone/kidney alkaline phosphatase gene: Two kinds of mRNA from a single gene. Biochem. Biophys. Res. Commun. 1990, 168, 993–1000. [Google Scholar] [CrossRef]
- Toh, Y.; Yamamoto, M.; Endo, H.; Misumi, Y.; Ikehara, Y. Sequence divergence of 5’ extremities in rat liver alkaline phosphatase mRNAs. J. Biochem. 1989, 105, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Zernik, J.; Thiede, M.A.; Twarog, K.; Stover, M.L.; Rodan, G.A.; Upholt, W.B.; Rowe, D.W. Cloning and analysis of the 5’ region of the rat bone/liver/kidney/placenta alkaline phosphatase gene. A dual-function promoter. Matrix 1990, 10, 38–47. [Google Scholar] [CrossRef]
- Terao, M.; Studer, M.; Gianní, M.; Garattini, E. Isolation and characterization of the mouse liver/bone/kidney-type alkaline phosphatase gene. Biochem. J. 1990, 268, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kotobuki, N.; Matsushima, A.; Kato, Y.; Kubo, Y.; Hirose, M.; Ohgushi, H. Small interfering RNA of alkaline phosphatase inhibits matrix mineralization. Cell Tissue Res. 2008, 332, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Lobe, C.G.; Koop, K.E.; Kreppner, W.; Lomeli, H.; Gertsenstein, M.; Nagy, A. Z/AP, a double reporter for cre-mediated recombination. Dev. Biol. 1999, 208, 281–292. [Google Scholar] [CrossRef]
- Acevedo-Arozena, A.; Wells, S.; Potter, P.; Kelly, M.; Cox, R.D.; Brown, S.D.M. ENU mutagenesis, a way forward to understand gene function. Annu. Rev. Genom. Hum. Genet. 2008, 9, 49–69. [Google Scholar] [CrossRef]
- Stottmann, R.; Beier, D.R. ENU mutagenesis in the mouse. Curr. Protoc. Hum. Genet. 2014, 82, 15.4.1–15.4.10. [Google Scholar] [CrossRef]
- Hough, T.A.; Polewski, M.; Johnson, K.; Cheeseman, M.; Nolan, P.M.; Vizor, L.; Rastan, S.; Boyde, A.; Pritzker, K.; Hunter, A.J.; et al. Novel mouse model of autosomal semidominant adult hypophosphatasia has a splice site mutation in the tissue nonspecific alkaline phosphatase gene AkpJ. Bone Miner. Res. 2007, 22, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Aigner, B.; Rathkolb, B.; Klaften, M.; Sedlmeier, R.; Klempt, M.; Wagner, S.; Michel, D.; Mayer, U.; Klopstock, T.; de Angelis, M.H. Generation of N-ethyl-N-nitrosourea-induced mouse mutants with deviations in plasma enzyme activities as novel organ-specific disease models. Exp. Physiol. 2009, 94, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sabrautzki, S.; Rubio-Aliaga, I.; Hans, W.; Fuchs, H.; Rathkolb, B.; Calzada-Wack, J.; Cohrs, C.M.; Klaften, M.; Seedorf, H.; Eck, S. New mouse models for metabolic bone diseases generated by genome-wide ENU mutagenesis. Mamm. Genome 2012, 23, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.L.; Sheen, C.R.; Hatch, N.E.; Liu, J.; Cory, E.; Narisawa, S.; Kiffer-Moreira, T.; Sah, R.L.; Whyte, M.P.; Somerman, M.J.; et al. Periodontal defects in the A116T knock-in murine model of odontohypophosphatasia. J. Dent. Res. 2015, 94, 706–714. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; Liu, S.; Liao, L.; Jin, F.; Shi, S.; Jin, Y. Alpl prevents bone ageing sensitivity by specifically regulating senescence and differentiation in mesenchymal stem cells. Bone Res. 2018, 6, 27. [Google Scholar] [CrossRef] [PubMed]
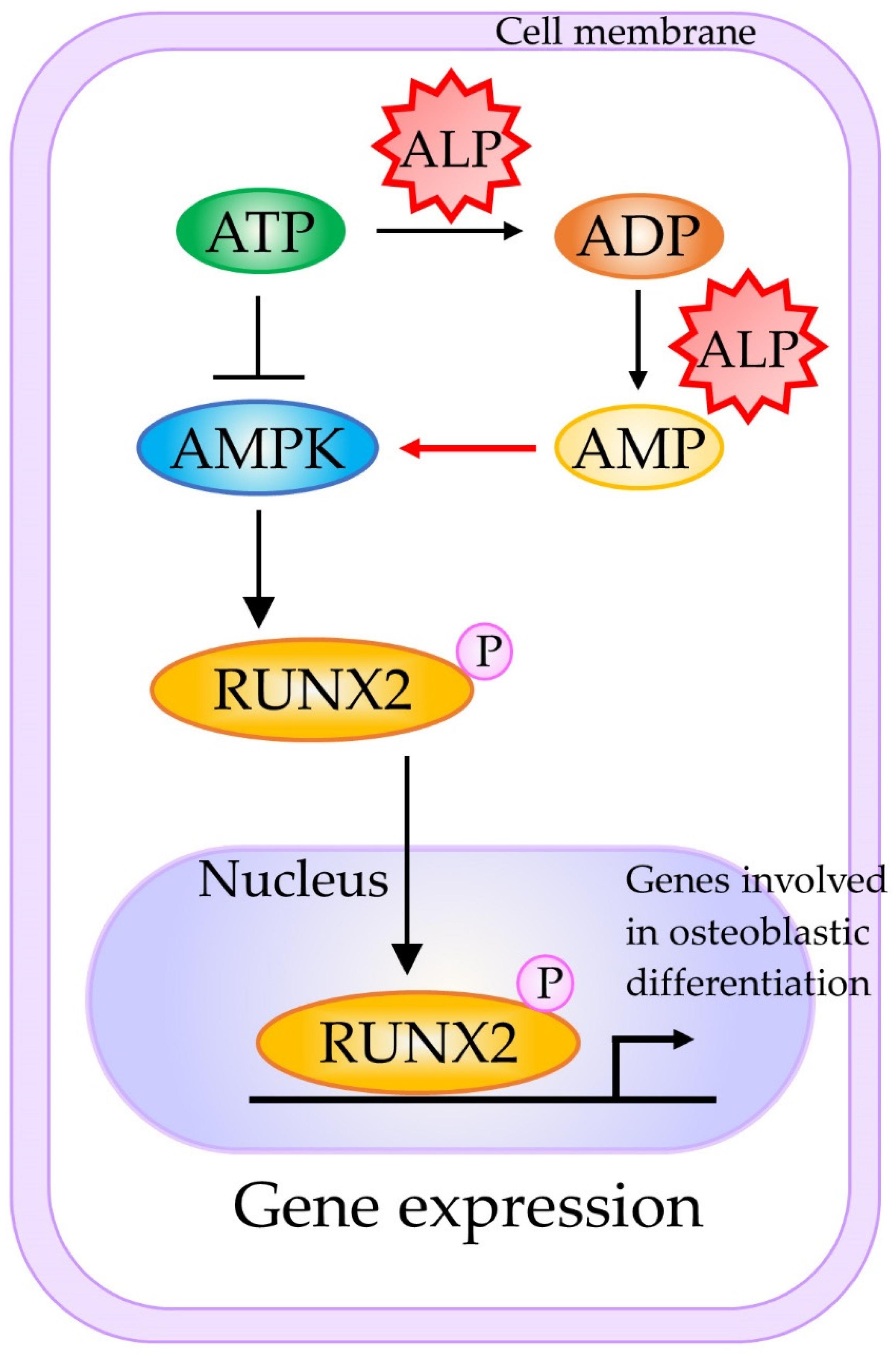
- Chava, S.; Chennakesavulu, S.; Gayatri, B.M.; Reddy, A.B.M. A novel phosphorylation by AMP-activated kinase regulates RUNX2 from ubiquitination in osteogenesis over adipogenesis. Cell Death Dis. 2018, 9, 754. [Google Scholar] [CrossRef]
- Hernández-Mosqueira, C.; Velez-delValle, C.; Kuri-Harcuch, W. Tissue alkaline phosphatase is involved in lipid metabolism and gene expression and secretion of adipokines in adipocytes. Biochim. Biophys. Acta 2015, 1850, 2485–2496. [Google Scholar] [CrossRef]
- Kermer, V.; Ritter, M.; Albuquerque, B.; Leib, C.; Stanke, M.; Zimmermann, H. Knockdown of tissue non-specific alkaline phosphatase impairs neural stem cell proliferation and differentiation. Neurosci. Lett. 2010, 485, 208–211. [Google Scholar] [CrossRef]
- Graser, S.; Mentrup, B.; Schneider, D.; Klein-Hitpass, L.; Jakob, F.; Hofmann, C. Overexpression of tissue-nonspecific alkaline phosphatase increases the expression of neurogenic differentiation markers in the human SH-SY5Y neuroblastoma cell line. Bone 2015, 79, 150–161. [Google Scholar] [CrossRef]
- García-Rozas, C.; Plaza, A.; Díaz-Espada, F.; Kreisler, M.; Martínez-Alonso, C. Alkaline phosphatase activity as a membrane marker for activated B cells. J. Immunol. 1982, 129, 52–55. [Google Scholar]
- Marquez, C.; Toribio, M.L.; Marcos, M.A.; de la Hera, A.; Barcena, A.; Pezzi, L.; Martinez, C. Expression of alkaline phosphatase in murine B lymphocytes. Correlation with B cell differentiation into Ig secretion. J. Immunol. 1989, 142, 3187–3192. [Google Scholar] [PubMed]
- Bauer, J.; Kachel, V. The increase of electrophoretic mobility and alkaline phosphatase activity are parallel events during B-cell maturation. Immunol. Invest. 1990, 19, 57–68. [Google Scholar] [CrossRef]
- Palit, S.; Kendrick, J. Vascular calcification in chronic kidney disease: Role of disordered mineral metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020, cvaa299. [Google Scholar] [CrossRef] [PubMed]
- Andleeb, H.; Hussain, M.; Ejaz, S.A.; Sevigny, J.; Farman, M.; Yasinzai, M.; Zhang, J.; Iqbal, J.; Hameed, S. Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h-TNAP): A therapeutic target against vascular calcification. Bioorganic Chem. 2020, 101, 103999. [Google Scholar] [CrossRef]
- Pinkerton, A.B.; Sergienko, E.; Bravo, Y.; Dahl, R.; Ma, C.-T.; Jackson, M.R.; Cosford, N.D.P.; Millán, J.L. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 31–34. [Google Scholar] [CrossRef]
- Sharma, U.; Pal, D.; Singh, S.K.; Kakkar, N.; Prasad, R. Reduced L/B/K alkaline phosphatase gene expression in renal cell carcinoma: Plausible role in tumorigenesis. Biochimie 2014, 104, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Hui, M.Z.; Sukhu, B.; Tenenbaum, H.C. Expression of tissue non-specific alkaline phosphatase stimulates differentiated behaviour in specific transformed cell populations. Anat. Rec. 1996, 244, 423–436. [Google Scholar] [CrossRef]
- Kwack, M.H.; Jang, Y.J.; Won, G.H.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Overexpression of alkaline phosphatase improves the hair-inductive capacity of cultured human dermal papilla spheres. J. Dermatol. Sci. 2019, 95, 126–129. [Google Scholar] [CrossRef]
- Najar, M.; Crompot, E.; van Grunsven, L.A.; Dollé, L.; Lagneaux, L. Aldehyde dehydrogenase activity in adipose tissue: Isolation and gene expression profile of distinct sub-population of mesenchymal stromal cells. Stem Cell Rev. Rep. 2018, 14, 599–611. [Google Scholar] [CrossRef]
- Sun, Y.; Rahbani, J.F.; Jedrychowski, M.P.; Riley, C.L.; Vidoni, S.; Bogoslavski, D.; Hu, B.; Dumesic, P.A.; Zeng, X.; Wang, A.B.; et al. Mitochondrial TNAP controls thermogenesis by hydrolysis of phosphocreatine. Nature 2021, 593, 580–585. [Google Scholar] [CrossRef]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jawnisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef]
- O’Connor, M.D.; Kardel, M.D.; Iosfina, I.; Youssef, D.; Lu, M.; Li, M.M.; Vercauteren, S.; Nagy, A.; Eaves, C. Alkaline phosphatase-positive colony formation is a sensitive, specific, and quantitative indicator of undifferentiated human embryonic stem cells. Stem Cells 2008, 26, 1109–1116. [Google Scholar] [CrossRef]
- David, L.; Polode, J.M. Phases of reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kopp, W.; Wu, G.; Heising, S.; Greber, B.; Stehling, M.; Araúzo-Bravo, M.J.; Boerno, S.T.; Timmermann, B.; Vingron, M.; et al. Esrrb unlocks silenced enhancers for reprogramming to naive pluripotency. Cell Stem Cell 2018, 23, 266–275.e6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
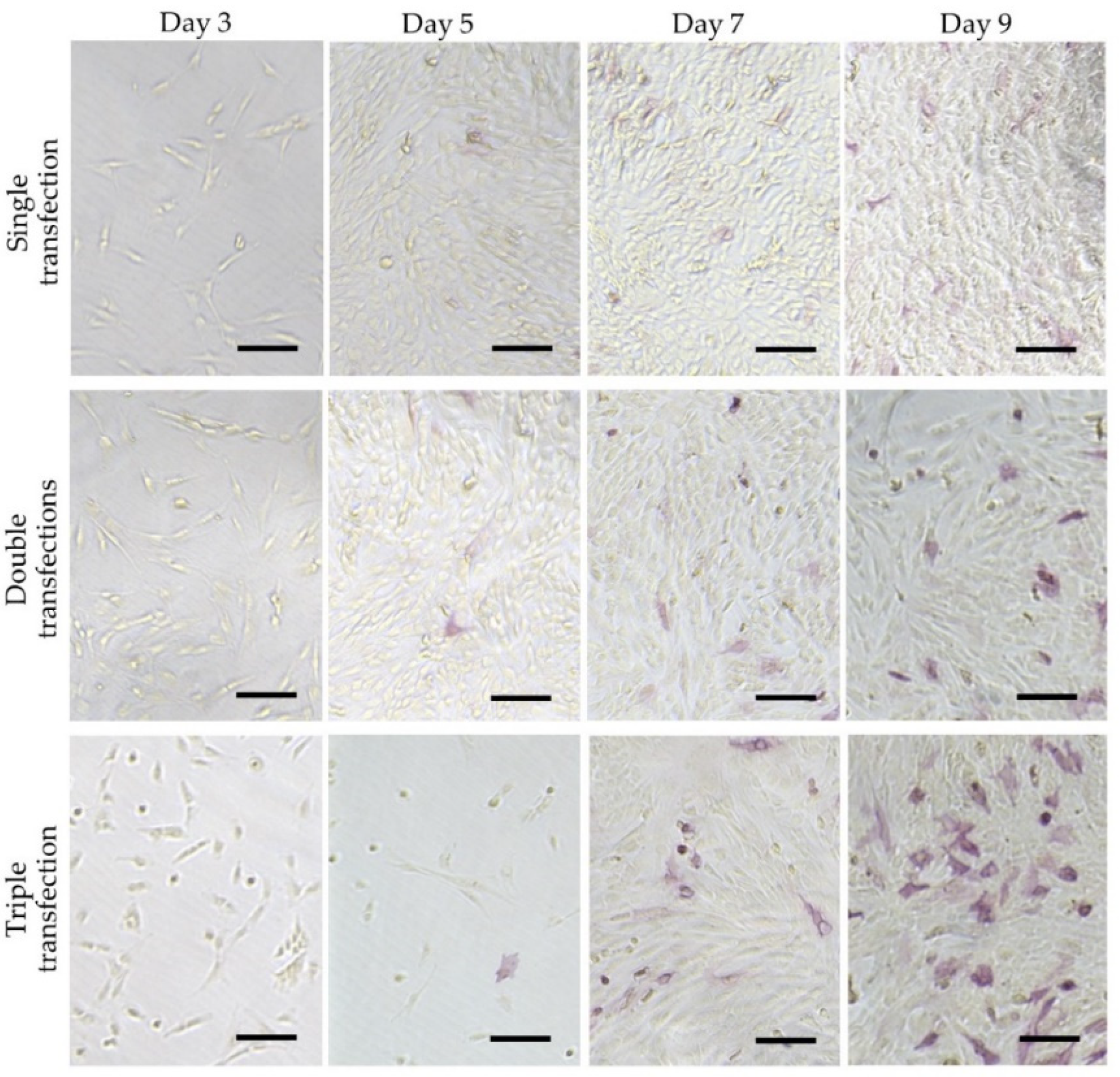
- Inada, E.; Saitoh, I.; Kubota, N.; Murakami, T.; Soda, M.; Matsueda, K.; Murakami, T.; Sawami, T.; Kagoshima, A.; Yamasaki, Y. Alkaline phosphatase and OCT-3/4 as useful markers for predicting susceptibility of human deciduous teeth-derived dental pulp cells to reprogramming factor-induced iPS cells. J. Investig. Clin. Dent. 2017, 8. [Google Scholar] [CrossRef]
- Soda, M.; Saitoh, I.; Murakami, T.; Inada, E.; Iwase, Y.; Noguchi, H.; Shibasaki, S.; Kurosawa, M.; Sawami, T.; Terunuma, M.; et al. Repeated human deciduous tooth-derived dental pulp cell reprogramming factor transfection yields multipotent intermediate cells with enhanced iPS cell formation capability. Sci. Rep. 2019, 9, 1490. [Google Scholar] [CrossRef]
- Štefková, K.; Procházková, J.; Pacherník, J. Alkaline phosphatase in stem cells. Stem Cells Int. 2015, 628368. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed]
- Wodarz, A.; Nusse, R. Mechanisms of Wnt signaling in development. Annu. Rev. Cell Dev. Biol. 1998, 14, 59–88. [Google Scholar] [CrossRef]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef] [PubMed]
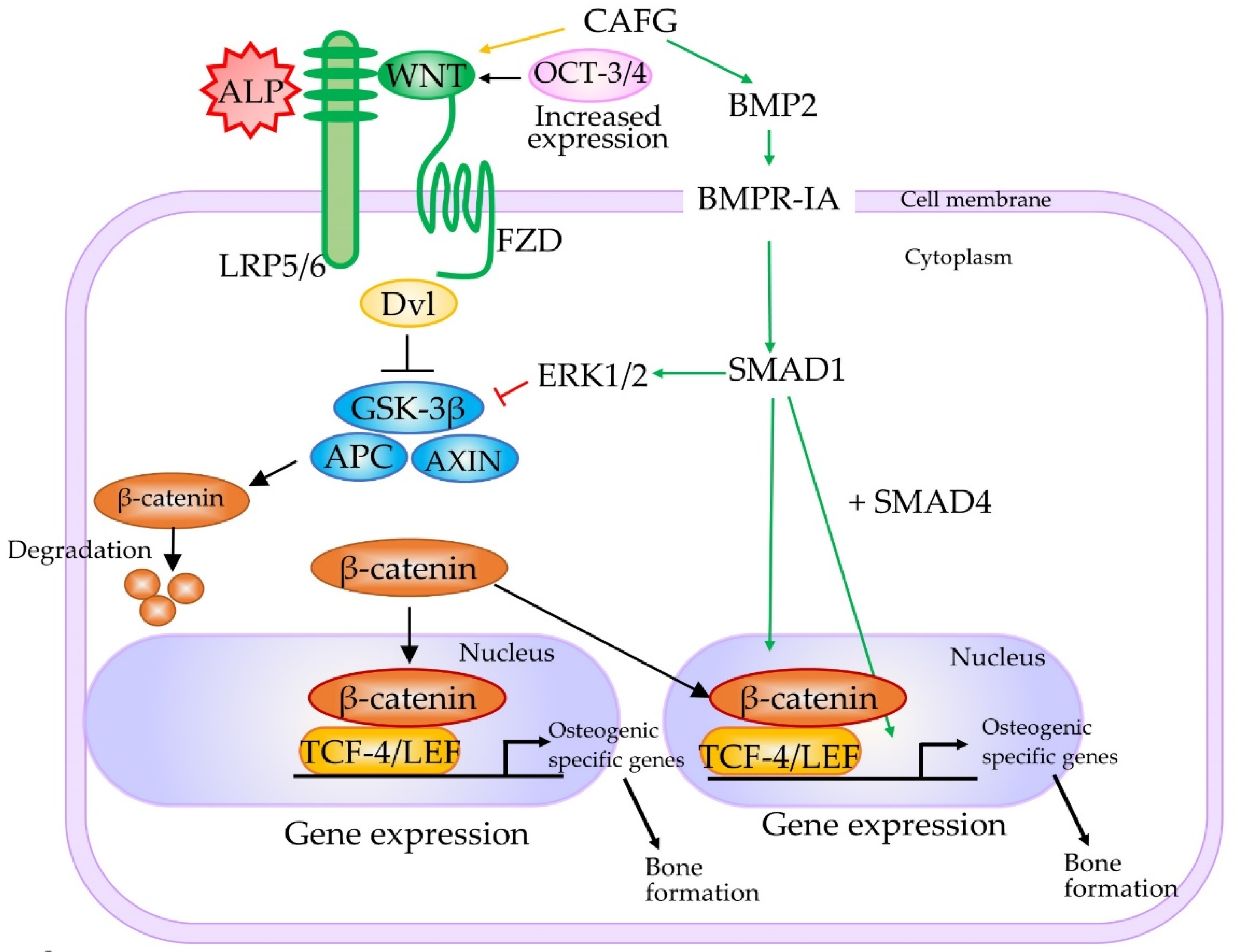
- Liu, W.; Zhang, L.; Xuan, K.; Hu, C.; Li, L.; Zhang, Y.; Jin, F.; Jin, Y. Alkaline phosphatase controls lineage switching of mesenchymal stem cells by regulating the LRP6/GSK3β complex in hypophosphatasia. Theranostics 2018, 8, 5575–5592. [Google Scholar] [CrossRef]
- Kimura, M.; Nakajima-Koyama, M.; Lee, J.; Nishida, E. Transient expression of WNT2 promotes somatic cell reprogramming by inducing β-catenin nuclear accumulation. Stem Cell Rep. 2016, 6, 834–843. [Google Scholar] [CrossRef] [PubMed]
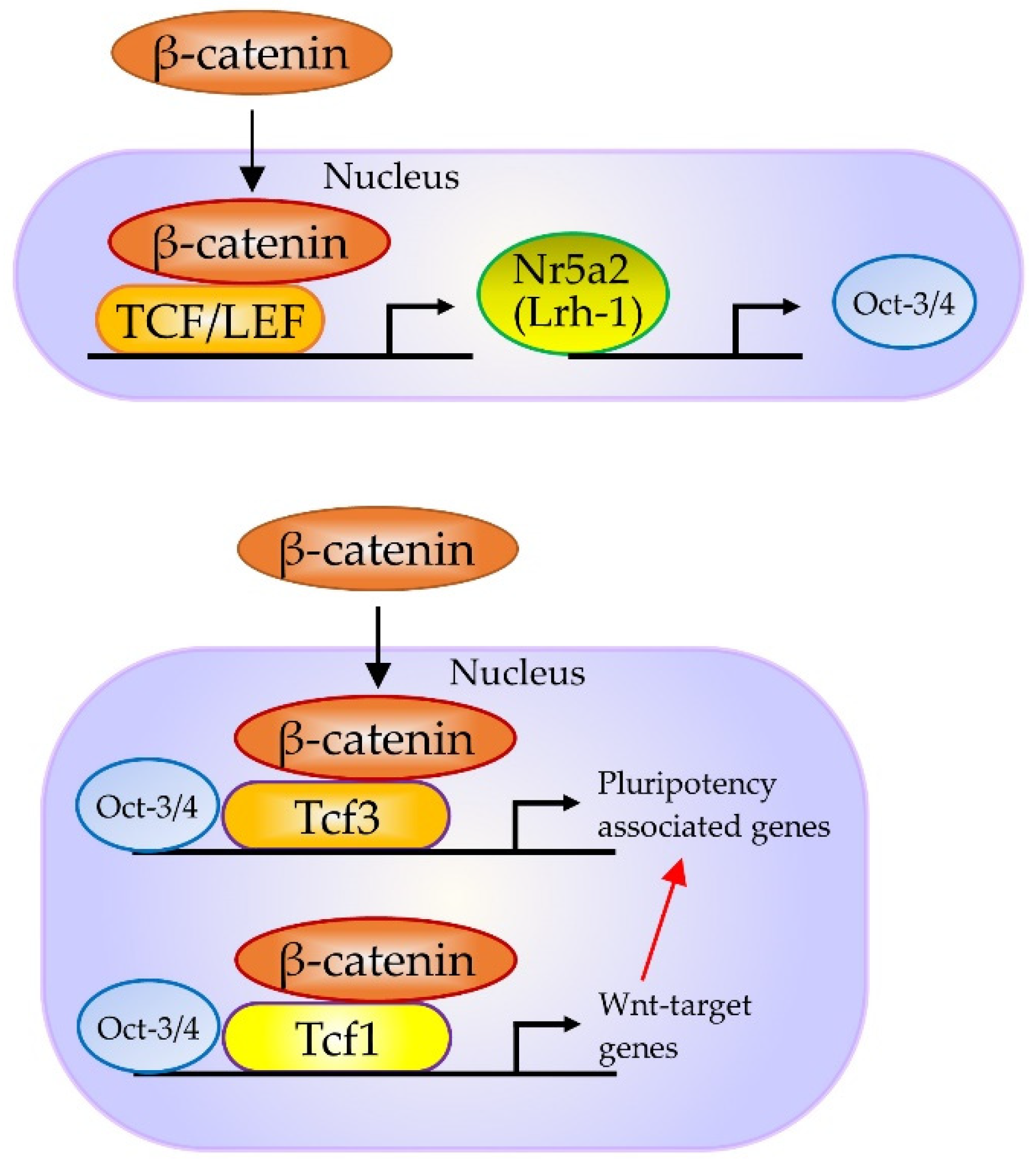
- Kelly, K.F.; Ng, D.Y.; Jayakumaran, G.; Wood, G.A.; Koide, H.; Doble, B.W. β-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 2011, 8, 214–227. [Google Scholar] [CrossRef]
- Wagner, R.; Xu, X.; Yi, F.; Merrill, B.; Cooney, A. Canonical Wnt/ß-catenin regulation of liver receptor homolog-1 mediates pluripotency gene expression. Stem Cells 2010, 28, 1794–1804. [Google Scholar] [CrossRef]
- Heng, J.; Feng, B.; Han, J.; Jiang, J.; Kraus, P.; Orlov, Y.; Huss, M.; Yang, L.; Lufkin, T.; Lim, B.; et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell 2010, 6, 167–174. [Google Scholar] [CrossRef]
- Tanaka, S.S.; Kojima, Y.; Yamaguchi, Y.L.; Nishinakamura, R.; Tam, P.P. Impact of WNT signaling on tissue lineage differentiation in the early mouse embryo. Dev. Growth Differ. 2011, 53, 843–856. [Google Scholar] [CrossRef]
- Si, W.; Kang, Q.; Luu, H.H.; Park, J.K.; Luo, Q.; Song, W.-X.; Jiang, W.; Luo, X.; Li, X.; Yin, H.; et al. CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol. Cell Biol. 2006, 26, 2955–2964. [Google Scholar] [CrossRef]
- Sakisaka, Y.; Tsuchiya, M.; Nakamura, T.; Tamura, M.; Shimauchi, H.; Nemoto, E. Wnt5a attenuates Wnt3a-induced alkaline phosphatase expression in dental follicle cells. Exp. Cell Res. 2015, 336, 85–93. [Google Scholar] [CrossRef]
- Zhao, X.-E.; Yang, Z.; Gao, Z.; Ge, J.; Wei, Q.; Ma, B. 6-Bromoindirubin-3′-oxime promotes osteogenic differentiation of canine BMSCs through inhibition of GSK3β activity and activation of the Wnt/β-catenin signaling pathway. An. Acad. Bras. Cienc. 2019, 91, e20180459. [Google Scholar] [CrossRef]
- Katagiri, T.; Yamaguchi, A.; Ikeda, T.; Yoshiki, S.; Wozney, J.M.; Rosen, V.; Wang, E.A.; Tanaka, H.; Omura, S.; Suda, T. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein-2. Biochem. Biophys. Res. Commun. 1990, 172, 295–299. [Google Scholar] [CrossRef]
- Takuwa, Y.; Ohse, C.; Wang, E.A.; Wozney, J.M.; Yamashita, K. Bone morphogenetic protein-2 stimulates alkaline phosphatase activity and collagen synthesis in cultured osteoblastic cells, MC3T3-E. Biochem. Biophys. Res. Commun. 1991, 174, 96–101. [Google Scholar] [CrossRef]
- Rawadi, G.; Vayssiere, B.; Dunn, F.; Baron, R.; Roman-Roman, S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Miner. Res. 2003, 18, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.N.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef]
- Zhang, P.; Chang, W.-H.; Fong, B.; Gao, F.; Liu, C.; Al Alam, D.; Bellusci, S.; Lu, W. Regulation of induced pluripotent stem (iPS) cell induction by Wnt/β-catenin signaling. J. Biol. Chem. 2014, 289, 9221–9232. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, Q.; He, C.; Li, F.; Sheng, H.; Shen, X.; Zhang, X.; Zhu, S.; Chen, H.; Chen, X.; et al. Activation of the Wnt pathway through Wnt2 promotes metastasis in pancreatic cancer. Am. J. Cancer Res. 2014, 4, 537–544. [Google Scholar]
- Katsube, Y.; Kotobuki, N.; Tadokoro, M.; Kanai, R.; Taketani, T.; Yamaguchi, S.; Ohgushi, H. Restoration of cellular function of mesenchymal stem cells from a hypophosphatasia patient. Gene Ther. 2010, 17, 494–502. [Google Scholar] [CrossRef]
- Nakamura, T.; Nakamura-Takahashi, A.; Kasahara, M.; Yamaguchi, A.; Azuma, T. Tissue-nonspecific alkaline phosphatase promotes the osteogenic differentiation of osteoprogenitor cells. Biochem. Biophys. Res. Commun. 2020, 524, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Kiledjian, M.; Kadesch, T. Analysis of the human liver/bone/kidney alkaline phosphatase promoter in vivo and in vitro. Nucleic Acids Res. 1990, 18, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Sugimoto, T.; Kanzawa, M.; Chihara, K. Identification of an enhancer sequence in 5′-flanking region of 1A exon of mouse liver/bone/kidney-type alkaline phosphatase gene. IUBMB Life 1998, 44, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, E.; Takahashi-Yanaga, F.; Miwa, Y.; Hirata, M.; Watanabe, Y.; Sato, N.; Morimoto, S.; Hirofuji, T.; Maeda, K.; Sasaguri, T. Differentiation-inducing factor-1 alters canonical Wnt signaling and suppresses alkaline phosphatase expression in osteoblast-like cell lines. J. Bone Miner. Res. 2006, 21, 1307–1316. [Google Scholar] [CrossRef]
- Hatta, M.; Daitoku, H.; Matsuzaki, H.; Deyama, Y.; Yoshimura, Y.; Suzuki, K.; Matsumoto, A.; Fukamizu, A. Regulation of alkaline phosphatase promoter activity by forkhead transcription factor FKHR. Int. J. Mol. Med. 2002, 9, 147–152. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Lee, M.-H.; Wozney, J.M.; Cho, J.-Y.; Ryoo, H.-M. Bone morphogenetic protein-2-induced alkaline phosphatase expression is stimulated by Dlx5 and repressed by Msx2. J. Biol. Chem. 2004, 279, 50773–50780. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.; Patel, P.J.; Gleicher, S.; Amer, K.; Himelman, E.; Goel, S.; Moran, E. p107-dependent recruitment of SWI/SNF to the alkaline phosphatase promoter during osteoblast differentiation. Bone 2014, 69, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Yusa, N.; Watanabe, K.; Yoshida, S.; Shirafuji, N.; Shimomura, S.; Tani, K.; Asano, S.; Sato, N. Transcription factor Sp3 activates the liver/bone/kidney-type alkaline phosphatase promoter in hematopoietic cells. J. Leukoc. Biol. 2000, 68, 772–777. [Google Scholar] [CrossRef]
- Lammers, W.J.; van Buuren, H.R.; Hirschfield, G.M.; Janssen, H.L.A.; Invernizzi, P.; Mason, A.L.; Ponsioen, C.Y.; Floreani, A.; Corpechot, C.; Mayo, M.J.; et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirhosis: An international follow-up study. Gastroenterology 2014, 147, 1338–1349.e5. [Google Scholar] [CrossRef]
- Saraç, F.; Saygılı, F. Causes of high bone alkaline phosphatase. Biotechnol. Biotechnol. Equip. 2007, 21, 194–197. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr. Rev. 1994, 15, 439–461. [Google Scholar] [CrossRef]
- Rader, B.A. Alkaline phosphatase, an unconventional immune protein. Front. Immunol. 2017, 8, 897. [Google Scholar] [CrossRef]
- Whyte, M.P.; Wenkert, D.; Zhang, F. Hypophosphatasia: Natural history study of 101 affected children investigated at one research center. Bone 2016, 93, 125–138. [Google Scholar] [CrossRef]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Asfotase Alfa in perinatal/infantile-onset and juvenile-onset hypophosphatasia: A guide to its use in the USA. BioDrugs 2016, 30, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Rush, E.T.; Arundel, P.; Bishop, N.; Dahir, K.; Fraser, W.; Harmatz, P.; Linglart, A.; Munns, C.F.; Nunes, M.E.; et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol. Genet. Metab. 2017, 122, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Hypophosphatasia: Enzyme replacement therapy brings new opportunities and new challenges. J. Bone Miner. Res. 2017, 32, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Nakano, C.; Kitabatake, Y.; Takeyari, S.; Ohata, Y.; Kubota, T.; Taketani, K.; Kogo, M.; Ozono, K. Genetic correction of induced pluripotent stem cells mediated by transcription activator-like effector nucleases targeting ALPL recovers enzyme activity and calcification in vitro. Mol. Genet. Metab. 2019, 127, 158–165. [Google Scholar] [CrossRef]
- Yamamoto, S.; Orimo, H.; Matsumoto, T.; Iijima, O.; Narisawa, S.; Maeda, T.; Millán, J.L.; Shimada, T. Prolonged survival and phenotypic correction of Akp2−/− hypophosphatasia mice by lentiviral gene therapy. J. Bone Miner. Res. 2011, 26, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Miyake, K.; Yamamoto, S.; Orimo, H.; Miyake, N.; Odagaki, Y.; Adachi, K.; Iijima, O.; Narisawa, S.; Millán, J.L.; et al. Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Hum. Gene Ther. 2011, 22, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Takahashi, A.; Miyake, K.; Watanabe, A.; Hirai, Y.; Iijima, O.; Miyake, N.; Adachi, K.; Nitahara-Kasahara, Y.; Kinoshita, H.; Noguchi, T.; et al. Treatment of hypophosphatasia by muscle-directed expression of bone-targeted alkaline phosphatase via self-complementary AAV8 vector. Mol. Ther. Methods Clin. Dev. 2016, 3, 15059. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Mohamed, F.F.; de Oliveira, F.A.; Narisawa, S.; Miyake, K.; Foster, B.L.; Millán, J.L. Gene therapy using adeno-associated virus serotype 8 encoding TNAP-D10 improves the skeletal and dentoalveolar phenotypes in Alpl−/− mice. J. Bone Miner. Res. 2021, 36, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.K.; Pinzón, C.; Huggins, S.; Pryor, J.H.; Falck, A.; Herman, F.; Oldeschulte, J.; Chavez, M.B.; Foster, B.L.; White, S.H.; et al. Genetic engineering a large animal model of human hypophosphatasia in sheep. Sci. Rep. 2018, 8, 16945. [Google Scholar] [CrossRef] [PubMed]
- Takinami, H.; Goseki-Sone, M.; Watanabe, H.; Orimo, H.; Hamatani, R.; Fukushi-Irie, M.; Ishikawa, I. The mutant (F310L and V365I) tissue-nonspecific alkaline phosphatase gene from hypophosphatasia. J. Med. Dent. Sci. 2004, 51, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Panetta, N.J.; Gupta, D.M.; Wilson, K.D.; Lee, A.; Jia, F.; Hu, S.; Cherry, A.M.; Robbins, R.C.; Longaker, M.T.; et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15720–15725. [Google Scholar] [CrossRef]
- Singh, U.; Quintanilla, R.H.; Grecian, S.; Gee, K.R.; Rao, M.S.; Lakshmipathy, U. Novel live alkaline phosphatase substrate for identification of pluripotent stem cells. Stem Cell Rev. Rep. 2012, 8, 1021–1029. [Google Scholar] [CrossRef]
- Kuang, Y.; Miki, K.; Parr, C.J.C.; Hayashi, K.; Takei, I.; Li, J.; Iwasaki, M.; Nakagawa, M.; Yoshida, Y.; Saito, H. Efficient, selective removal of human pluripotent stem cells via ecto-alkaline phosphatase-mediated aggregation of synthetic peptides. Cell Chem. Biol. 2017, 24, 685–694.e4. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Accession Numbers | Protein Names | Tissue Distribution | Function |
|---|---|---|---|---|
| Human genes ALPL | NM_000478 | TNAP, TNSALP, L/B/K ALP | Developing nervous system, skeletal tissues, liver and kidney | Skeletal mineralization |
| ALPP | NM_001632 | PLAP, PLALP, Regan isoenzyme | Syncytiotrophoblast, a variety of tumors | Unknown |
| ALPP2 | NM_031313 | GCAP, GCALP, NAGAO isozyme | Testis, malignant trophoblasts, testicular cancer | Unknown |
| ALP1 | NM_001631 | IAP, IALP, Kasahara isoenzyme | Gut | Fat absorption, Detoxification of lipopolysaccharide |
| Mouse genes Akp2 (Alpl) | NM_007431 | TNAP, TNSALP, L/B/K ALP | Developing nervous system, skeletal tissues and kidney | Skeletal mineralization |
| Akp3 | NM_007432 | IAP, IALP, dIALP | Gut | Fat absorption, detoxification of lipopolysaccharide |
| Akp5 (Alpl2) | NM_007433 | EAP | Preimplantation embryo, testis, gut | Early embryogenesis |
| Akp-ps1 | NG 001340 | ALP pseudogene, pseudoALP | Not described | |
| Akp6 (Alpi) | AK008000 | gIALP | Gut | Under investigation |
| Methods | Target Gene | Outcome (Note) | Reference |
|---|---|---|---|
| Overexpression | ALPP | Tg mice overexpressing human PLALP systemically were produced. There were no adverse effects on mouse development or viability. | [20,21] |
| Gene targeting for KO | ALP1 | Homozygous mutants (called Akp3−/−) were histologically normal and fertile. However, accelerated transport of fat droplets through the intestinal epithelium and elevation of serum triglyceride levels were discernible, which was associated with higher intestinal Ca2+ uptake. | [22,23] |
| Overexpression | ALPP2 | Tg mouse lines harboring human ALPP2 linked to short or long promoter region of ALPP2 were produced. A 450 bp promoter sequence directs GCALP expression to the intestine and endothelial cells, while a 1.7 kb promoter sequence directs GCALP expression to the spermatogenic lineage and to the eight-cell embryos through the blastocysts. | [24] |
| Gene targeting for KO | Akp5 (Alpl2) | No obvious phenotypic abnormalities. Normal reproductive activity with acquisition of live offspring, indicating the nonessential role of EAP during embryonic development. | [25] |
| Gene targeting for KO | Akp5 (Alpl2) | In homozygous mutants (called EAP.ko), preimplantation embryos exhibited slower development and higher rates of degeneration, delayed parturition, and reduced litter size. | [26] |
| Gene targeting for KI | ALPL | In homozygous mutants (called Alpltm1Sor), primordial germ cells appear unaffected indicating that ALPL is not essential for their development or migration. At first, the mice exhibited normal skeletal development; however, homozygous mutant mice developed seizures and apnea at approximately two weeks after birth, and died before weaning. Rescued animals subsequently develop defective dentition. TNSALP modulates T lymphocyte function (specifically T cell-dependent colitis) when examined using heterozygous Alpltm1Sor mice. | [27,28,29] |
| Gene targeting for KO | ALPL | In homozygous mutants (called Alpltm1Jlm or Akp2−/−), abnormal bone mineralization was evident. Morphological changes in the osteoblasts, aberrant development of the lumbar nerve roots, disturbances in intestinal physiology, increased apoptosis in the thymus, and abnormal spleens are also discernible. Loss of ALPL causes myelin abnormalities and synaptic dysfunction. | [25,30,31] |
| Gene targeting for KI | ALPL | KI of Cre expression unit was carried out into the region between exons 6 and 7 of ALPL gene. The resulting line was called Alpltm1(cre)Nagy. After crossed Alpltm1(cre)Nagy line with the double-reporter line, Z/AP, human ALP expression is discernible in PGCs at E9.5–10.5. After mid-gestation, however, it was also expressed in the labyrinthine region of the placenta, the intestine and the neural tube. | [32] |
| Gene targeting for KI | ALPL | KI of an around 12 kb genomic sequence of Akp2 in which two loxP sites (located in introns 2 and 4, respectively) and a cassette containing neomycin resistance gene expression unit into the endogenous Akp2 locus. This floxed mouse (called Alplflox/flox) is normal in the absence of Cre expression. However, in the presence of Cre, the deletion of exons 3 and 4 should occur, which may result in the ablation of endogenous TNSALP expression. | [33] |
| Overexpression | ALPL | Tg mouse line (called “Col1a1-Tnap”) expressing human ALPL under control of an osteoblast-specific collagen type I α1 chain (Col1a1) promoter was produced. This line is healthy and exhibits increased bone mineralization. | [34] |
| Overexpression | ALPL | Tg mice carrying human ALPL under the vascular smooth muscle cell-specific transgelin (Tagln) promoter were produced. They developed severe arterial medial calcification and reduced viability. | [35] |
| Overexpression | ALPL | Tg mice (celled “Endothelial TNSALP mice”) carrying ALPL under the endothelial-specific tunica intima endothelial kinase 2 (Tie2) promoter were produced. They survived well into adulthood and displayed generalized arterial calcification together with elevated blood pressure and compensatory left ventricular hypertrophy. | [36] |
| Method 1 | Type of Cells Used | Genes Upregulated | Genes Downregulated | Genes Unaltered | References |
|---|---|---|---|---|---|
| siRNA | High-ALP-expressing cells derived from human osteoblast-like cells (HOS) | COL1A1 RUNX2 | OCN | [48] | |
| siRNA | 3T3-F442A adipocytes | GAPDH ADIPOQ FABP4 | LEP | [58] | |
| Revamisol | T3-F442A adipocytes | PPARG LEP ADIPOQ ATGL | TIP47 SCARB1 | [58] | |
| Revamisol | Murine osteoblast precursor cells | RUNX2 SP7 BGLAP2 DMP1 | [71] | ||
| Overexpression | Tg mice overexpressing ALPL gene | OPN BMP2 MGP SOX9 SLC20A1 | ACTA2 TAGLN | ANK | [35] |
| Overexpression | Dermal papilla | BMP4 AXIN2 VCAN LEF1 SOX2 | BMP2 NOGFGF7 | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, M.; Saitoh, I.; Kiyokawa, Y.; Iwase, Y.; Kubota, N.; Ibano, N.; Noguchi, H.; Yamasaki, Y.; Inada, E. Tissue-Nonspecific Alkaline Phosphatase, a Possible Mediator of Cell Maturation: Towards a New Paradigm. Cells 2021, 10, 3338. https://doi.org/10.3390/cells10123338
Sato M, Saitoh I, Kiyokawa Y, Iwase Y, Kubota N, Ibano N, Noguchi H, Yamasaki Y, Inada E. Tissue-Nonspecific Alkaline Phosphatase, a Possible Mediator of Cell Maturation: Towards a New Paradigm. Cells. 2021; 10(12):3338. https://doi.org/10.3390/cells10123338
Chicago/Turabian StyleSato, Masahiro, Issei Saitoh, Yuki Kiyokawa, Yoko Iwase, Naoko Kubota, Natsumi Ibano, Hirofumi Noguchi, Youichi Yamasaki, and Emi Inada. 2021. "Tissue-Nonspecific Alkaline Phosphatase, a Possible Mediator of Cell Maturation: Towards a New Paradigm" Cells 10, no. 12: 3338. https://doi.org/10.3390/cells10123338
APA StyleSato, M., Saitoh, I., Kiyokawa, Y., Iwase, Y., Kubota, N., Ibano, N., Noguchi, H., Yamasaki, Y., & Inada, E. (2021). Tissue-Nonspecific Alkaline Phosphatase, a Possible Mediator of Cell Maturation: Towards a New Paradigm. Cells, 10(12), 3338. https://doi.org/10.3390/cells10123338