
Acetylation in Mitochondria Dynamics and Neurodegeneration


Abstract
:1. Introduction
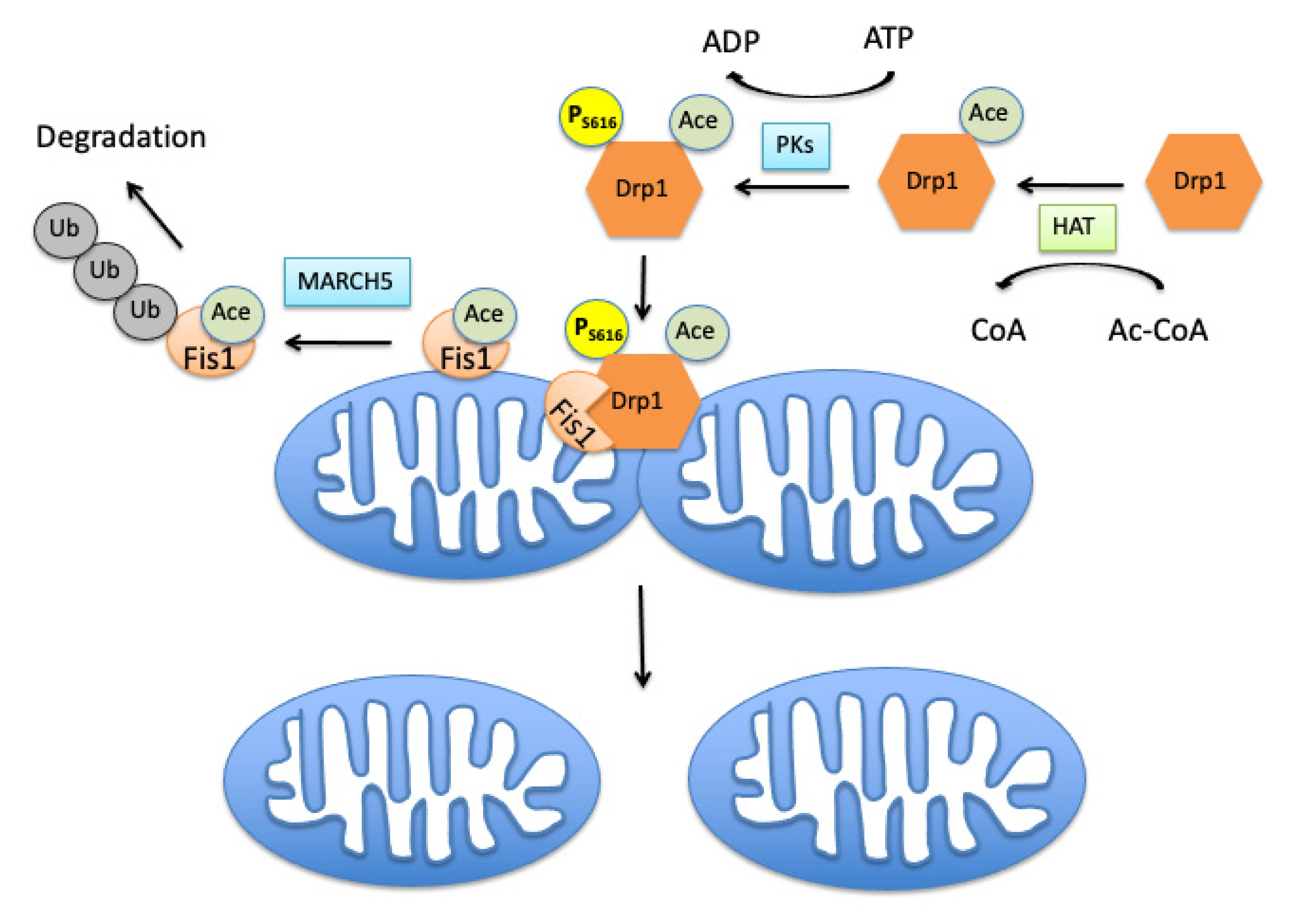
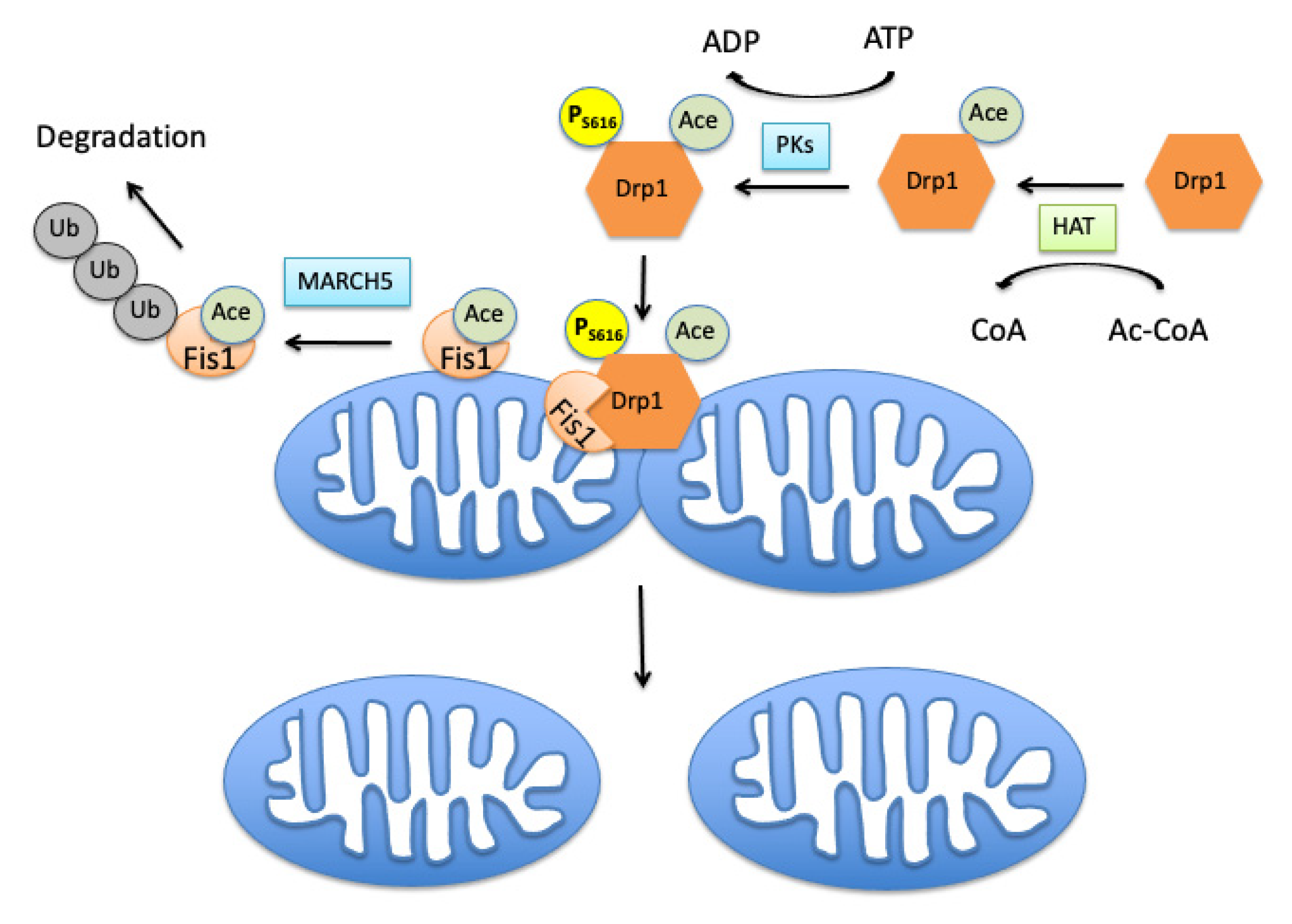
2. Mitochondrial Fission Controlling Machinery
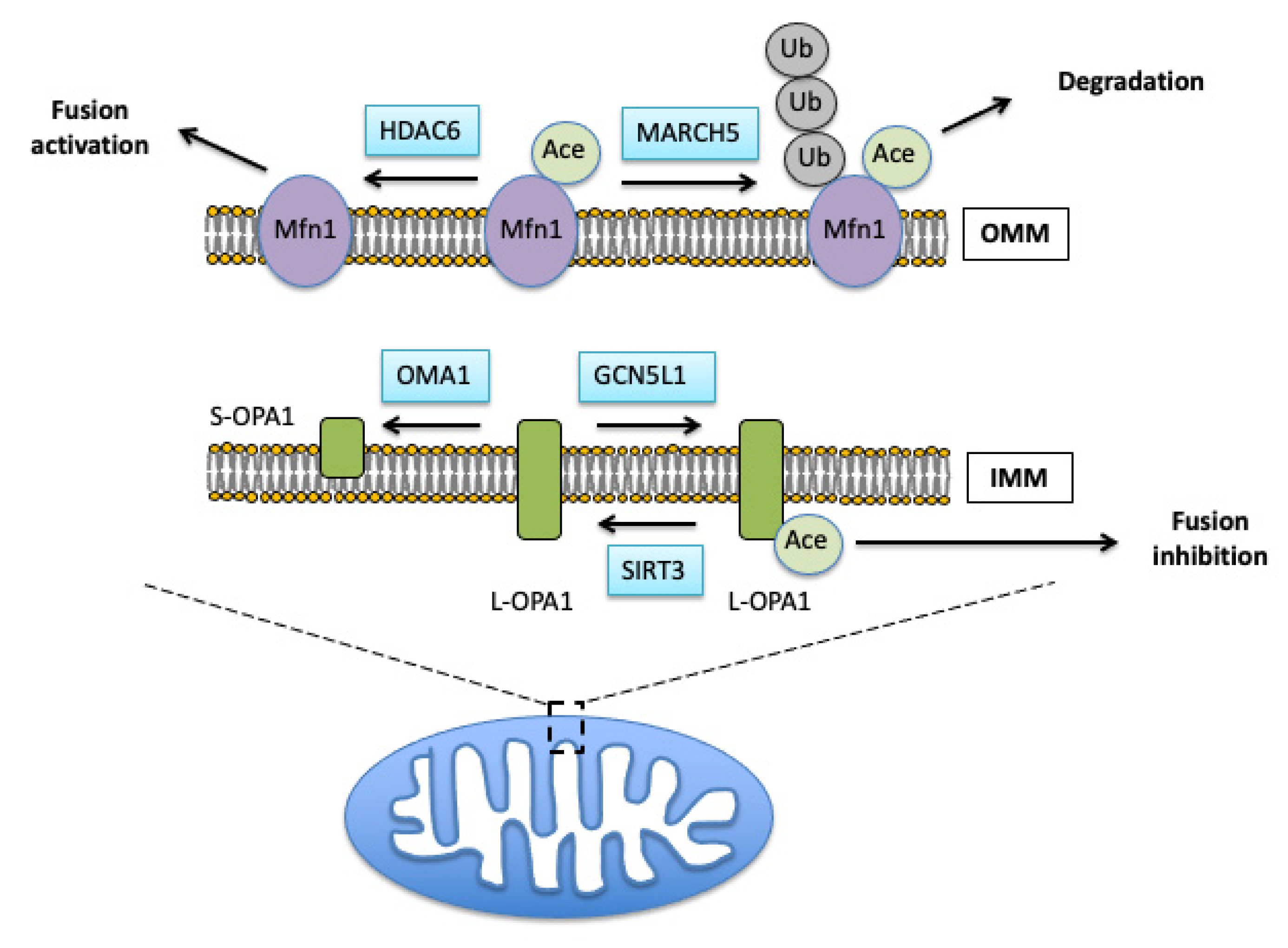
Proteins Regulating the Fusion Process
3. Tight Regulation of Fission and Fusion Proteins Activity by Post-Translational Modifications
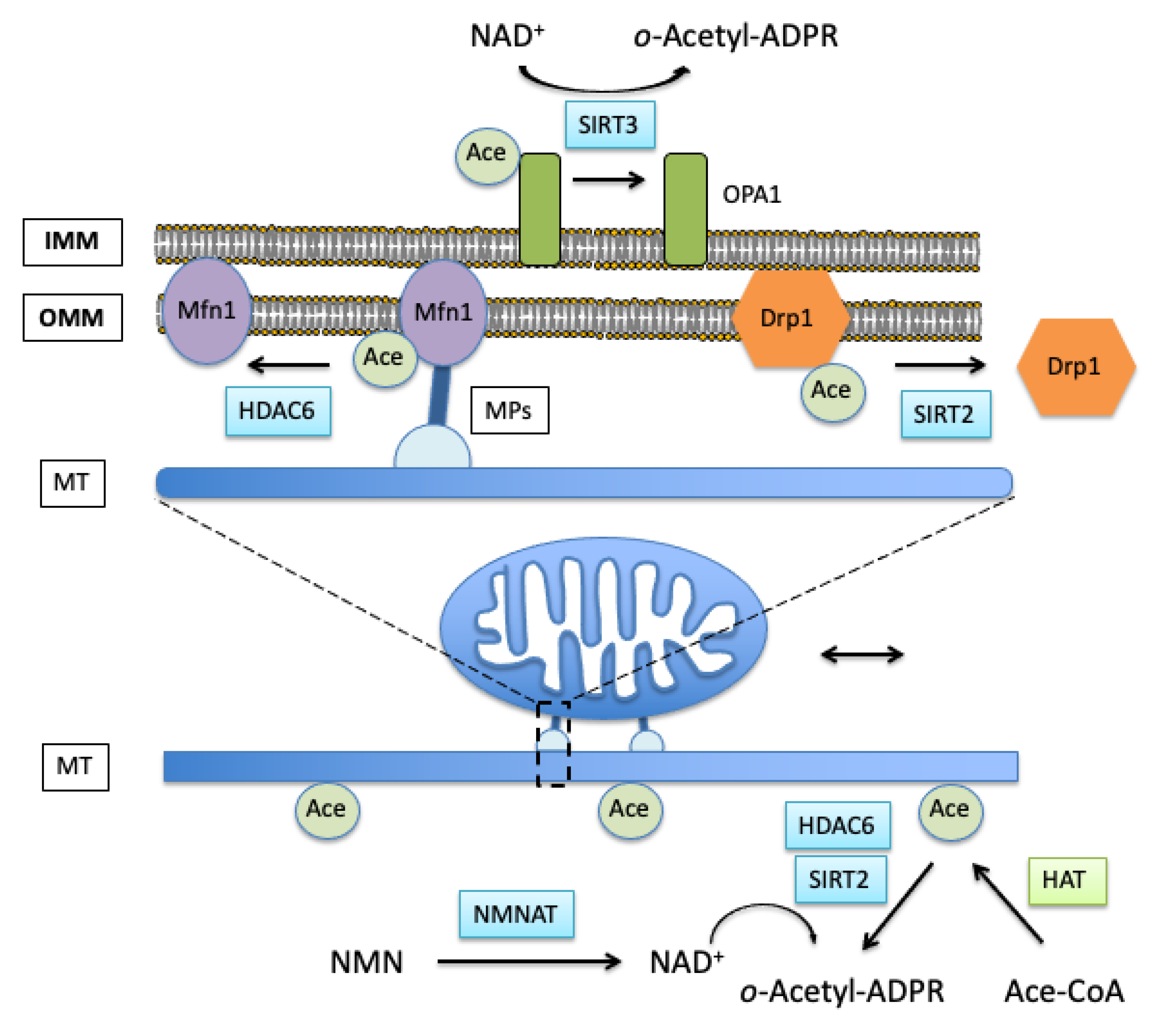
3.1. Role of Acetylation in Control of Mitochondrial Dynamics
3.2. Effects of Mitochondrial Protein Acetylation on Free Radical-Induced Fission and Fusion Dynamics
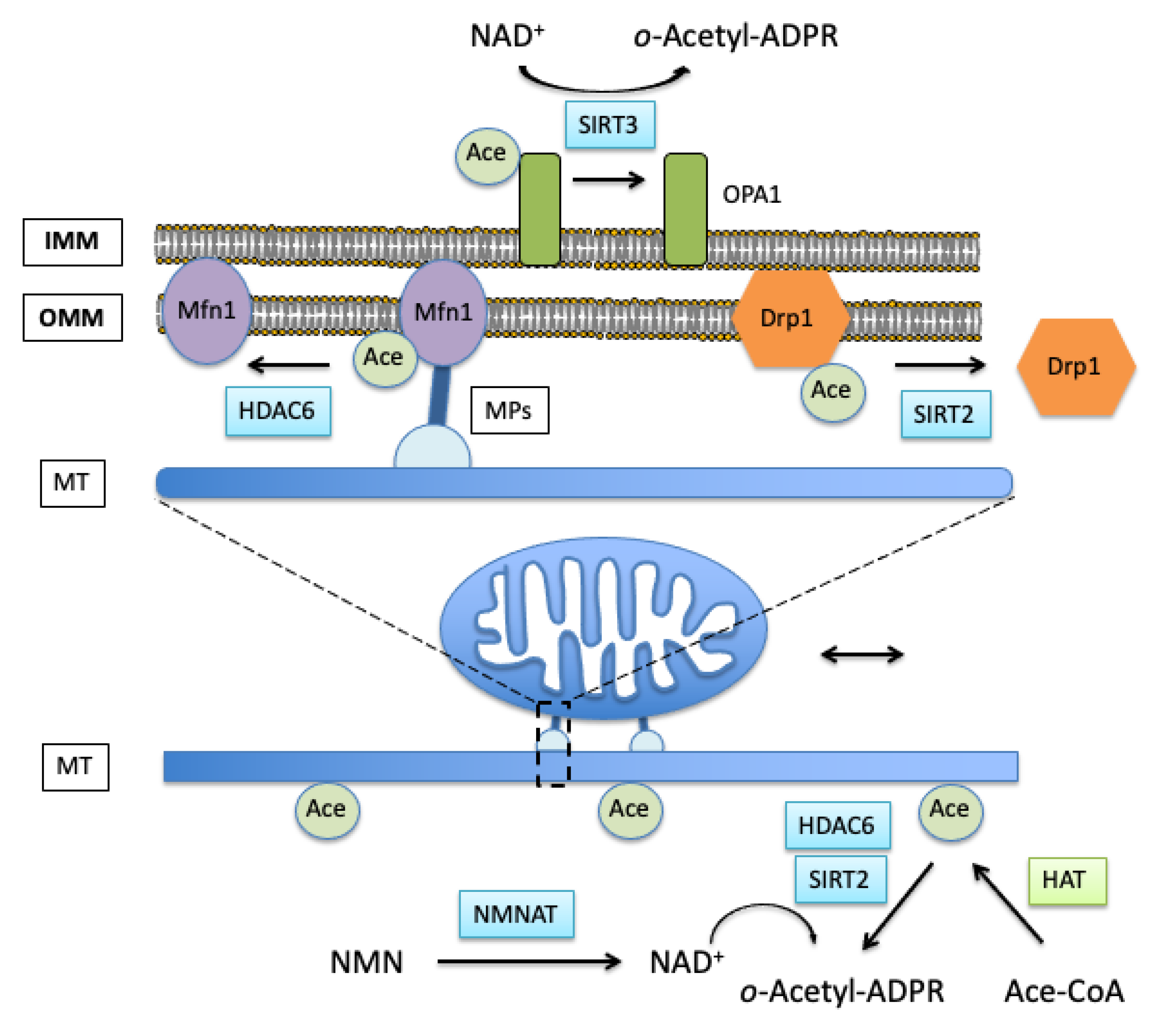
4. Role of Cellular Cytoskeleton in Mitochondrial Fission/Fusion Dynamics
4.1. Actin Cytoskeleton Acetylation and Mitochondrial Fission
4.2. Mitochondrial Movement and Tubulin Acetylation
5. Pathophysiology of Mitochondrial Dynamics Dysfunction
Role of NAD+ in the Regulation of Mitochondrial Dynamics in Acute Brain Injury
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henze, K.; Martin, W. Evolutionary biology: Essence of mitochondria. Nature 2003, 426, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Hoffmeister, M.; Rotte, C.; Henze, K. An overview of endosymbiotic models for the origins of eukaryotes, their ATP-producing organelles (mitochondria and hydrogenosomes), and their heterotrophic lifestyle. Biol. Chem. 2001, 382, 1521–1539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5, 7949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Bernardi, P.; Pozzan, T. Mitochondria as all-round players of the calcium game. J. Physiol. 2000, 529 Pt 1, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kristian, T.; Siesjo, B.K. Calcium-related damage in ischemia. Life Sci. 1996, 59, 357–367. [Google Scholar] [CrossRef]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Kristian, T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 2004, 36, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, J.M.; Brustovetsky, N.; Pinelis, V.; Kristal, B.S.; Herman, C.; Li, X. The mitochondrial permeability transition: The brain’s point of view. Biochem. Soc. Symp. 1999, 66, 75–84. [Google Scholar] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Balog, J.; Mehta, S.L.; Vemuganti, R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J. Cereb. Blood Flow Metab. 2016, 36, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Cagalinec, M.; Safiulina, D.; Liiv, M.; Liiv, J.; Choubey, V.; Wareski, P.; Veksler, V.; Kaasik, A. Principles of the mitochondrial fusion and fission cycle in neurons. J. Cell Sci. 2013, 126, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, M.P. The machinery of mitochondrial inheritance and behavior. Science 1999, 283, 1493–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimova, N.; Long, A.; Kristian, T. Significance of Mitochondrial Protein Post-translational Modifications in Pathophysiology of Brain Injury. Transl. Stroke Res. 2018, 9, 223–237. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Anderson, C.A.; Blackstone, C. SUMO wrestling with Drp1 at mitochondria. EMBO J. 2013, 32, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.; Wang, C.; Cho, J.H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell 2014, 25, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojansky, R.; Cha, M.Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016, 5, e17896. [Google Scholar] [CrossRef] [PubMed]
- Glauser, L.; Sonnay, S.; Stafa, K.; Moore, D.J. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J. Neurochem. 2011, 118, 636–645. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, J.; Wu, R.; Ma, J.; Du, G.; Jiao, R.; Tian, Y.; Zheng, Z.; Yuan, Z. DJ-1 promotes the proteasomal degradation of Fis1: Implications of DJ-1 in neuronal protection. Biochem. J. 2012, 447, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.L.; Castro, I.G.; Camoes, F.; Schrader, T.A.; McNeall, D.; Yang, J.; Giannopoulou, E.A.; Gomes, S.; Pogenberg, V.; Bonekamp, N.A.; et al. Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J. Cell Sci. 2017, 130, 1675–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koirala, S.; Guo, Q.; Kalia, R.; Bui, H.T.; Eckert, D.M.; Frost, A.; Shaw, J.M. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl. Acad. Sci. USA 2013, 110, E1342–E1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Loson, O.C.; Meng, S.; Ngo, H.; Liu, R.; Kaiser, J.T.; Chan, D.C. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. 2015, 24, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation-induced apoptosis. FASEB J. 2016, 30, 466–476. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Long, Q.; Liu, J.; Tang, H.; Li, Y.; Bao, F.; Qin, D.; Pei, D.; Liu, X. Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell. Mol. Life Sci. 2015, 72, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Kimura, M.; Okano, Y. Human Misato regulates mitochondrial distribution and morphology. Exp. Cell Res. 2007, 313, 1393–1404. [Google Scholar] [CrossRef]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Varnai, P.; Gyuris, T.; Horvath, A.; Nagy, L.; Seifert, E.L.; et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017, 9, 967–984. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Hamamoto, T.; Seo, N.; Kagawa, Y.; Endo, H. Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem. Biophys. Res. Commun. 2003, 300, 482–493. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Stiburek, L.; Cesnekova, J.; Kostkova, O.; Fornuskova, D.; Vinsova, K.; Wenchich, L.; Houstek, J.; Zeman, J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 2012, 23, 1010–1023. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Baburamani, A.A.; Hurling, C.; Stolp, H.; Sobotka, K.; Gressens, P.; Hagberg, H.; Thornton, C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2015, 16, 22509–22526. [Google Scholar] [CrossRef] [Green Version]
- Jahani-Asl, A.; Pilon-Larose, K.; Xu, W.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Slack, R.S. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J. Biol. Chem. 2011, 286, 4772–4782. [Google Scholar] [CrossRef] [Green Version]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Baricault, L.; Segui, B.; Guegand, L.; Olichon, A.; Valette, A.; Larminat, F.; Lenaers, G. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 2007, 313, 3800–3808. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Cherok, E.; Das, S.; Li, S.; Roelofs, B.A.; Ge, S.X.; Polster, B.M.; Boyman, L.; Lederer, W.J.; Wang, C.; et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol. Biol. Cell 2016, 27, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Norvell, A.; McMahon, S.B. Cell biology. Rise of the rival. Science 2010, 327, 964–965. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. HATs and HDACs in neurodegeneration: A tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006, 13, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borra, M.T.; Langer, M.R.; Slama, J.T.; Denu, J.M. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry 2004, 43, 9877–9887. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Quant. Biol. 2011, 76, 267–277. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta 2009, 1789, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Webster, B.R.; Li, J.H.; Sack, M.N. Identification of a molecular component of the mitochondrial acetyltransferase programme: A novel role for GCN5L1. Biochem. J. 2012, 443, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Zhang, H.; Gutierrez Cortes, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef]
- Sever, S.; Muhlberg, A.B.; Schmid, S.L. Impairment of dynamin’s GAP domain stimulates receptor-mediated endocytosis. Nature 1999, 398, 481–486. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, T.; Wang, L.; Cai, Y.; Zhong, X.; He, X.; Hu, L.; Tian, S.; Wu, M.; Hui, L.; et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017, 36, 1330–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kapur, M.; Li, M.; Choi, M.C.; Choi, S.; Kim, H.J.; Kim, I.; Lee, E.; Taylor, J.P.; Yao, T.P. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 2014, 127, 4954–4963. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.Y.; Nguyen, O.T.; Kang, H.; Cho, H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014, 5, e1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Galkin, A. Brain Ischemia/Reperfusion Injury and Mitochondrial Complex I Damage. Biochemistry 2019, 84, 1411–1423. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Inarrea, P.; Moini, H.; Han, D.; Rettori, D.; Aguilo, I.; Alava, M.A.; Iturralde, M.; Cadenas, E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu,Zn-superoxide dismutase activity: Implications for mitochondrial energy metabolism and apoptosis. Biochem. J. 2007, 405, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowe, D.F.; Camara, A.K. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimova, N.; Long, A.; Kristian, T. Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3 dependent mechanism in male mice. J. Neurosci. Res. 2019, 97, 975–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, L.; Wang, P.; Li, X.; Qiu, D.; Li, L.; Zhang, J.; Hou, X.; Han, L.; Ge, J.; et al. Sirt3-dependent deacetylation of SOD2 plays a protective role against oxidative stress in oocytes from diabetic mice. Cell Cycle 2017, 16, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lu, H.F.; Alano, C.C. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS ONE 2011, 6, e14731. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anesti, V.; Scorrano, L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 2006, 1757, 692–699. [Google Scholar] [CrossRef]
- Bartolak-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Moritz, M.; Agard, D.A. Gamma-tubulin complexes and microtubule nucleation. Curr. Opin. Struct. Biol. 2001, 11, 174–181. [Google Scholar] [CrossRef]
- Hammond, J.W.; Cai, D.; Verhey, K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008, 20, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E. Structural insights into microtubule function. Annu. Rev. Biochem. 2000, 69, 277–302. [Google Scholar] [CrossRef]
- Cai, Q.; Davis, M.L.; Sheng, Z.H. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci. Res. 2011, 70, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Paturle, L.; Wehland, J.; Margolis, R.L.; Job, D. Complete separation of tyrosinated, detyrosinated, and nontyrosinatable brain tubulin subpopulations using affinity chromatography. Biochemistry 1989, 28, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Paturle-Lafanechere, L.; Manier, M.; Trigault, N.; Pirollet, F.; Mazarguil, H.; Job, D. Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J. Cell Sci. 1994, 107 Pt 6, 1529–1543. [Google Scholar] [CrossRef]
- Suryavanshi, S.; Edde, B.; Fox, L.A.; Guerrero, S.; Hard, R.; Hennessey, T.; Kabi, A.; Malison, D.; Pennock, D.; Sale, W.S.; et al. Tubulin glutamylation regulates ciliary motility by altering inner dynein arm activity. Curr. Biol. 2010, 20, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, T.; Yanagisawa, H.A.; Yagi, T.; Hirono, M.; Kamiya, R. Tubulin polyglutamylation regulates axonemal motility by modulating activities of inner-arm dyneins. Curr. Biol. 2010, 20, 441–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 2015, 4, e08828. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Ji, W.K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehklau, K.; Hoffmann, L.; Gurniak, C.B.; Ott, M.; Witke, W.; Scorrano, L.; Culmsee, C.; Rust, M.B. Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis. 2017, 8, e3063. [Google Scholar] [CrossRef]
- Varland, S.; Vandekerckhove, J.; Drazic, A. Actin Post-translational Modifications: The Cinderella of Cytoskeletal Control. Trends Biochem. Sci. 2019, 44, 502–516. [Google Scholar] [CrossRef] [Green Version]
- Mu, A.; Fung, T.S.; Kettenbach, A.N.; Chakrabarti, R.; Higgs, H.N. A complex containing lysine-acetylated actin inhibits the formin INF2. Nat. Cell Biol. 2019, 21, 592–602. [Google Scholar] [CrossRef]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef]
- Cueva, J.G.; Hsin, J.; Huang, K.C.; Goodman, M.B. Posttranslational acetylation of alpha-tubulin constrains protofilament number in native microtubules. Curr. Biol. 2012, 22, 1066–1074. [Google Scholar] [CrossRef] [Green Version]
- Topalidou, I.; Keller, C.; Kalebic, N.; Nguyen, K.C.; Somhegyi, H.; Politi, K.A.; Heppenstall, P.; Hall, D.H.; Chalfie, M. Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr. Biol. 2012, 22, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, J.W.; Huang, C.F.; Kaech, S.; Jacobson, C.; Banker, G.; Verhey, K.J. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 2010, 21, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Perdiz, D.; Mackeh, R.; Pous, C.; Baillet, A. The ins and outs of tubulin acetylation: More than just a post-translational modification? Cell Signal. 2011, 23, 763–771. [Google Scholar] [CrossRef]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an alpha-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.W.; Li, L.; Ghorbani, M.; You, L.; Yang, X.J. Mice lacking alpha-tubulin acetyltransferase 1 are viable but display alpha-tubulin acetylation deficiency and dentate gyrus distortion. J. Biol. Chem. 2013, 288, 20334–20350. [Google Scholar] [CrossRef] [Green Version]
- Mackeh, R.; Lorin, S.; Ratier, A.; Mejdoubi-Charef, N.; Baillet, A.; Bruneel, A.; Hamai, A.; Codogno, P.; Pous, C.; Perdiz, D. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate alpha-tubulin acetyltransferase-1 (alphaTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J. Biol. Chem. 2014, 289, 11816–11828. [Google Scholar] [CrossRef] [Green Version]
- Nahhas, F.; Dryden, S.C.; Abrams, J.; Tainsky, M.A. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 2007, 303, 221–230. [Google Scholar] [CrossRef]
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 2011, 20, 3986–3996. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Southwood, C.M.; Peppi, M.; Dryden, S.; Tainsky, M.A.; Gow, A. Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 2007, 32, 187–195. [Google Scholar] [CrossRef]
- Fukada, M.; Hanai, A.; Nakayama, A.; Suzuki, T.; Miyata, N.; Rodriguiz, R.M.; Wetsel, W.C.; Yao, T.P.; Kawaguchi, Y. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS ONE 2012, 7, e30924. [Google Scholar] [CrossRef]
- Chen, S.; Owens, G.C.; Makarenkova, H.; Edelman, D.B. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE 2010, 5, e10848. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Kamemura, K.; Ogawa, M.; Ohkubo, S.; Ohtsuka, Y.; Shitara, Y.; Komiya, T.; Maeda, S.; Ito, A.; Yoshida, M. Depression of mitochondrial metabolism by downregulation of cytoplasmic deacetylase, HDAC6. FEBS Lett. 2012, 586, 1379–1383. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Choi, H.; Jung, E.S.; Lee, W.; Oh, S.; Jeon, N.L.; Mook-Jung, I. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS ONE 2012, 7, e42983. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Nagashima, S.; Tabara, L.C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef]
- Tabara, L.C.; Morris, J.L.; Prudent, J. The Complex Dance of Organelles during Mitochondrial Division. Trends Cell Biol. 2021, 31, 241–253. [Google Scholar] [CrossRef]
- Biel, T.G.; Lee, S.; Flores-Toro, J.A.; Dean, J.W.; Go, K.L.; Lee, M.H.; Law, B.K.; Law, M.E.; Dunn, W.A., Jr.; Zendejas, I.; et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 2016, 23, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Guedes-Dias, P.; Oliveira, J.M. Lysine deacetylases and mitochondrial dynamics in neurodegeneration. Biochim. Biophys. Acta 2013, 1832, 1345–1359. [Google Scholar] [CrossRef]
- Nakamura, T.; Cho, D.H.; Lipton, S.A. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp. Neurol. 2012, 238, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.K.; Gotz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Anzell, A.R.; Fogo, G.M.; Gurm, Z.; Raghunayakula, S.; Wider, J.M.; Maheras, K.J.; Emaus, K.J.; Bryson, T.D.; Wang, M.; Neumar, R.W.; et al. Mitochondrial fission and mitophagy are independent mechanisms regulating ischemia/reperfusion injury in primary neurons. Cell Death Dis. 2021, 12, 475. [Google Scholar] [CrossRef]
- Kumari, S.; Anderson, L.; Farmer, S.; Mehta, S.L.; Li, P.A. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl. Stroke Res. 2012, 3, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Tian, F.; Kurata, T.; Morimoto, N.; Abe, K. Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J. Neurosci. Res. 2012, 90, 1183–1189. [Google Scholar] [CrossRef]
- Zhang, L.; He, Z.; Zhang, Q.; Wu, Y.; Yang, X.; Niu, W.; Hu, Y.; Jia, J. Exercise pretreatment promotes mitochondrial dynamic protein OPA1 expression after cerebral ischemia in rats. Int. J. Mol. Sci. 2014, 15, 4453–4463. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Klimova, N.; Fearnow, A.; Kristian, T. Role of NAD(+)-Modulated Mitochondrial Free Radical Generation in Mechanisms of Acute Brain Injury. Brain Sci. 2020, 10, 449. [Google Scholar] [CrossRef]
- Xiong, T.Q.; Guo, C.Y.; Tan, B.H.; Gui, Y.; Li, Y.C. The temporal and spatial changes of microtubule cytoskeleton in the CA1 stratum radiatum following global transient ischemia. J. Chem. Neuroanat. 2019, 101, 101682. [Google Scholar] [CrossRef]
- Guo, C.Y.; Xiong, T.Q.; Tan, B.H.; Gui, Y.; Ye, N.; Li, S.L.; Li, Y.C. The temporal and spatial changes of actin cytoskeleton in the hippocampal CA1 neurons following transient global ischemia. Brain Res. 2019, 1720, 146297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sirtuin | Localization | Activity | Reference |
|---|---|---|---|
| SIRT1 | Cytosol, nucleus | deacetylase | [88] |
| SIRT2 | Cytosol | deacetylase | [89] |
| SIRT3 | Mitochondria | deacetylase | [90] |
| SIRT4 | Mitochondria | ADP-ribosyl transferase | [85] |
| SIRT5 | Mitochondria | desuccinylase | [91] |
| SIRT6 | nucleus | ADP-ribosyl transferase | [92] |
| SIRT7 | nucleus | deacetylase | [93] |
| Dynamics Protein | Acetylated Lysine | Acetylation | Reference |
|---|---|---|---|
| Drp1 | K642 | NAD+-dependent | [90] |
| Fis1 | ND | TSA and Nam 1 | [95] |
| Mfn1 | ND | SIRT1/SIRT2 | [93] |
| Mfn2 | ND | SIRT1/SIRT2 | [164] |
| OPA1 | ND | SIRT3 | [92] |
| α-tubulin | K40 | αTAT1/SIRT2/HDAC6 | [134,136] |
| actin | K50, K61, K328 | HDAC6 | [139] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waddell, J.; Banerjee, A.; Kristian, T. Acetylation in Mitochondria Dynamics and Neurodegeneration. Cells 2021, 10, 3031. https://doi.org/10.3390/cells10113031
Waddell J, Banerjee A, Kristian T. Acetylation in Mitochondria Dynamics and Neurodegeneration. Cells. 2021; 10(11):3031. https://doi.org/10.3390/cells10113031
Chicago/Turabian StyleWaddell, Jaylyn, Aditi Banerjee, and Tibor Kristian. 2021. "Acetylation in Mitochondria Dynamics and Neurodegeneration" Cells 10, no. 11: 3031. https://doi.org/10.3390/cells10113031
APA StyleWaddell, J., Banerjee, A., & Kristian, T. (2021). Acetylation in Mitochondria Dynamics and Neurodegeneration. Cells, 10(11), 3031. https://doi.org/10.3390/cells10113031