
Platelets in ITP: Victims in Charge of Their Own Fate?

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Hypothesis 1: Platelets and Their Immune Functions in ITP
2.1. Platelet Activation and Immune Functions
2.2. Platelets and Inflammation in ITP
2.3. Platelets and (Auto)-Immune Responses in ITP
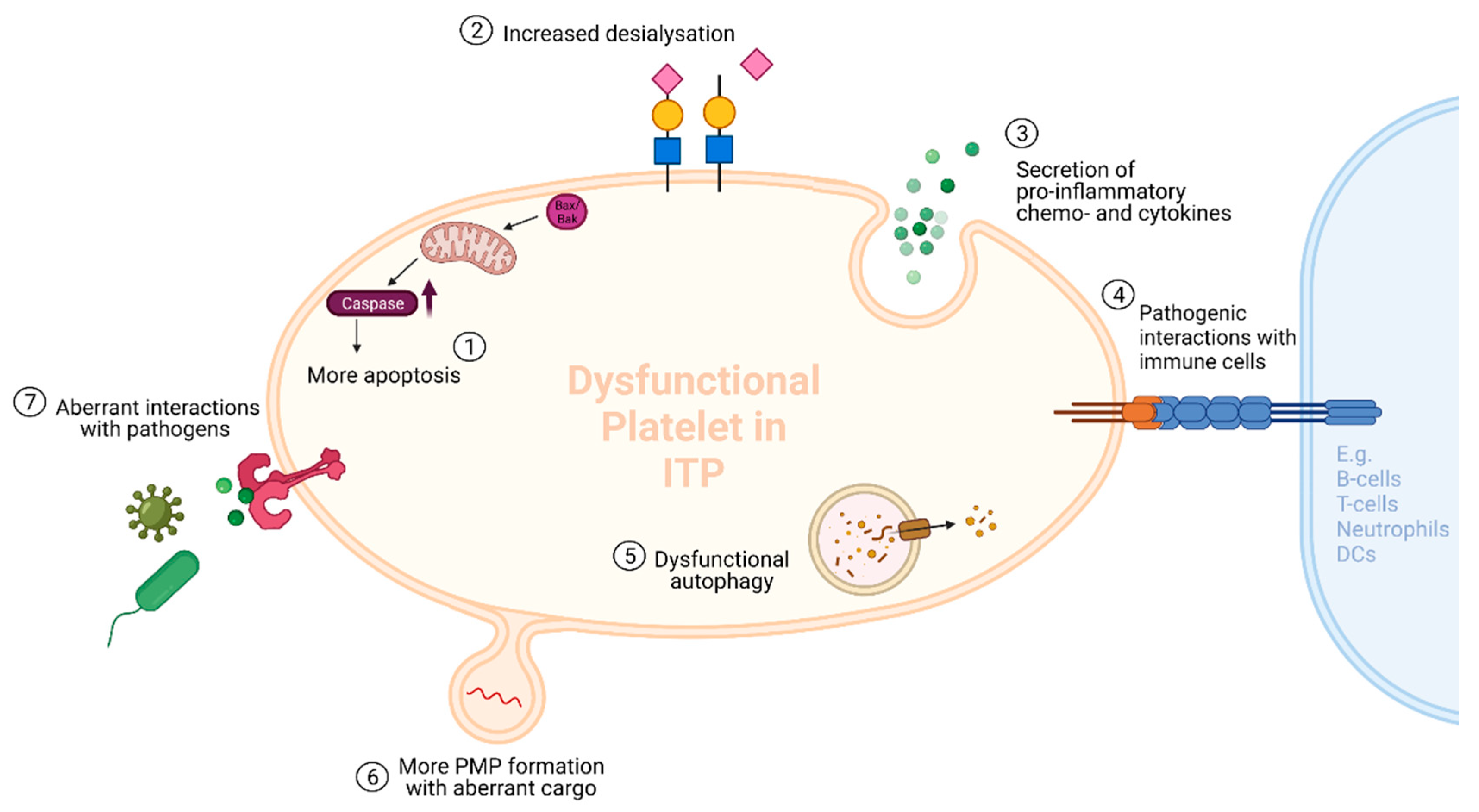
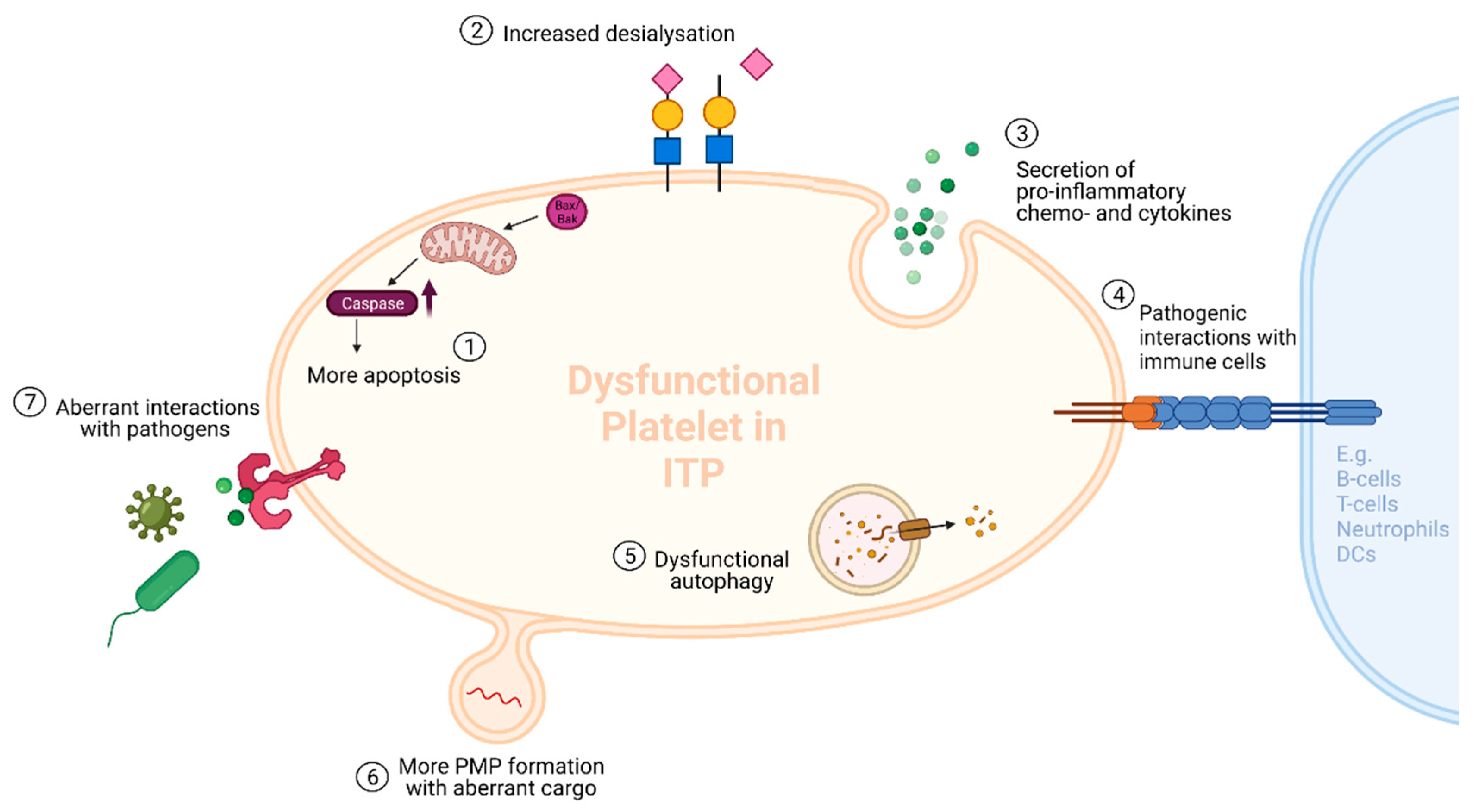
3. Hypothesis 2: Platelet Microparticles in ITP
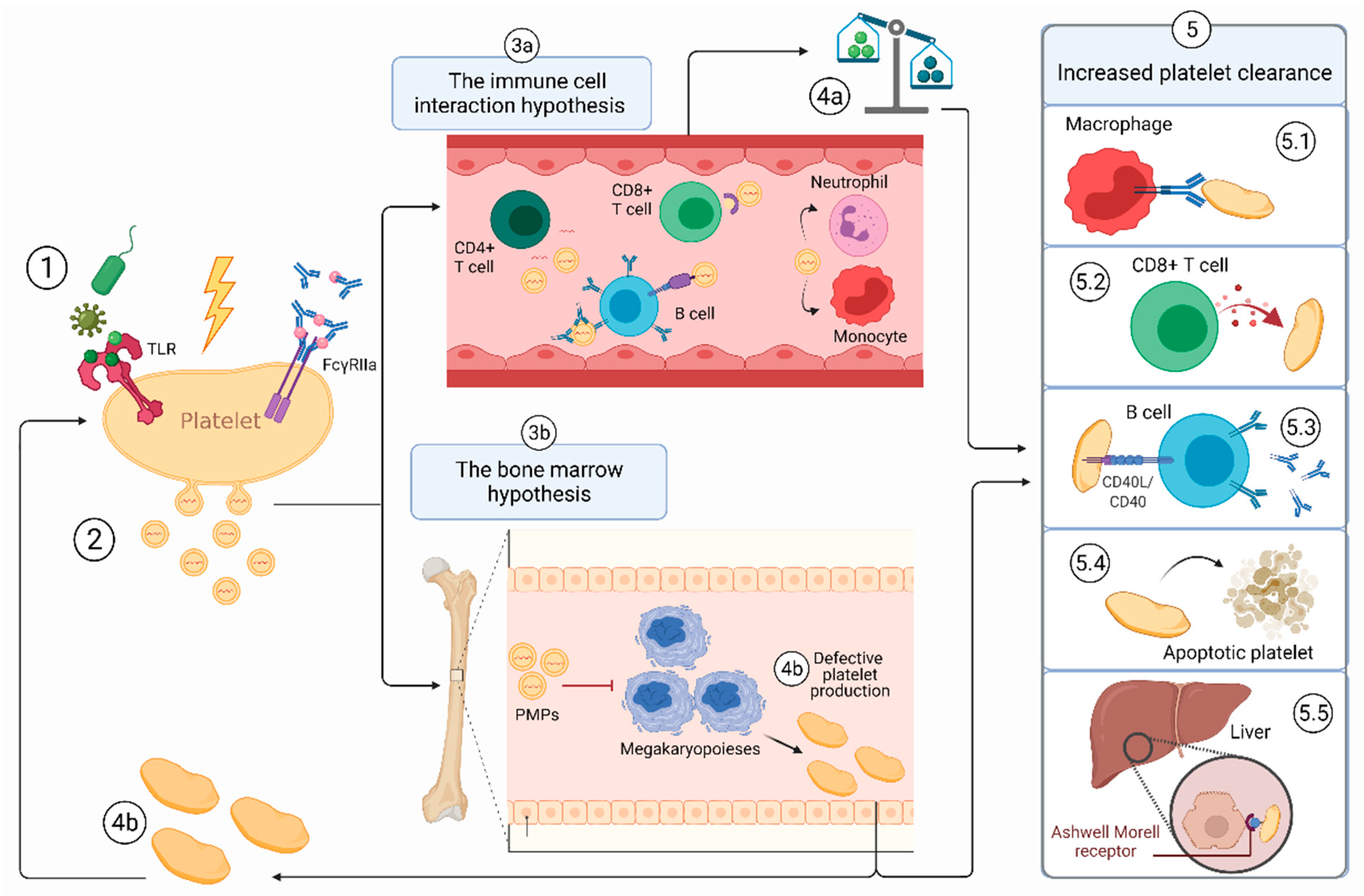
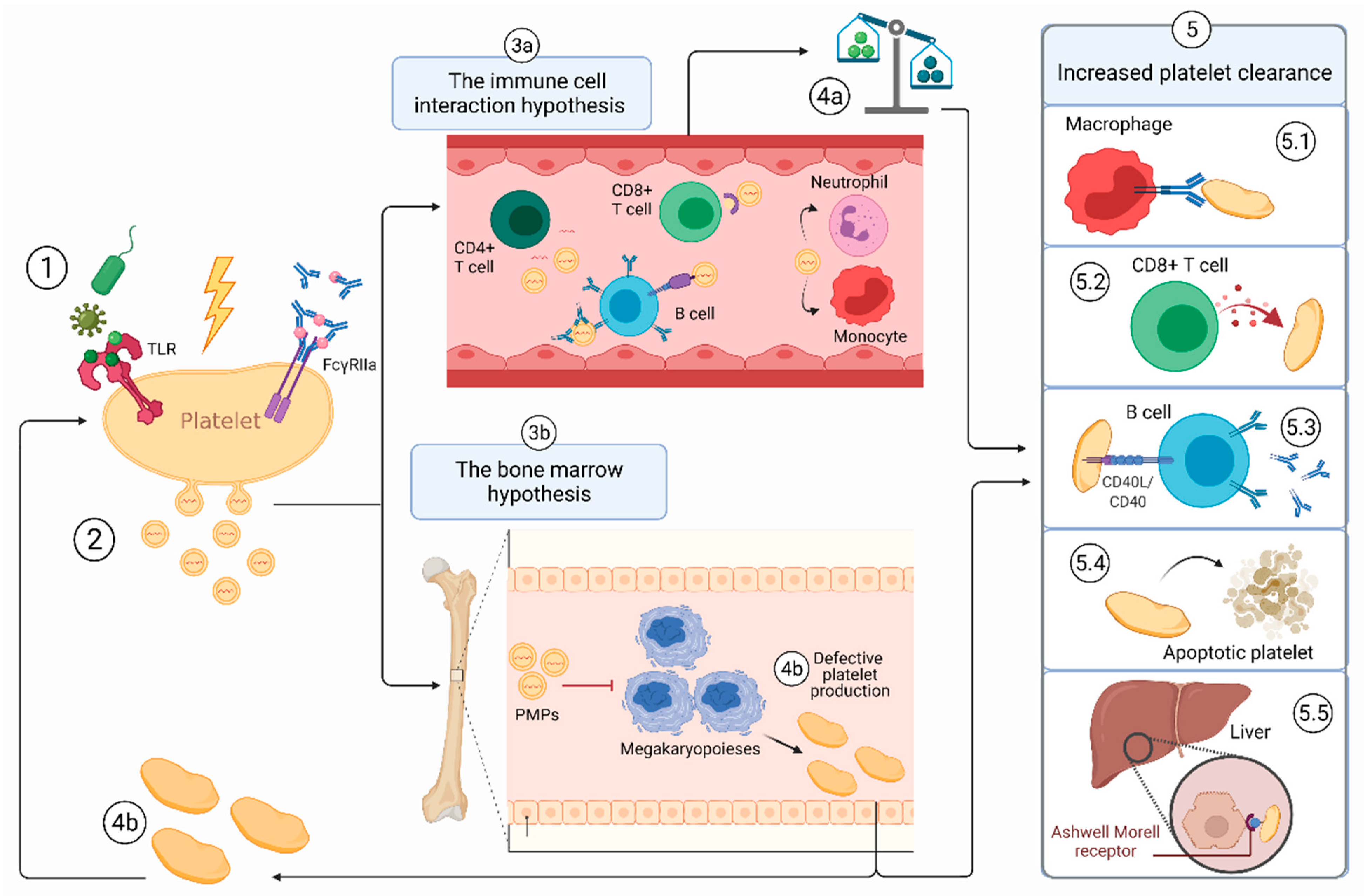
3.1. The First Hit
3.2. ITP-PMPs: How and Why Are They so Different?
3.2.1. Increased Levels of PMPs in ITP
3.2.2. The Different Cargos of ITP-PMPs
3.3. Hypothesis 2a: The PMP-Immune Cell Interaction Hypothesis
3.4. Hypothesis 2b: The PMP-Bone Marrow Hypothesis
4. Final Recommendations
Author Contributions
Funding
Conflicts of Interest
References
- Semple, J.W.; Rebetz, J.; Maouia, A.; Kapur, R. An update on the pathophysiology of immune thrombocytopenia. Curr. Opin. Hematol. 2020, 27, 423–429. [Google Scholar] [CrossRef]
- Zufferey, A.; Kapur, R.; Semple, J.W. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef]
- Marini, I.; Bakchoul, T. Pathophysiology of Autoimmune Thrombocytopenia: Current Insight with a Focus on Thrombopoiesis. Hämostaseologie 2019, 39, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, N.; Ba, A.K.; Ba, C.K.; Watson, S.; Morgan, M.; Provan, D.; Ghanima, W.; Arnold, D.M.; Tomiyama, Y.; Santoro, C.; et al. Immune thrombocytopenia (ITP) World Impact Survey (I-WISh): Impact of ITP on health-related quality of life. Am. J. Hematol. 2020, 96, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrbensky, J.R.; Nazy, I.; Clare, R.; Larché, M.; Arnold, D.M. T cell–mediated autoimmunity in immune thrombocytopenia. Eur. J. Haematol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.B.; Andemariam, B.; Yin, W.; Bussel, J.B. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. Br. J. Haematol. 2010, 148, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Porcelijn, L.; Huiskes, E.; Oldert, G.; Schipperus, M.; Zwaginga, J.J.; De Haas, M. Detection of platelet autoantibodies to identify immune thrombocytopenia: State of the art. Br. J. Haematol. 2018, 182, 423–426. [Google Scholar] [CrossRef]
- Olsson, B.; Andersson, P.-O.; Jernås, M.; Jacobsson, S.; Carlsson, B.; Carlsson, L.M.S.; Wadenvik, H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat. Med. 2003, 9, 1123–1124. [Google Scholar] [CrossRef]
- Li, J.; Sullivan, J.A.; Ni, H. Pathophysiology of immune thrombocytopenia. Curr. Opin. Hematol. 2018, 25, 373–381. [Google Scholar] [CrossRef]
- Khodadi, E.; Aminasnafi, A.; Shahrabi, S.; Shahjahani, M.; Saki, N. Bone marrow niche in immune thrombocytopenia: A focus on megakaryopoiesis. Ann. Hematol. 2016, 95, 1765–1776. [Google Scholar] [CrossRef]
- Maouia, A.; Rebetz, J.; Kapur, R.; Semple, J.W. The Immune Nature of Platelets Revisited. Transfus. Med. Rev. 2020, 34, 209–220. [Google Scholar] [CrossRef]
- Semple, J.W.; Kapur, R. Platelet immunology from the inside out. ISBT Sci. Ser. 2020, 15, 315–319. [Google Scholar] [CrossRef]
- Kapur, R.; Zufferey, A.; Boilard, E.; Semple, J.W. Nouvelle Cuisine: Platelets Served with Inflammation. J. Immunol. 2015, 194, 5579–5587. [Google Scholar] [CrossRef] [Green Version]
- Kapur, R.; Semple, J.W. The nonhemostatic immune functions of platelets. Semin. Hematol. 2016, 53, S2–S6. [Google Scholar] [CrossRef]
- Scherlinger, M.; Guillotin, V.; Truchetet, M.-E.; Contin-Bordes, C.; Sisirak, V.; Duffau, P.; Lazaro, E.; Richez, C.; Blanco, P. Systemic lupus erythematosus and systemic sclerosis: All roads lead to platelets. Autoimmun. Rev. 2018, 17, 625–635. [Google Scholar] [CrossRef]
- Habets, K.L.L.; Huizinga, T.W.J.; Toes, R. Platelets and autoimmunity. Eur. J. Clin. Investig. 2013, 43, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Semple, J.W. Platelets as immune-sensing cells. Blood Adv. 2016, 1, 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, M.L.; Garabet, L.; Pérez, M.P.F.; Reyes-García, A.M.D.L.; Diaz-Lozano, P.; Garcia-Barbera, N.; Aguila, S.; Vicente, V.; Ghanima, W.; Martinez, C.; et al. Platelet activation and neutrophil extracellular trap (NET) formation in immune thrombocytopenia: Is there an association? Platelets 2019, 31, 906–912. [Google Scholar] [CrossRef]
- Manzano, E.M.; Román, M.T.; Sanz, R.J.; Bello, I.F.; Hernández-Flórez, D.; Salces, M.M.; Valor, L.; Pollmar, I.R.; Butta, N.V.; Yuste, V.J. Platelet and immune characteristics of immune thrombocytopaenia patients non-responsive to therapy reveal severe immune dysregulation. Br. J. Haematol. 2020, 189, 943–953. [Google Scholar] [CrossRef]
- Middelburg, R.A.; Carbaat-Ham, J.C.; Hesam, H.; Ragusi, M.A.; Zwaginga, J.J. Platelet function in adult ITP patients can be either increased or decreased, compared to healthy controls, and is associated with bleeding risk. Hematology 2016, 21, 549–551. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Bussel, J.B.; Frelinger, A.L.; Babula, B.; Linden, M.; Li, Y.; Barnard, M.R.; Tate, C.; Feldman, E.J.; Michelson, A.D. Differences in platelet function in patients with acute myeloid leukemia and myelodysplasia compared to equally thrombocytopenic patients with immune thrombocytopenia. J. Thromb. Haemost. 2011, 9, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, A.A.; Demina, I.A.; Ptushkin, V.V.; Khaspekova, S.G.; Shustova, O.N.; Pankrashkina, M.M.; Ryabykh, A.A.; Obydennyi, S.I.; Strelkova, O.S.; Polokhov, D.; et al. Evolution of platelet function in adult patients with chronic immune thrombocytopenia on romiplostim treatment. Br. J. Haematol. 2019, 187, e38–e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frelinger, I.A.L.; Grace, F.; Gerrits, A.J.; Berny-Lang, M.; Brown, T.; Carmichael, S.L.; Neufeld, E.; Michelson, A.D. Platelet function tests, independent of platelet count, are associated with bleeding severity in ITP. Blood 2015, 126, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kronbichler, A.; Park, D.D.-Y.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Garabet, L.; Henriksson, C.E.; Lozano, M.L.; Ghanima, W.; Bussel, J.; Brodin, E.; Fernández-Pérez, M.P.; Martínez, C.; González-Conejero, R.; Mowinckel, M.-C.; et al. Markers of endothelial cell activation and neutrophil extracellular traps are elevated in immune thrombocytopenia but are not enhanced by thrombopoietin receptor agonists. Thromb. Res. 2019, 185, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.A.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel Anti-bacterial Activities of β-defensin 1 in Human Platelets: Suppression of Pathogen Growth and Signaling of Neutrophil Extracellular Trap Formation. PLOS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef]
- Swinkels, M.; Rijkers, M.; Voorberg, J.; Vidarsson, G.; Leebeek, F.W.G.; Jansen, A.J.G. Emerging Concepts in Immune Thrombocytopenia. Front. Immunol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.A.; Nestel, F.P.; Ni, H.; Lazarus, A.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, J.W.; Aslam, R.; Kim, M.; Speck, E.R.; Freedman, J. Platelet-bound lipopolysaccharide enhances Fc receptor–mediated phagocytosis of IgG-opsonized platelets. Blood 2007, 109, 4803–4805. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, Y.; Li, G.; Feng, Q.; Hou, M.; Peng, J. Reduced intracellular antioxidant capacity in platelets contributes to primary immune thrombocytopenia via ROS-NLRP3-caspase-1 pathway. Thromb. Res. 2020, 199, 1–9. [Google Scholar] [CrossRef]
- Kapur, R.; Heitink-Pollé, K.M.J.; Porcelijn, L.; Bentlage, A.E.H.; Bruin, M.C.A.; Visser, R.; Roos, D.; Schasfoort, R.B.M.; De Haas, M.; Van Der Schoot, C.E.; et al. C-reactive protein enhances IgG-mediated phagocyte responses and thrombocytopenia. Blood 2015, 125, 1793–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanilla, A.; Pasquet, J.-M.; Viallard, J.-F.; Contin, C.; Grosset, C.; Déchanet-Merville, J.; Dupouy, M.; Landry, M.; Belloc, F.; Nurden, P.; et al. Platelet-associated CD154 in immune thrombocytopenic purpura. Blood 2005, 105, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Nomura, S.; Kanazawa, S.; Ozaki, Y.; Kagawa, H.; Fukuhara, S. Significance of chemokines and soluble CD40 ligand in patients with autoimmune thrombocytopenic purpura. Eur. J. Haematol. 2002, 69, 303–308. [Google Scholar] [CrossRef]
- Patel, V.L.; Schwartz, J.; Bussel, J.B. The effect of anti-CD40 ligand in immune thrombocytopenic purpura. Br. J. Haematol. 2008, 141, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2018, 141, 92–103. [Google Scholar] [CrossRef]
- Audia, S.; Bonnotte, B. Emerging Therapies in Immune Thrombocytopenia. J. Clin. Med. 2021, 10, 1004. [Google Scholar] [CrossRef]
- Zufferey, A.; Speck, E.R.; Machlus, K.R.; Aslam, R.; Guo, L.; McVey, M.J.; Kim, M.; Kapur, R.; Boilard, E.; Italiano, J.E.; et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. 2017, 1, 1773–1785. [Google Scholar] [CrossRef]
- Guo, L.; Shen, S.; Rowley, J.W.; Tolley, N.D.; Jia, W.; Manne, B.K.; McComas, K.N.; Bolingbroke, B.; Kosaka, Y.; Krauel, K.; et al. Platelet MHC class I mediates CD8+ T-cell suppression during sepsis. Blood 2021, 138, 401–416. [Google Scholar] [CrossRef]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M.; Morrell, C.N. Platelets Present Antigen in the Context of MHC Class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Battaglia, M.; Lee, S.H.; Sun, Q.-H.; Aster, R.H.; Visentin, G.P. Platelet Factor 4 Differentially Modulates CD4+CD25+(Regulatory) versus CD4+CD25−(Nonregulatory) T Cells. J. Immunol. 2005, 174, 2680–2686. [Google Scholar] [CrossRef] [Green Version]
- Aboud, N.; Depré, F.; Salama, A. Is Autoimmune Thrombocytopenia Itself the Primary Disease in the Presence of Second Diseases? Data from a Long-Term Observation. Transfus. Med. Hemother. 2016, 44, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Kailashiya, J. Platelet-derived microparticles analysis: Techniques, challenges and recommendations. Anal. Biochem. 2018, 546, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Baffour, S.; Adjei, J.; Aryeh, C.; Kyeremeh, R.; Kyei, F.; Seidu, M.A. Understanding the biosynthesis of platelets-derived extracellular vesicles. Immunity Inflamm. Dis. 2015, 3, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; Ritzel, E.; Liehn, E.A.; Hristov, M.; Bidzhekov, K.; Müller-Newen, G.; Soehnlein, O.; Weber, C. Platelet Microparticles Enhance the Vasoregenerative Potential of Angiogenic Early Outgrowth Cells After Vascular Injury. Circulation 2010, 122, 495–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantawy, A.A.G.; Matter, R.M.; Hamed, A.A.; Telbany, M.A.S.E.D.E. Platelet microparticles in immune thrombocytopenic purpura in pediatrics. Pediatr. Hematol. Oncol. 2010, 27, 283–296. [Google Scholar] [CrossRef]
- Sewify, E.M.; Sayed, D.; Aal, R.F.A.; Ahmad, H.M.; Abdou, M.A. Increased circulating red cell microparticles (RMP) and platelet microparticles (PMP) in immune thrombocytopenic purpura. Thromb. Res. 2013, 131, e59–e63. [Google Scholar] [CrossRef]
- Wu, W.-C.; Song, S.-J.; Zhang, Y.; Li, X. Role of Extracellular Vesicles in Autoimmune Pathogenesis. Front. Immunol. 2020, 11, 579043. [Google Scholar] [CrossRef] [PubMed]
- Scherlinger, M.; Sisirak, V.; Richez, C.; Lazaro, E.; Duffau, P.; Blanco, P. New Insights on Platelets and Platelet-Derived Microparticles in Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2017, 19, 48. [Google Scholar] [CrossRef]
- French, S.L.; Butov, K.R.; Allaeys, I.; Canas, J.; Morad, G.; Davenport, P.; Laroche, A.; Trubina, N.M.; Italiano, J.E.; Moses, M.A.; et al. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020, 4, 3011–3023. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Zou, X.; Fang, F.; Wang, S.; Xu, L.; Zeng, Q.; Fan, Z.; Chen, L.; Yue, W.; Xie, X.; et al. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat. Commun. 2020, 11, 4964. [Google Scholar] [CrossRef]
- Grodzielski, M.; Goette, N.P.; Glembotsky, A.C.; Pietto, M.C.B.; Méndez-Huergo, S.P.; Pierdominici, M.S.; Montero, V.S.; Rabinovich, G.A.; Molinas, F.C.; Heller, P.G.; et al. Multiple concomitant mechanisms contribute to low platelet count in patients with immune thrombocytopenia. Sci. Rep. 2019, 9, 2208. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Ma, S.; Bi, S.-J.; Su, L.; Huang, S.-Y.; Miao, J.-Y.; Ma, C.-H.; Gao, C.-J.; Hou, M.; Peng, J. Enhancing autophagy protects platelets in immune thrombocytopenia patients. Ann. Transl. Med. 2019, 7, 134. [Google Scholar] [CrossRef]
- Cines, D.B.; Bussel, J.B.; Liebman, H.A.; Prak, E.T.L. The ITP syndrome: Pathogenic and clinical diversity. Blood 2009, 113, 6511–6521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.E.; Heitink-Pollé, K.M.J.; Mertens, B.; Porcelijn, L.; Kapur, R.; van der Schoot, C.E.; Vidarsson, G.; van der Bom, J.G.; Bruin, M.C.A.; de Haas, M. Biological stratification of clinical disease courses in childhood immune thrombocytopenia. J. Thromb. Haemost. 2021, 19, 1071–1081. [Google Scholar] [CrossRef]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide Signaling without a Nucleus: Kinase Cascades Stimulate Platelet Shedding of Proinflammatory IL-1β–Rich Microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Allaeys, I.; Marcoux, G.; Machlus, K.; Mailhot, B.; Zufferey, A.; Levesque, T.; Becker, Y.; Tessandier, N.; Melki, I.; et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc. Natl. Acad. Sci. USA 2018, 115, E1550–E1559. [Google Scholar] [CrossRef] [Green Version]
- Beutier, H.; Hechler, B.; Godon, O.; Wang, Y.; Gillis, C.M.; de Chaisemartin, L.; Gouel-Chéron, A.; Magnenat, S.; Macdonald, L.E.; Murphy, A.J.; et al. Platelets expressing IgG receptor FcγRIIA/CD32A determine the severity of experimental anaphylaxis. Sci. Immunol. 2018, 3, eaan5997. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, J. Pathogenesis in immune thrombocytopenia: New insights. Hematology 2012, 2012, 306–312. [Google Scholar] [CrossRef]
- Chen, M.; Yan, R.; Zhou, K.; Li, X.; Zhang, Y.; Liu, C.; Jiang, M.; Ye, H.; Meng, X.; Pang, N.; et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc. Natl. Acad. Sci. USA 2018, 115, E10682–E10691. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, G.; Laroche, A.; Hasse, S.; Bellio, M.; Mbarik, M.; Tamagne, M.; Allaeys, I.; Zufferey, A.; Lévesque, T.; Rebetz, J.; et al. Platelet EVs contain an active proteasome involved in protein processing for antigen presentation via MHC-I molecules. Blood 2021. [Google Scholar] [CrossRef]
- Rana, A.; Rana, A.; Westein, E.; Westein, E.; Niego, B.; Niego, B.; Hagemeyer, C.E.; Hagemeyer, C.E.; Rana, A.; Rana, A.; et al. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Boylan, B.; Chen, H.; Rathore, V.; Paddock, C.; Salacz, M.; Friedman, K.D.; Curtis, B.R.; Stapleton, M.; Newman, D.K.; Kahn, M.L.; et al. Anti-GPVI–associated ITP: An acquired platelet disorder caused by autoantibody-mediated clearance of the GPVI/FcRγ-chain complex from the human platelet surface. Blood 2004, 104, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Rabbolini, D.J.; Gardiner, E.E.; Morel-Kopp, M.; Dunkley, S.; Jahangiri, A.; Lee, C.S.; Stevenson, W.S.; Ward, C.M. Anti-glycoprotein VI mediated immune thrombocytopenia: An under-recognized and significant entity? Res. Pract. Thromb. Haemost. 2017, 1, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Burbano, C.; Villar-Vesga, J.; Orejuela, J.; Muñoz, C.; Vanegas, A.; Vásquez, G.; Rojas, M.; Castaño, D. Potential Involvement of Platelet-Derived Microparticles and Microparticles Forming Immune Complexes during Monocyte Activation in Patients with Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 322. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Østergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H.H. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Mause, S.F.; Koenen, R.R. Microvesicles from platelets: Novel drivers of vascular inflammation. Thromb. Haemost. 2015, 114, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.K.; Abdel-Monem, H.; Niravath, P.; Le, A.; Bellera, R.V.; Langlois, K.; Nagata, S.; Rumbaut, R.E.; Thiagarajan, P. Lactadherin and clearance of platelet-derived microvesicles. Blood 2009, 113, 1332–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, J.E.; Mairuhu, A.T.; Flaumenhaft, R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr. Opin. Hematol. 2010, 17, 578–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Boilard, E.; Duchez, A.-C.; Brisson, A. The diversity of platelet microparticles. Curr. Opin. Hematol. 2015, 22, 437–444. [Google Scholar] [CrossRef]
- Vrbensky, J.R.; Nazy, I.; Toltl, L.J.; Ross, C.; Ivetic, N.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Megakaryocyte apoptosis in immune thrombocytopenia. Platelets 2018, 29, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.; Esselink, M.T.; Koornstra, J.J.; Smit, J.W.; Louwes, H.; Vellenga, E.; De Wolf, J.T.M. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004, 103, 500–506. [Google Scholar] [CrossRef]
- Iraqi, M.; Perdomo, J.; Yan, F.; Choi, P.Y.-I.; Chong, B.H. Immune thrombocytopenia: Antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica 2015, 100, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Kapur, R.; Aslam, R.; Hunt, K.; Hou, Y.; Zufferey, A.; Speck, E.R.; Rondina, M.T.; Lazarus, A.H.; Ni, H.; et al. Antiplatelet antibody-induced thrombocytopenia does not correlate with megakaryocyte abnormalities in murine immune thrombocytopenia. Scand. J. Immunol. 2018, 88, e12678. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, Y.; Meng, G.; Han, P.; Zhao, Y.; Wang, H.; Xu, M.; Wang, Y.; Qiu, J.; Peng, J.; et al. Proteomic analysis and microRNA expression profiling of plasma-derived exosomes in primary immune thrombocytopenia. Br. J. Haematol. 2021, 194, 1045–1052. [Google Scholar] [CrossRef]
- Garabet, L.; Ghanima, W.; Rangberg, A.; Teruel-Montoya, R.; Martinez, C.; Lozano, M.L.; Nystrand, C.F.; Bussel, J.B.; Sandset, P.M.; Jonassen, C.M. Circulating microRNAs in patients with immune thrombocytopenia before and after treatment with thrombopoietin-receptor agonists. Platelets 2019, 31, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Edelstein, L. MicroRNAs in Platelet Physiology and Function. Semin. Thromb. Hemost. 2016, 42, 215–222. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Su, Y.; Wang, C.; Zhang, G.; Liu, X.; Chen, Q.; Lv, M.; Chang, Y.; Peng, J.; et al. miRNA-98-5p Targeting IGF2BP1 Induces Mesenchymal Stem Cell Apoptosis by Modulating PI3K/Akt and p53 in Immune Thrombocytopenia. Mol. Ther.—Nucleic Acids 2020, 20, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Yu, S.; He, Y.; Sun, T.; Liang, W.; Yu, L.; Xu, D.; Li, Q.; Zhang, R. MicroRNA profiling of platelets from immune thrombocytopenia and target gene prediction. Mol. Med. Rep. 2017, 16, 2835–2843. [Google Scholar] [CrossRef] [Green Version]
- Qian, C.; Yan, W.; Li, T.; Cui, Q.; Liu, P.; Gu, M.; Guo, J.; Zhang, W.; Ren, C.; Wu, T.; et al. Differential Expression of MiR-106b-5p and MiR-200c-3p in Newly Diagnosed Versus Chronic Primary Immune Thrombocytopenia Patients Based on Systematic Analysis. Cell. Physiol. Biochem. 2018, 45, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Bay, A.; Coskun, E.; Oztuzcu, S.; Ergun, S.; Yilmaz, F.; Aktekin, E. Plasma microRNA profiling of pediatric patients with immune thrombocytopenic purpura. Blood Coagul. Fibrinolysis 2014, 25, 379–383. [Google Scholar] [CrossRef]
- Jolink, A.-T.; Nelson, V.; Schipperus, M.; Amini, S.; Vidarsson, G.; van der Schoot, C.; Porcelijn, L.; de Haas, M.; Kapur, R. Potential Diagnostic Approaches for Prediction of Therapeutic Responses in Immune Thrombocytopenia. J. Clin. Med. 2021, 10, 3403. [Google Scholar] [CrossRef]
- Zuo, B.; Zhai, J.; You, L.; Zhao, Y.; Yang, J.; Weng, Z.; Dai, L.; Wu, Q.; Ruan, C.; He, Y. Plasma microRNAs characterising patients with immune thrombo cytopenic purpura. Thromb. Haemost. 2017, 117, 1420–1431. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2019, 16, 3–17. [Google Scholar] [CrossRef]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Fontana, V.; Jy, W.; Ahn, E.; Dudkiewicz, P.; Horstman, L.; Duncan, R.; Ahn, Y. Increased procoagulant cell-derived microparticles (C-MP) in splenectomized patients with ITP. Thromb. Res. 2008, 122, 599–603. [Google Scholar] [CrossRef]
- Qi, Q.; Yang, B.; Li, H.; Bao, J.; Li, H.; Wang, B.; Mei, Q. Platelet Microparticles Regulate Neutrophil Extracellular Traps in Acute Pancreatitis. Pancreas 2020, 49, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Slomka, A.; Urban, S.K.; Lukacs-Kornek, V.; Żekanowska, E.; Kornek, M. Large Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front. Immunol. 2018, 9, 2723. [Google Scholar] [CrossRef] [PubMed]
- Sprague, D.L.; Elzey, B.D.; Crist, S.A.; Waldschmidt, T.J.; Jensen, R.J.; Ratliff, T.L. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 2008, 111, 5028–5036. [Google Scholar] [CrossRef] [Green Version]
- Dinkla, S.; Van Cranenbroek, B.; Van Der Heijden, W.A.; He, X.; Wallbrecher, R.; Dumitriu, I.E.; Van Der Ven, A.J.; Bosman, G.J.C.G.M.; Koenen, H.J.P.M.; Joosten, I. Platelet microparticles inhibit IL-17 production by regulatory T cells through P-selectin. Blood 2016, 127, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ji, D.; Lu, W.; Li, F.; Huang, X.; Huang, R.; Chen, G. Bone marrow mesenchymal stem cell-derived exosomes induce the Th17/Treg imbalance in immune thrombocytopenia through miR-146a-5p/IRAK1 axis. Hum. Cell 2021, 34, 1360–1374. [Google Scholar] [CrossRef]
- Miao, W.; Song, B.; Shi, B.; Wan, Q.; Lv, Q.; Chen, H.; Zhu, M.; Zhang, L.; Han, Y.; Wu, D. Immune Thrombocytopenia Plasma-Derived Exosomes Impaired Megakaryocyte and Platelet Production through an Apoptosis Pathway. Thromb. Haemost. 2020, 121, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles. Arter. Thromb. Vasc. Biol. 2020, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Uçar, C.; Ören, H.; I˙ rken, G.; Ates, H.; Atabay, B.; Türker, M.; Vergin, C.; Yaprak, I. Investigation of megakaryocyte apoptosis in children with acute and chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2003, 70, 347–352. [Google Scholar] [CrossRef]
- Liu, Z.; Mei, T. Immune thrombocytopenia induces autophagy and suppresses apoptosis in megakaryocytes. Mol. Med. Rep. 2018, 18, 4016–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.-J.; Shan, N.-N. Megakaryocytic dysfunction in immune thrombocytopenia is linked to autophagy. Cancer Cell Int. 2019, 19, 59. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Tan, X.; Jing, H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012, 8, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, L.; Aslam, R.; Speck, E.R.; Kim, M.; Cridland, N.; Webster, M.L.; Chen, P.; Sahib, K.; Ni, H.; Lazarus, A.; et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell–mediated responses that are differentially sensitive to therapy. Blood 2010, 115, 1247–1253. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Key Hypotheses in the Pathogenesis of ITP |
|---|
| Hypothesis 1: Platelets and their immune functions in ITP |
| 1. Platelets promote inflammation and drive pathogenic immuno-modulatory responses. |
| Hypothesis 2: PMPs in ITP |
| 2a. Platelets shed PMPs which interact with immune cells and stimulate pathogenic immuno-modulatory responses |
| 2b. Platelets shed PMPs which impair bone marrow megakaryopoiesis and cause deficits in platelet number and function. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, V.S.; Jolink, A.-T.C.; Amini, S.N.; Zwaginga, J.J.; Netelenbos, T.; Semple, J.W.; Porcelijn, L.; de Haas, M.; Schipperus, M.R.; Kapur, R. Platelets in ITP: Victims in Charge of Their Own Fate? Cells 2021, 10, 3235. https://doi.org/10.3390/cells10113235
Nelson VS, Jolink A-TC, Amini SN, Zwaginga JJ, Netelenbos T, Semple JW, Porcelijn L, de Haas M, Schipperus MR, Kapur R. Platelets in ITP: Victims in Charge of Their Own Fate? Cells. 2021; 10(11):3235. https://doi.org/10.3390/cells10113235
Chicago/Turabian StyleNelson, Vivianne S., Anne-Tess C. Jolink, Sufia N. Amini, Jaap Jan Zwaginga, Tanja Netelenbos, John W. Semple, Leendert Porcelijn, Masja de Haas, Martin R. Schipperus, and Rick Kapur. 2021. "Platelets in ITP: Victims in Charge of Their Own Fate?" Cells 10, no. 11: 3235. https://doi.org/10.3390/cells10113235
APA StyleNelson, V. S., Jolink, A.-T. C., Amini, S. N., Zwaginga, J. J., Netelenbos, T., Semple, J. W., Porcelijn, L., de Haas, M., Schipperus, M. R., & Kapur, R. (2021). Platelets in ITP: Victims in Charge of Their Own Fate? Cells, 10(11), 3235. https://doi.org/10.3390/cells10113235