
PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes


Abstract
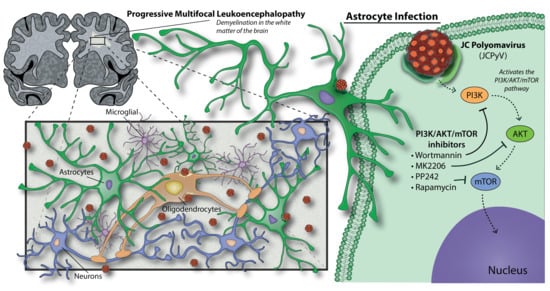
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. JCPyV Infection
2.3. Chemical Inhibitor Treatments
2.4. Indirect Immunofluorescence Staining and Quantitation of JCPyV Infection
2.5. ICW Assay to Measure Protein Expression Using LI-COR Software
2.6. RNA-seq and Pathway Analysis
2.7. Statistical Analysis and Graphing in RStudio
3. Results
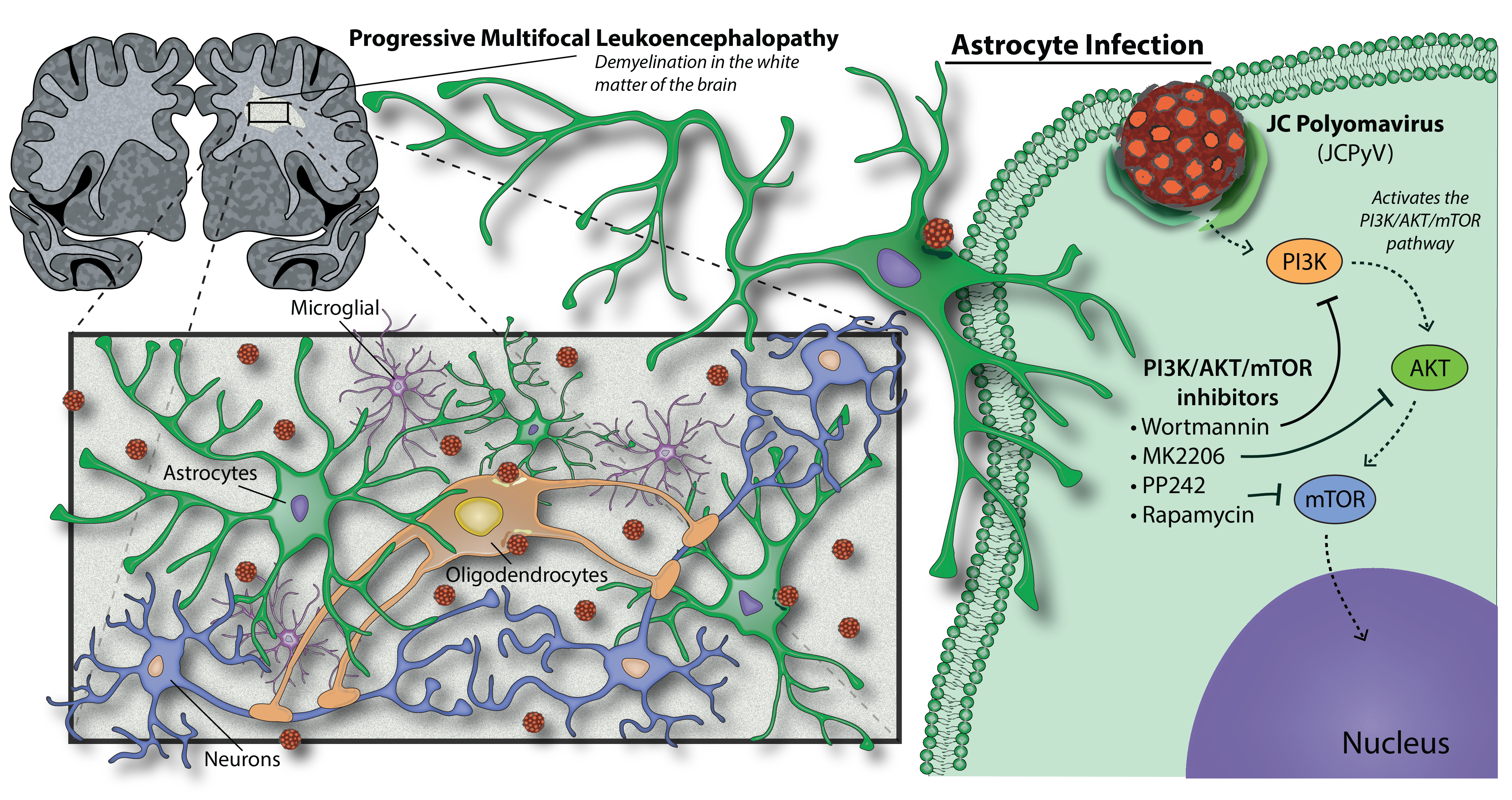
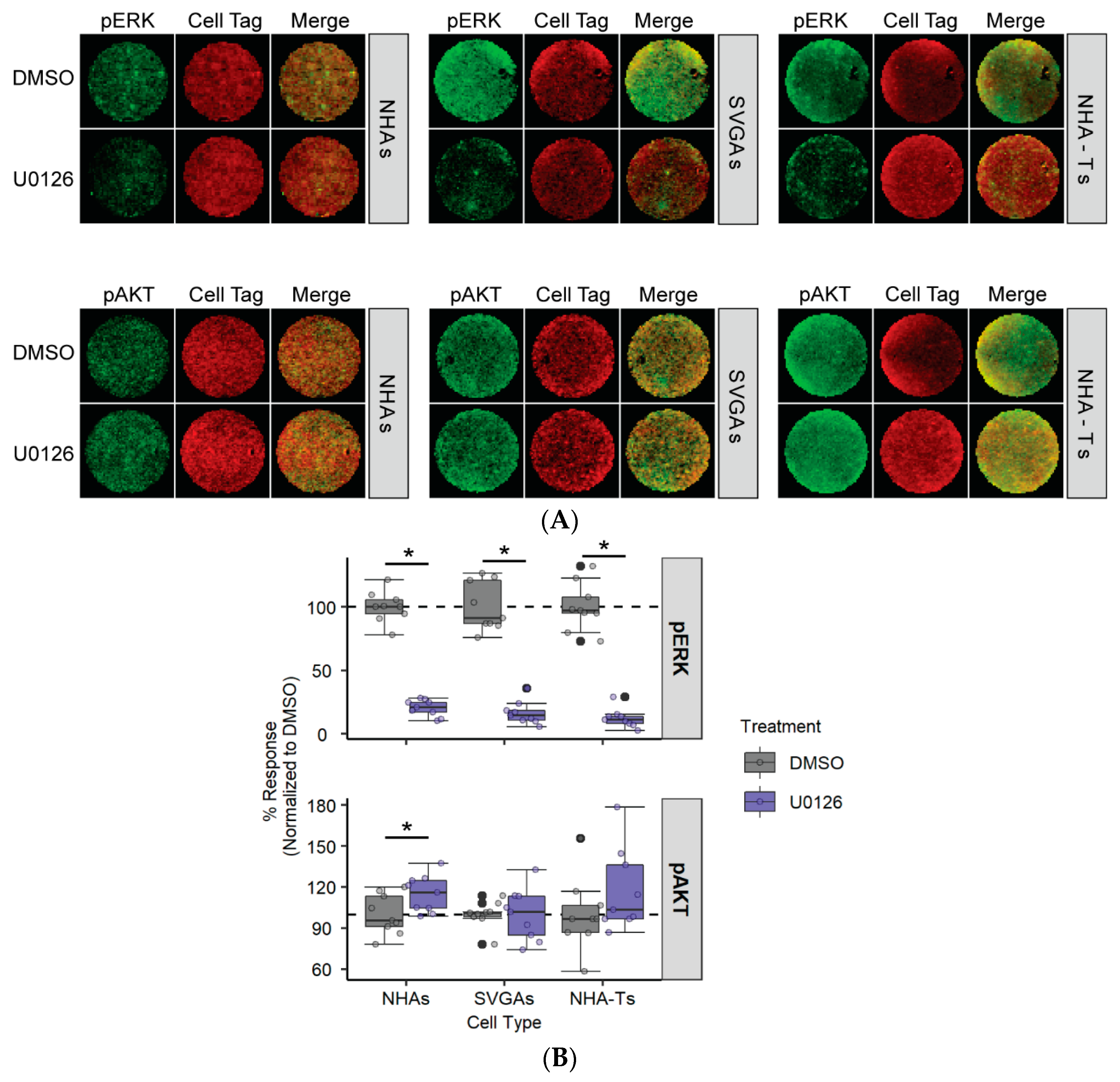
3.1. U0126, a Common MEK Inhibitor, Does Not Reduce JCPyV Infection in Primary Astrocytes
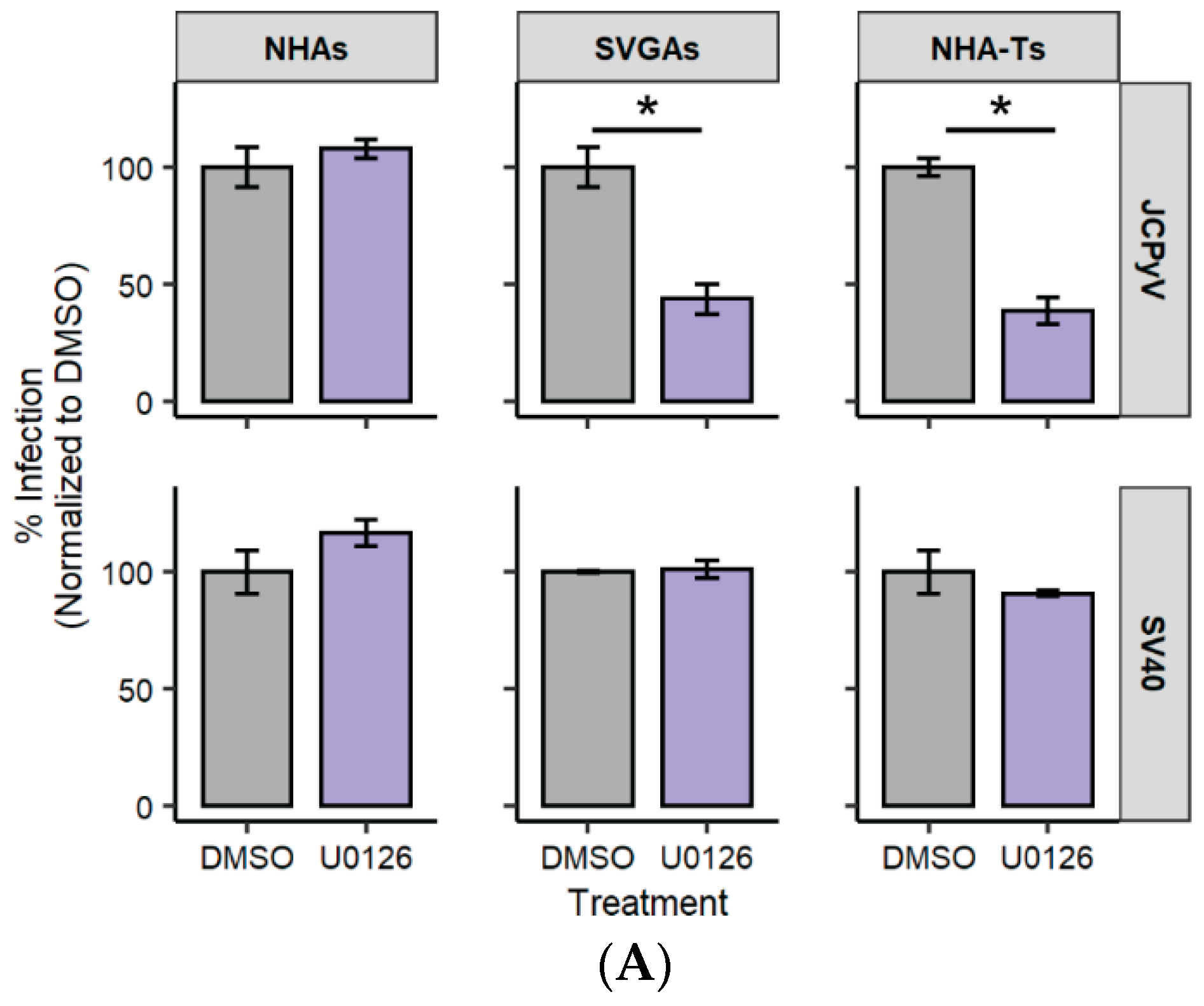
3.2. AKT Phosphorylation Is Moderately Increased in NHAs during U0126 Treatment Compared to SVGAs and NHA-Ts
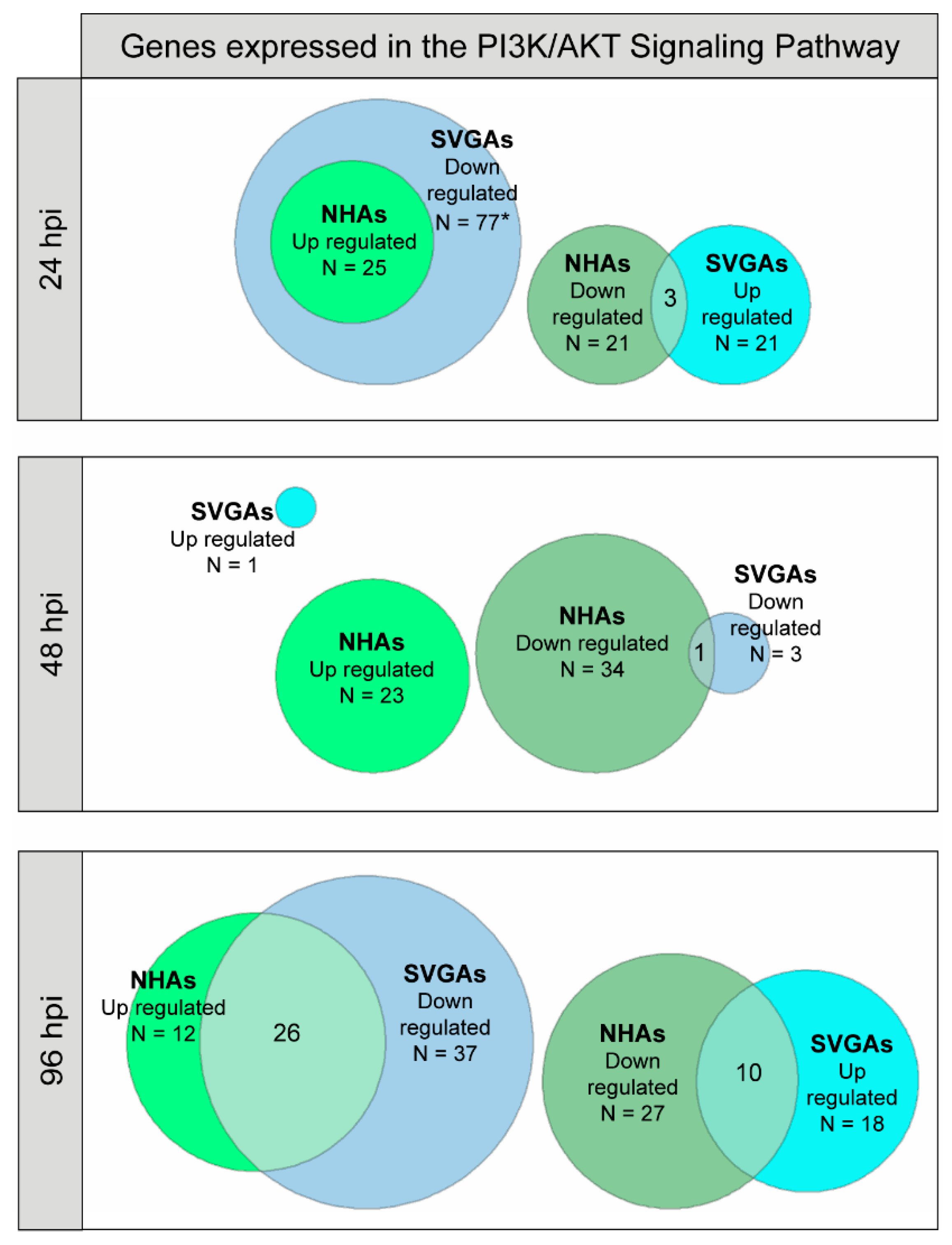
3.3. PI3K/AKT Signaling Pathway Genes Are Upregulated during JCPyV Infection in NHAs
3.4. AKT Is Differentially Expressed and Required for JCPyV Infection in NHAs
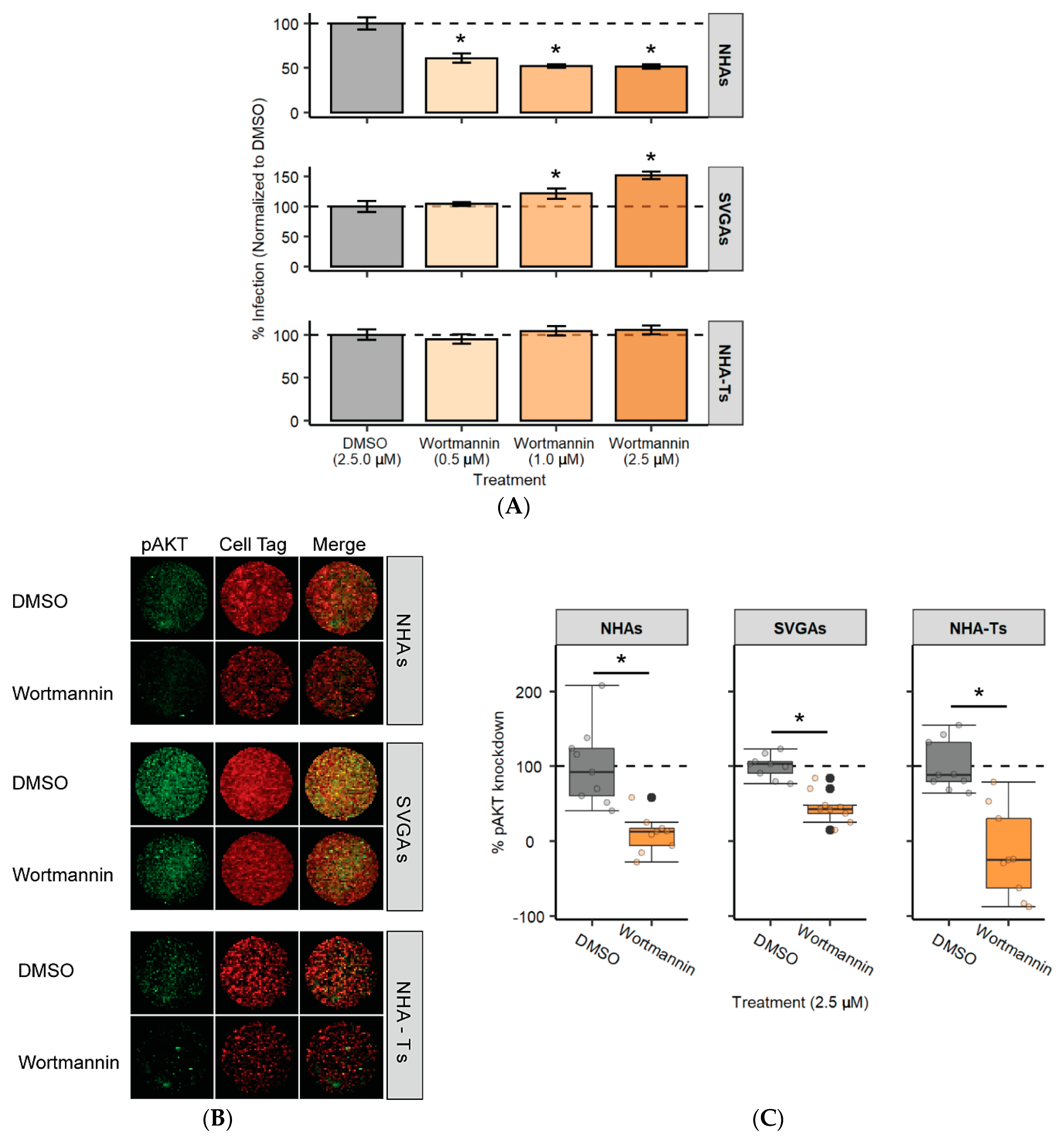
3.5. PI3K Is Required for JCPyV Infection in NHAs
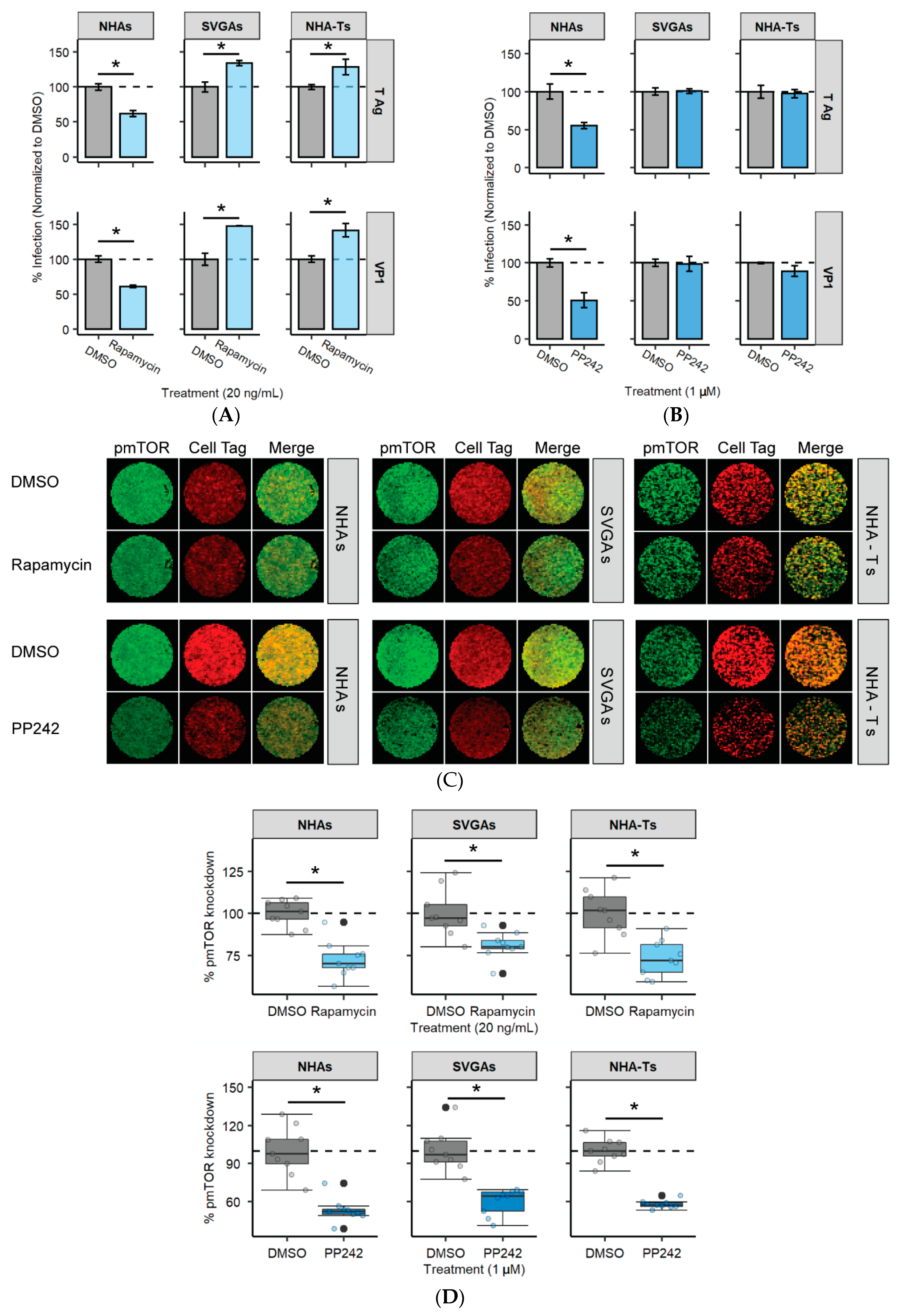
3.6. mTOR Inhibition Significantly Reduces JCPyV Infection in NHAs
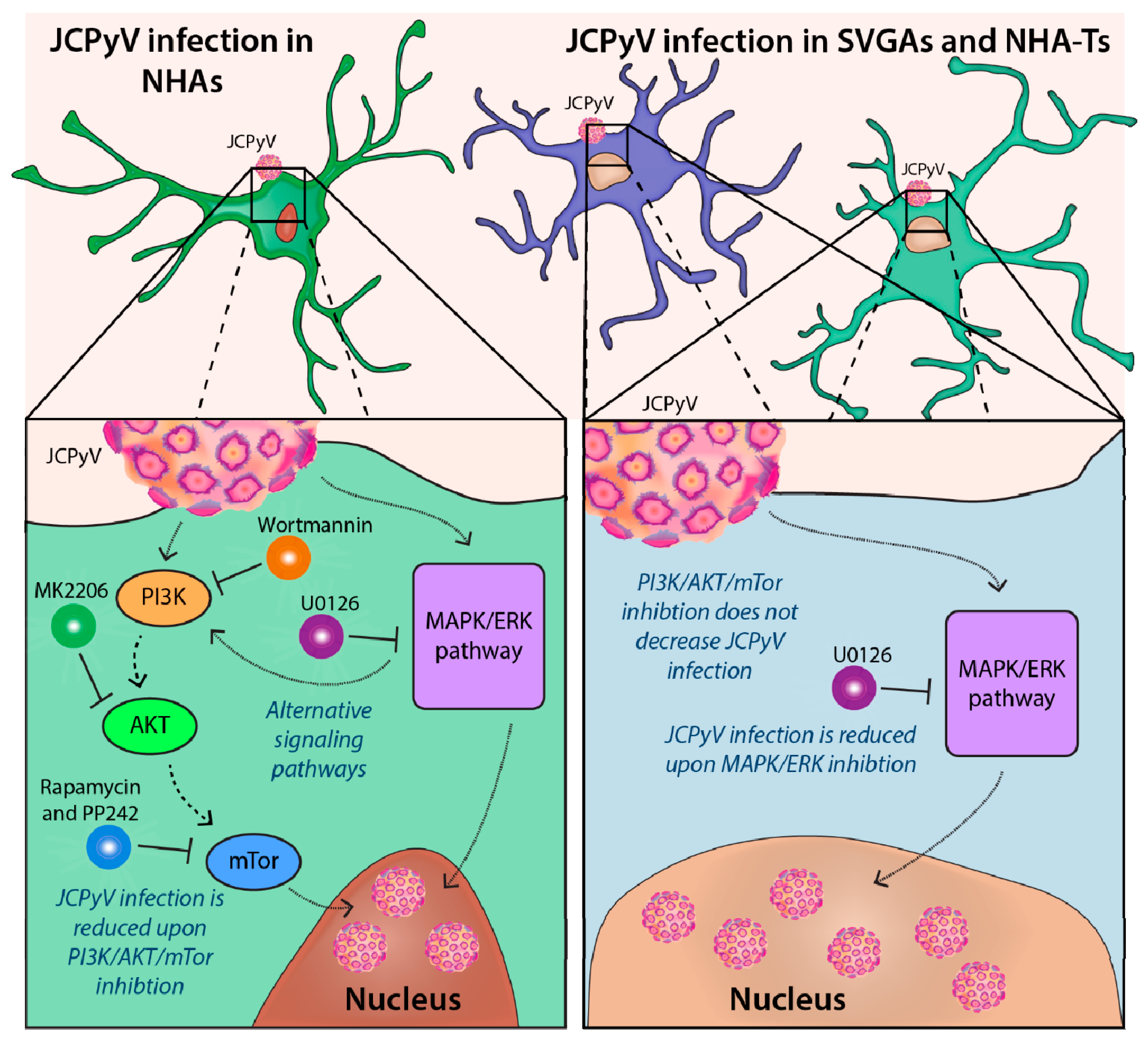
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The Human JC Polyomavirus (JCPyV): Virological Background and Clinical Implications. APMIS 2013, 121, 685–727. [Google Scholar] [CrossRef] [PubMed]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of Papova-like Virus from Human Brain with Progressive Multifocal Leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Silverman, L.; Rubinstein, L.J. Electron Microscopic Observations on a Case of Progressive Multifocal Leukoencephalopathy. Acta Neuropathol. 1965, 5, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Zurhein, G.; Chou, S.M. Particles Resembling Papova Viruses in Human Cerebral Demyelinating Disease. Science 1965, 148, 1477–1479. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of Human Polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC Virus Infection of Hematopoietic Progenitor Cells, Primary B Lymphocytes, and Tonsillar Stromal Cells: Implications for Viral Latency. J. Virol. 1996, 70, 7004–7012. [Google Scholar] [CrossRef]
- Monaco, M.C.G.; Jensen, P.N.; Hou, J.; Durham, L.C.; Major, E.O. Detection of JC Virus DNA in Human Tonsil Tissue: Evidence for Site of Initial Viral Infection. J. Virol. 1998, 72, 9918–9923. [Google Scholar] [CrossRef]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and Reactivation of JC Virus in Peripheral Blood of Human Immunodeficiency Virus Type 1-Infected Patients. J. Clin. Microbiol. 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Chapagain, M.L.; Nerurkar, V.R. Human Polyomavirus JC (JCV) Infection of Human B Lymphocytes: A Possible Mechanism for JCV Transmigration across the Blood-Brain Barrier. J. Infect. Dis. 2010, 202, 184–191. [Google Scholar] [CrossRef]
- White, M.K.; Khalili, K. Pathogenesis of Progressive Multifocal Leukoencephalopathy—Revisited. J. Infect. Dis. 2011, 203, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, P.; Caldarelli-Stefano, R.; Omodeo-Zorini, E.; Vago, L.; Boldorini, R.; Costanzi, G. PCR Detection of JC Virus DNA in Brain Tissue from Patients with and without Progressive Multifocal Leukoencephalopathy. J. Med. Virol. 1995, 47, 219–225. [Google Scholar] [CrossRef]
- Gorelik, L.; Reid, C.; Testa, M.; Brickelmaier, M.; Bossolasco, S.; Pazzi, A.; Bestetti, A.; Carmillo, P.; Wilson, E.; McAuliffe, M.; et al. Progressive Multifocal Leukoencephalopathy (PML) Development Is Associated with Mutations in JC Virus Capsid Protein VP1 That Change Its Receptor Specificity. J. Infect. Dis. 2011, 204, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Elzi, L.; Mueller, N.J.; Garzoni, C.; Cavassini, M.; Fux, C.A.; Vernazza, P.; Bernasconi, E.; Battegay, M.; Hirsch, H.H.; et al. Incidence and Outcome of Progressive Multifocal Leukoencephalopathy over 20 Years of the Swiss HIV Cohort Study. Clin. Infect. Dis. 2009, 48, 1459–1466. [Google Scholar] [CrossRef]
- Anand, P.; Hotan, G.C.; Vogel, A.; Venna, N.; Mateen, F.J. Progressive Multifocal Leukoencephalopathy: A 25-Year Retrospective Cohort Study. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e618. [Google Scholar] [CrossRef]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive Multifocal Leukoencephalopathy and the Spectrum of JC Virus-Related Disease. Nat. Rev. Neurol. 2021, 17, 37–51. [Google Scholar] [CrossRef]
- Carson, K.R.; Evens, A.M.; Richey, E.A.; Habermann, T.M.; Focosi, D.; Seymour, J.F.; Laubach, J.; Bawn, S.D.; Gordon, L.I.; Winter, J.N.; et al. Progressive Multifocal Leukoencephalopathy after Rituximab Therapy in HIV-Negative Patients: A Report of 57 Cases from the Research on Adverse Drug Events and Reports Project. Blood 2009, 113, 4834–4840. [Google Scholar] [CrossRef] [PubMed]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of Natalizumab-Associated Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef]
- Pavlovic, D.; Patera, A.C.; Nyberg, F.; Gerber, M.; Liu, M.; Leukeoncephalopathy, C.P.M. Progressive Multifocal Leukoencephalopathy: Current Treatment Options and Future Perspectives. Ther. Adv. Neurol. Diso. 2015, 8, 255–273. [Google Scholar] [CrossRef]
- Tan, I.L.; Koralnik, I.J.; Rumbaugh, J.A.; Burger, P.C.; King-Rennie, A.; McArthur, J.C. Progressive Multifocal Leukoencephalopathy in a Patient without Immunodeficiency. Neurology 2011, 77, 297–299. [Google Scholar] [CrossRef]
- Vermersch, P.; Kappos, L.; Gold, R.; Foley, J.F.; Olsson, T.; Cadavid, D.; Bozic, C.; Richman, S. Clinical Outcomes of Natalizumab-Associated Progressive Multifocal Leukoencephalopathy(Podcast). Neurology 2011, 76, 1697–1704. [Google Scholar] [CrossRef]
- Prosperini, L.; de Rossi, N.; Scarpazza, C.; Moiola, L.; Cosottini, M.; Gerevini, S.; Capra, R.; Italian PML Study Group. Study Natalizumab-Related Progressive Multifocal Leukoencephalopathy in Multiple Sclerosis: Findings from an Italian Independent Registry. PLoS ONE 2016, 11, e0168376. [Google Scholar] [CrossRef] [PubMed]
- Balduzzi, A.; Lucchini, G.; Hirsch, H.H.; Basso, S.; Cioni, M.; Rovelli, A.; Zincone, A.; Grimaldi, M.; Corti, P.; Bonanomi, S.; et al. Polyomavirus JC-Targeted T-Cell Therapy for Progressive Multiple Leukoencephalopathy in a Hematopoietic Cell Transplantation Recipient. Bone Marrow Transpl. 2011, 46, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, M.; Olson, A.; Marin, D.; Ahmed, S.; Mulanovich, V.; Tummala, S.; Chi, T.L.; Ferrajoli, A.; Kaur, I.; Li, L.; et al. Allogeneic BK Virus–Specific T Cells for Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2018, 379, 1443–1451. [Google Scholar] [CrossRef]
- Cortese, I.; Muranski, P.; Enose-Akahata, Y.; Ha, S.-K.; Smith, B.; Monaco, M.; Ryschkewitsch, C.; Major, E.O.; Ohayon, J.; Schindler, M.K.; et al. Pembrolizumab Treatment for Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2019, 380, 1597–1605. [Google Scholar] [CrossRef]
- Kondo, Y.; Windrem, M.S.; Zou, L.; Chandler-Militello, D.; Schanz, S.J.; Auvergne, R.M.; Betstadt, S.J.; Harrington, A.R.; Johnson, M.; Kazarov, A.; et al. Human Glial Chimeric Mice Reveal Astrocytic Dependence of JC Virus Infection. J. Clin. Investig. 2014, 124, 5323–5336. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Bernards, R.; Friend, S.H.; Gooding, L.R.; Hassell, J.A.; Major, E.O.; Pipas, J.M.; Vandyke, T.; Harlow, E. Large T Antigens of Many Polyomaviruses Are Able to Form Complexes with the Retinoblastoma Protein. J. Virol. 1990, 64, 1353–1356. [Google Scholar] [CrossRef]
- Valle, L.D.; Gordon, J.; Assimakopoulou, M.; Enam, S.; Geddes, J.F.; Varakis, J.N.; Katsetos, C.D.; Croul, S.; Khalili, K. Detection of JC Virus DNA Sequences and Expression of the Viral Regulatory Protein T-Antigen in Tumors of the Central Nervous System. Cancer Res. 2001, 61, 4287–4293. [Google Scholar]
- Dickmanns, A.; Zeitvogel, A.; Simmersbach, F.; Weber, R.; Arthur, A.K.; Dehde, S.; Wildeman, A.G.; Fanning, E. The Kinetics of Simian Virus 40-Induced Progression of Quiescent Cells into S Phase Depend on Four Independent Functions of Large T Antigen. J. Virol. 1994, 68, 5496–5508. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Marshall, L.J.; Nelson, C.D.; Atwood, W.J.; Nath, A.; Khalili, K.; Major, E.O. Molecular Biology, Epidemiology, and Pathogenesis of Progressive Multifocal Leukoencephalopathy, the JC Virus-Induced Demyelinating Disease of the Human Brain. Clin. Microbiol. Rev. 2012, 25, 471–506. [Google Scholar] [CrossRef]
- Wilczek, M.P.; DuShane, J.K.; Armstrong, F.J.; Maginnis, M.S. JC Polyomavirus Infection Reveals Delayed Progression of the Infectious Cycle in Normal Human Astrocytes. J. Virol. 2019, 94, e01331-19. [Google Scholar] [CrossRef]
- Lynch, K.J.; Frisque, R.J. Factors Contributing to the Restricted DNA Replicating Activity of JC Virus. Virology 1991, 180, 306–317. [Google Scholar] [CrossRef]
- Sock, E.; Wegner, M.; Fortunato, E.A.; Grummt, F. Large T-Antigen and Sequences within the Regulatory Region of JC Virus Both Contribute to the Features of JC Virus DNA Replication. Virology 1993, 197, 537–548. [Google Scholar] [CrossRef]
- Major, E.O.; Miller, A.E.; Mourrain, P.; Traub, R.G.; de Widt, E.; Sever, J. Establishment of a Line of Human Fetal Glial Cells That Supports JC Virus Multiplication. Proc. Natl. Acad. Sci. USA 1985, 82, 1257–1261. [Google Scholar] [CrossRef]
- Ariza, A.; Mate, J.L.; Isamat, M.; Calatrava, A.; Fernandez-Vasalo, A.; Navas-Palacios, J.J. Overexpression of Ki-67 and Cyclins A and B1 in JC Virus-Infected Cells of Progressive Multifocal Leukoencephalopathy. J. Neuropathol. Exp. Neurol. 1998, 57, 226–230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez, V.; McElroy, A.K.; Spector, D.H. Mechanisms Governing Maintenance of Cdk1/Cyclin B1 Kinase Activity in Cells Infected with Human Cytomegalovirus. J. Virol. 2003, 77, 13214–13224. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Rushbrook, S.; Davies, S.E.; Morris, L.S.; Scott, I.S.; Vowler, S.L.; Coleman, N.; Alexander, G. Relation between Hepatocyte G1 Arrest, Impaired Hepatic Regeneration, and Fibrosis in Chronic Hepatitis C Virus Infection. Gastroenterology 2005, 128, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 Large T Antigen Targets Multiple Cellular Pathways to Elicit Cellular Transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt Pathway in Cell Cycle Progression, Apoptosis, and Neoplastic Transformation: A Target for Cancer Chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef]
- Choudhury, G.G.; Karamitsos, C.; Hernandez, J.; Gentilini, A.; Bardgette, J.; Abboud, H.E. PI-3-Kinase and MAPK Regulate Mesangial Cell Proliferation and Migration in Response to PDGF. Am. J. Physiol. Renal. 1997, 273, F931–F938. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen Synthase Kinase-3β Regulates Cyclin D1 Proteolysis and Subcellular Localization. Gene. Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Gille, H.; Downward, J. Multiple Ras Effector Pathways Contribute to G1Cell Cycle Progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead Transcription Factors Mediate Cell-Cycle Regulation by Ras and PKB through P27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Muise-Helmericks, R.C.; Grimes, H.L.; Bellacosa, A.; Malstrom, S.E.; Tsichlis, P.N.; Rosen, N. Cyclin D Expression Is Controlled Post-Transcriptionally via a Phosphatidylinositol 3-Kinase/Akt-Dependent Pathway*. J. Biol. Chem. 1998, 273, 29864–29872. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.G. Akt Serine Threonine Kinase Regulates Platelet-Derived Growth Factor-Induced DNA Synthesis in Glomerular Mesangial Cells: Regulation of c-Fos and P27Kip1 Gene Expression. J. Biol. Chem. 2001, 276, 35636–35643. [Google Scholar] [CrossRef]
- Yu, Y.; Alwine, J.C. Human Cytomegalovirus Major Immediate-Early Proteins and Simian Virus 40 Large T Antigen Can Inhibit Apoptosis through Activation of the Phosphatidylinositide 3′-OH Kinase Pathway and the Cellular Kinase Akt. J. Virol. 2002, 76, 3731–3738. [Google Scholar] [CrossRef]
- Yu, Y.; Alwine, J.C. Interaction between Simian Virus 40 Large T Antigen and Insulin Receptor Substrate 1 Is Disrupted by the K1 Mutation, Resulting in the Loss of Large T Antigen-Mediated Phosphorylation of Akt. J. Virol. 2008, 82, 4521–4526. [Google Scholar] [CrossRef]
- Clark, P.; Gee, G.V.; Albright, B.S.; Assetta, B.; Han, Y.; Atwood, W.J.; DiMaio, D. Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection. Viruses 2020, 12, 1190. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Orellana, J.A.; Kwun, H.J.; Artusi, S.; Chang, Y.; Moore, P.S. Sirolimus and Other MTOR Inhibitors Directly Activate Latent Pathogenic Human Polyomavirus Replication. J. Infect. Dis. 2020, 7, 224. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Oki, Y.; Fanale, M.; Romaguera, J.; Fayad, L.; Fowler, N.; Copeland, A.; Samaniego, F.; Kwak, L.W.; Neelapu, S.; Wang, M.; et al. Phase II Study of an AKT Inhibitor MK2206 in Patients with Relapsed or Refractory Lymphoma. Brit. J. Haematol. 2015, 171, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Rapamycin for Longevity: Opinion Article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef]
- Augustine, J.J.; Bodziak, K.A.; Hricik, D.E. Use of Sirolimus in Solid Organ Transplantation. Drugs 2007, 67, 369–391. [Google Scholar] [CrossRef]
- Kastrati, A.; Mehilli, J.; von Beckerath, N.; Dibra, A.; Hausleiter, J.; Pache, J.; Schühlen, H.; Schmitt, C.; Dirschinger, J.; Schömig, A.; et al. Sirolimus-Eluting Stent or Paclitaxel-Eluting Stent vs Balloon Angioplasty for Prevention of Recurrences in Patients with Coronary In-Stent Restenosis: A Randomized Controlled Trial. JAMA 2005, 293, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor Activity of Rapamycin in a Phase I Trial for Patients with Recurrent PTEN-Deficient Glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s Disease: Time for a Clinical Trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Peterson, J.N.; Lin, B.; Shin, J.; Phelan, P.J.; Tsichlis, P.; Schwob, J.E.; Bullock, P.A. The Replication of JCV DNA in the G144 Oligodendrocyte Cell Line Is Dependent Upon Akt. J. Virol. 2017, 91, e00735-17. [Google Scholar] [CrossRef] [PubMed]
- Vacante, D.A.; Traub, R.; Major, E.O. Extension of JC Virus Host Range to Monkey Cells by Insertion of a Simian Virus 40 Enhancer into the JC Virus Regulatory Region. Virology 1989, 170, 353–361. [Google Scholar] [CrossRef]
- DuShane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK Is a Critical Regulator of JC Polyomavirus Infection. J. Virol. 2018, 92, e01529-17. [Google Scholar] [CrossRef]
- Mayberry, C.L.; Wilczek, M.P.; Fong, T.M.; Nichols, S.L.; Maginnis, M.S. GRK2 Mediates β-Arrestin Interactions with 5-HT 2 Receptors for JC Polyomavirus Endocytosis. J. Virol. 2021, 95, e02139-20. [Google Scholar] [CrossRef] [PubMed]
- Mayberry, C.L.; Soucy, A.N.; Lajoie, C.R.; DuShane, J.K.; Maginnis, M.S. JC Polyomavirus Entry by Clathrin-Mediated Endocytosis Is Driven by β-Arrestin. J. Virol. 2019, 93, e01948-18. [Google Scholar] [CrossRef] [PubMed]
- DuShane, J.K.; Mayberry, C.L.; Wilczek, M.P.; Nichols, S.L.; Maginnis, M.S. JCPyV-Induced MAPK Signaling Activates Transcription Factors during Infection. Int. J. Mol. Sci. 2019, 20, 4779. [Google Scholar] [CrossRef] [PubMed]
- DuShane, J.K.; Wilczek, M.P.; Crocker, M.A.; Maginnis, M.S. High-Throughput Characterization of Viral and Cellular Protein Expression Patterns During JC Polyomavirus Infection. Front. Microbiol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, M.P.; Armstrong, F.J.; Geohegan, R.P.; Mayberry, C.L.; DuShane, J.K.; King, B.L.; Maginnis, M.S. The MAPK/ERK Pathway and the Role of DUSP1 in JCPyV Infection of Primary Astrocytes. Viruses 2021, 13, 1834. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2020, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2020, 49, D884–D891. [Google Scholar] [CrossRef]
- Larsson, J. Eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R Package Version 6.1.1, 2020. Available online: https://CRAN.R-project.org/package=eulerr (accessed on 1 June 2021).
- Shaul, Y.D.; Seger, R. The MEK/ERK Cascade: From Signaling Specificity to Diverse Functions. Biochim. Biophys. Acta BBA Mol Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Querbes, W.; Benmerah, A.; Tosoni, D.; Fiore, P.P.D.; Atwood, W.J. A JC Virus-Induced Signal Is Required for Infection of Glial Cells by a Clathrin- and Eps15-Dependent Pathway. J. Virol. 2003, 78, 250–256. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lee, J.T.; Steelman, L.S.; Blalock, W.L.; Moye, P.W.; Chang, F.; Pearce, M.; Shelton, J.G.; White, M.K.; Franklin, R.A.; et al. Interactions between the PI3K and Raf Signaling Pathways Can Result in the Transformation of Hematopoietic Cells. Cancer Detect Prev. 2001, 25, 375–393. [Google Scholar]
- Dangoria, N.S.; Breau, W.C.; Anderson, H.A.; Cishek, D.M.; Norkin, L.C. Extracellular Simian Virus 40 Induces an ERK/MAP Kinase-Independent Signalling Pathway That Activates Primary Response Genes and Promotes Virus Entry. J. Gen. Virol. 1996, 77, 2173–2182. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Collins, C.; Fried, M. Polyoma and SV40 Proteins Differentially Regulate PP2A to Activate Distinct Cellular Signaling Pathways Involved in Growth Control. Proc. Natl. Acad. Sci. USA 2006, 103, 19290–19295. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-MTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Tsuchiya, Y.; Nakayama, K.; Satoh, T.; Nishida, E. Down-regulation of the PI3-kinase/Akt Pathway by ERK MAP Kinase in Growth Factor Signaling. Genes Cells 2008, 13, 941–947. [Google Scholar] [CrossRef] [PubMed]
- heon Rhim, J.; Luo, X.; Gao, D.; Xu, X.; Zhou, T.; Li, F.; Wang, P.; Wong, S.T.C.; Xia, X. Cell Type-Dependent Erk-Akt Pathway Crosstalk Regulates the Proliferation of Fetal Neural Progenitor Cells. Sci. Rep 2016, 6, 26547. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK Inhibition Leads to PI3K/AKT Activation by Relieving a Negative Feedback on ERBB Receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef]
- Link, A.; Shin, S.K.; Nagasaka, T.; Balaguer, F.; Koi, M.; Jung, B.; Boland, C.R.; Goel, A. JC Virus Mediates Invasion and Migration in Colorectal Metastasis. PLoS ONE 2009, 4, e8146. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; and Hirsch, E. PI3K/AKT Signaling Pathway and Cancer: An Updated Review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.B.; White, J.R.; Jee, J.; Argilés, G.; Wan, J.C.M.; Rousseau, B.; Pessin, M.S.; Diaz, L.A. Association of Antineoplasic Therapy with Decreased SARS-CoV-2 Infection Rates in Patients with Cancer. JAMA Oncol. 2021, 7. [Google Scholar] [CrossRef]
- Karam, B.S.; Morris, R.S.; Bramante, C.T.; Puskarich, M.; Zolfaghari, E.J.; Lotfi-Emran, S.; Ingraham, N.E.; Charles, A.; Odde, D.J.; Tignanelli, C.J. MTOR Inhibition in COVID-19: A Commentary and Review of Efficacy in RNA Viruses. J. Med. Virol. 2021, 93, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral Potential of ERK/MAPK and PI3K/AKT/MTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef]
- Appelberg, S.; Gupta, S.; Akusjärvi, S.S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/MTOR/HIF-1 Signaling Identified by Proteo-Transcriptomics of SARS-CoV-2 Infected Cells. Emerg. Microbes Infec. 2020, 9, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Liacini, A.; Seamone, M.E.; Muruve, D.A.; Tibbles, L.A. Anti-BK Virus Mechanisms of Sirolimus and Leflunomide Alone and in Combination: Toward a New Therapy for BK Virus Infection. Transplantation 2010, 90, 1450–1457. [Google Scholar] [CrossRef]
- Andrabi, S.; Gjoerup, O.V.; Kean, J.A.; Roberts, T.M.; Schaffhausen, B. Protein Phosphatase 2A Regulates Life and Death Decisions via Akt in a Context-Dependent Manner. Proc. Natl. Acad. Sci. USA 2007, 104, 19011–19016. [Google Scholar] [CrossRef]
- Bollag, B.; Hofstetter, C.A.; Reviriego-Mendoza, M.M.; Frisque, R.J. JC Virus Small t Antigen Binds Phosphatase PP2A and Rb Family Proteins and Is Required for Efficient Viral DNA Replication Activity. PLoS ONE 2010, 5, e10606. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Yakhontova, K.; Lu, M.; Manzetti, J. BK Polyomavirus Replication in Renal Tubular Epithelial Cells Is Inhibited by Sirolimus, but Activated by Tacrolimus Through a Pathway Involving FKBP-12. Am. J. Transplant. 2016, 16, 821–832. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Randhawa, P.; the AST Infectious Diseases Community of Practice. BK Polyomavirus in Solid Organ Transplantation. Am. J. Transplant. 2013, 13, 179–188. [Google Scholar] [CrossRef]
- Campistol, J.M.; Eris, J.; Oberbauer, R.; Friend, P.; Hutchison, B.; Morales, J.M.; Claesson, K.; Stallone, G.; Russ, G.; Rostaing, L.; et al. Sirolimus Therapy after Early Cyclosporine Withdrawal Reduces the Risk for Cancer in Adult Renal Transplantation. J. Am. Soc. Nephrol. 2006, 17, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. MTOR: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Louis, N.; Evelegh, C.; Graham, F.L. Cloning and Sequencing of the Cellular–Viral Junctions from the Human Adenovirus Type 5 Transformed 293 Cell Line. Virology 1997, 233, 423–429. [Google Scholar] [CrossRef]
- Berk, A.J. Recent Lessons in Gene Expression, Cell Cycle Control, and Cell Biology from Adenovirus. Oncogene 2005, 24, 7673–7685. [Google Scholar] [CrossRef]
- Moran, E. DNA Tumor Virus Transforming Proteins and the Cell Cycle. Curr. Opin. Genet. Dev. 1993, 3, 63–70. [Google Scholar] [CrossRef]
- Nevins, J.R. E2F: A Link between the Rb Tumor Suppressor Protein and Viral Oncoproteins. Science 1992, 258, 424–429. [Google Scholar] [CrossRef]
- Frisch, S.M.; Mymryk, J.S. Adenovirus-5 E1A: Paradox and Paradigm. Nat. Rev. Mol. Cell Biol. 2002, 3, 441–452. [Google Scholar] [CrossRef]
- Levine, A.J. The P53 Protein and Its Interactions with the Oncogene Products of the Small DNA Tumor Viruses. Virology 1990, 177, 419–426. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Hahn, W.C. Involvement of PP2A in Viral and Cellular Transformation. Oncogene 2005, 24, 7746–7755. [Google Scholar] [CrossRef]
- Sariyer, I.K.; Khalili, K.; Safak, M. Dephosphorylation of JC Virus Agnoprotein by Protein Phosphatase 2A: Inhibition by Small t Antigen. Virology 2008, 375, 464–479. [Google Scholar] [CrossRef]
- Jhanwar-Uniya, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of MTOR Complexes: MTORC1 and MTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of MTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-Site Inhibitors of MTOR Target Rapamycin-Resistant Outputs of MTORC1 and MTORC2. PLoS Biol. 2009, 7, e1000038. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-Mediated Viral Latency Is a Novel Mechanism for Merkel Cell Polyomavirus Persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Su, H.; Yu, Z.; Xi, S.; Guo, C.; Hu, Z.; Qu, Y.; Cai, H.; Zhao, Y.; Zhao, H.; et al. Skp2 Modulates Proliferation, Senescence and Tumorigenesis of Glioma. Cancer Cell Int. 2020, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C. F-Box Protein Skp2: A Novel Transcriptional Target of E2F. Oncogene 2006, 25, 2615–2627. [Google Scholar] [CrossRef]
- Assetta, B.; Maginnis, M.S.; Ahufinger, I.G.; Haley, S.A.; Gee, G.V.; Nelson, C.D.S.; O’Hara, B.A.; Ramdial, S.A.A.; Atwood, W.J. 5-HT2 Receptors Facilitate JC Polyomavirus Entry. J. Virol. 2013, 87, 13490–13498. [Google Scholar] [CrossRef]
- Wipf, P.; Halter, R.J. Chemistry and Biology of Wortmannin. Org. Biomol. Chem. 2005, 3, 2053–2061. [Google Scholar] [CrossRef]
- Pittini, Á.; Casaravilla, C.; Allen, J.E.; Díaz, Á. Pharmacological Inhibition of PI3K Class III Enhances the Production of Pro- and Anti-Inflammatory Cytokines in Dendritic Cells Stimulated by TLR Agonists. Int. Immunopharmacol. 2016, 36, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Williams, J.; Sabers, C.; Wiederrecht, G.; Lawrence, J.C.; Abraham, R.T. Direct Inhibition of the Signaling Functions of the Mammalian Target of Rapamycin by the Phosphoinositide 3-kinase Inhibitors, Wortmannin and LY294002. EMBO J. 1996, 15, 5256–5267. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus Infected Choroid Plexus Epithelial Cells Produce Extracellular Vesicles That Infect Glial Cells Independently of the Virus Attachment Receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Huang, K.; Gao, C.; Lau, Q.C.; Pan, H.; Xie, K.; Li, J.; Liu, R.; Zhang, T.; Xie, N.; et al. Proteomics Identification of ITGB3 as a Key Regulator in Reactive Oxygen Species-Induced Migration and Invasion of Colorectal Cancer Cells. Mol. Cell Proteom. 2011, 10, M110.005397. [Google Scholar] [CrossRef]
- Fuentes, P.; Sesé, M.; Guijarro, P.J.; Emperador, M.; Sánchez-Redondo, S.; Peinado, H.; Hümmer, S.; y Cajal, S.R. ITGB3-Mediated Uptake of Small Extracellular Vesicles Facilitates Intercellular Communication in Breast Cancer Cells. Nat. Commun. 2020, 11, 4261. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat. Cell. Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Zhang, N.; Ma, D.; Wang, L.; Zhu, X.; Pan, Q.; Zhao, Y.; Zhu, W.; Zhou, J.; Wang, L.; Chai, Z.; et al. Insufficient Radiofrequency Ablation Treated Hepatocellular Carcinoma Cells Promote Metastasis by Up-Regulation ITGB3. J. Cancer 2017, 8, 3742–3754. [Google Scholar] [CrossRef]
- Anthony, D.C.; Ferguson, B.; Matyzak, M.K.; Miller, K.M.; Esiri, M.M.; Perry, V.H. Differential Matrix Metalloproteinase Expression in Cases of Multiple Sclerosis and Stroke. Neuropath. Appl. Neurobiol. 1997, 23, 406–415. [Google Scholar] [CrossRef]
- Anthony, D.C.; Miller, K.M.; Fearn, S.; Townsend, M.J.; Opdenakker, G.; Wells, G.M.A.; Clements, J.M.; Chandler, S.; Gearing, A.J.H.; Perry, V.H. Matrix Metalloproteinase Expression in an Experimentally-Induced DTH Model of Multiple Sclerosis in the Rat CNS. J. Neuroimmunol. 1998, 87, 62–72. [Google Scholar] [CrossRef]
- Maeda, A.; Sobel, R.A. Matrix Metalloproteinases in the Normal Human Central Nervous System, Microglial Nodules, and Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 1996, 55, 300–309. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | 1° Antibody (Dilution, Source) | 2° Antibody (Dilution, Manufacturer) |
|---|---|---|
| JCPyV T Ag | PAB962 (1:5, hybridoma, Tevethia Lab, Penn State University, State College, PA, USA) | anti-mouse Alexa Fluor 594 (1:1000, Thermo Fisher Scientific (Waltham, MA, USA)) |
| JCPyV VP1 | Ab34756 (1:1000, Abcam) | |
| pERK (P-p44/42 MAPK at T202/Y204) | 9101S (1:750, CST) | anti-rabbit IRDye 800CW (1:10,000, LI-COR, Lincoln, NE, USA) |
| pAKT (S473) | 4060S (1:400, CST) | |
| pmTOR (S2448) | 44-1125G (1:1000, Invitrogen, Waltham, MA, USA) | |
| CST, Cell Signaling Technology; ICW, In-Cell Western assay | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilczek, M.P.; Armstrong, F.J.; Mayberry, C.L.; King, B.L.; Maginnis, M.S. PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes. Cells 2021, 10, 3218. https://doi.org/10.3390/cells10113218
Wilczek MP, Armstrong FJ, Mayberry CL, King BL, Maginnis MS. PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes. Cells. 2021; 10(11):3218. https://doi.org/10.3390/cells10113218
Chicago/Turabian StyleWilczek, Michael P., Francesca J. Armstrong, Colleen L. Mayberry, Benjamin L. King, and Melissa S. Maginnis. 2021. "PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes" Cells 10, no. 11: 3218. https://doi.org/10.3390/cells10113218
APA StyleWilczek, M. P., Armstrong, F. J., Mayberry, C. L., King, B. L., & Maginnis, M. S. (2021). PI3K/AKT/mTOR Signaling Pathway Is Required for JCPyV Infection in Primary Astrocytes. Cells, 10(11), 3218. https://doi.org/10.3390/cells10113218