
Treatment for Viral Hepatitis as Secondary Prevention for Hepatocellular Carcinoma


Abstract
:1. Introduction
2. HBV
3. HCV
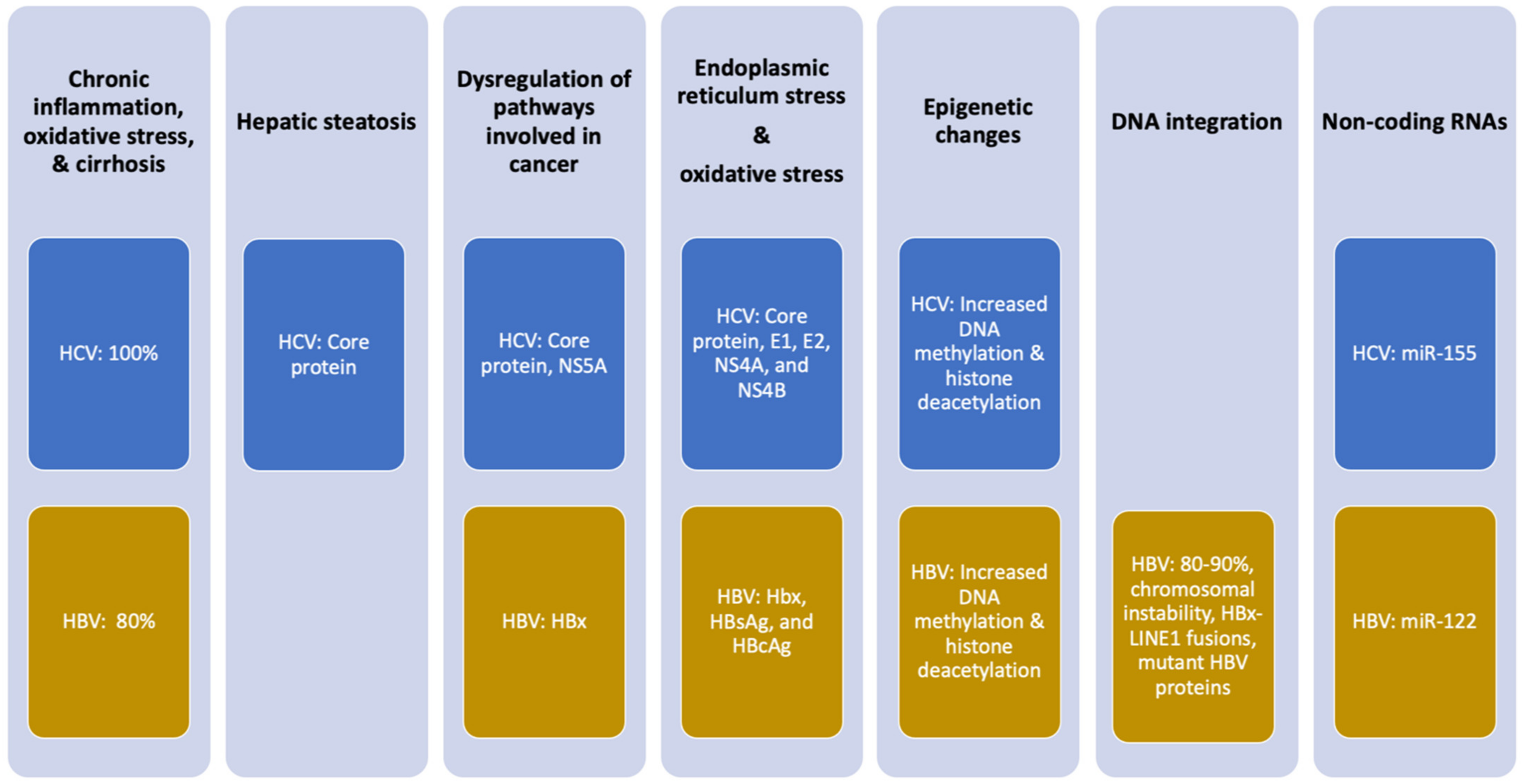
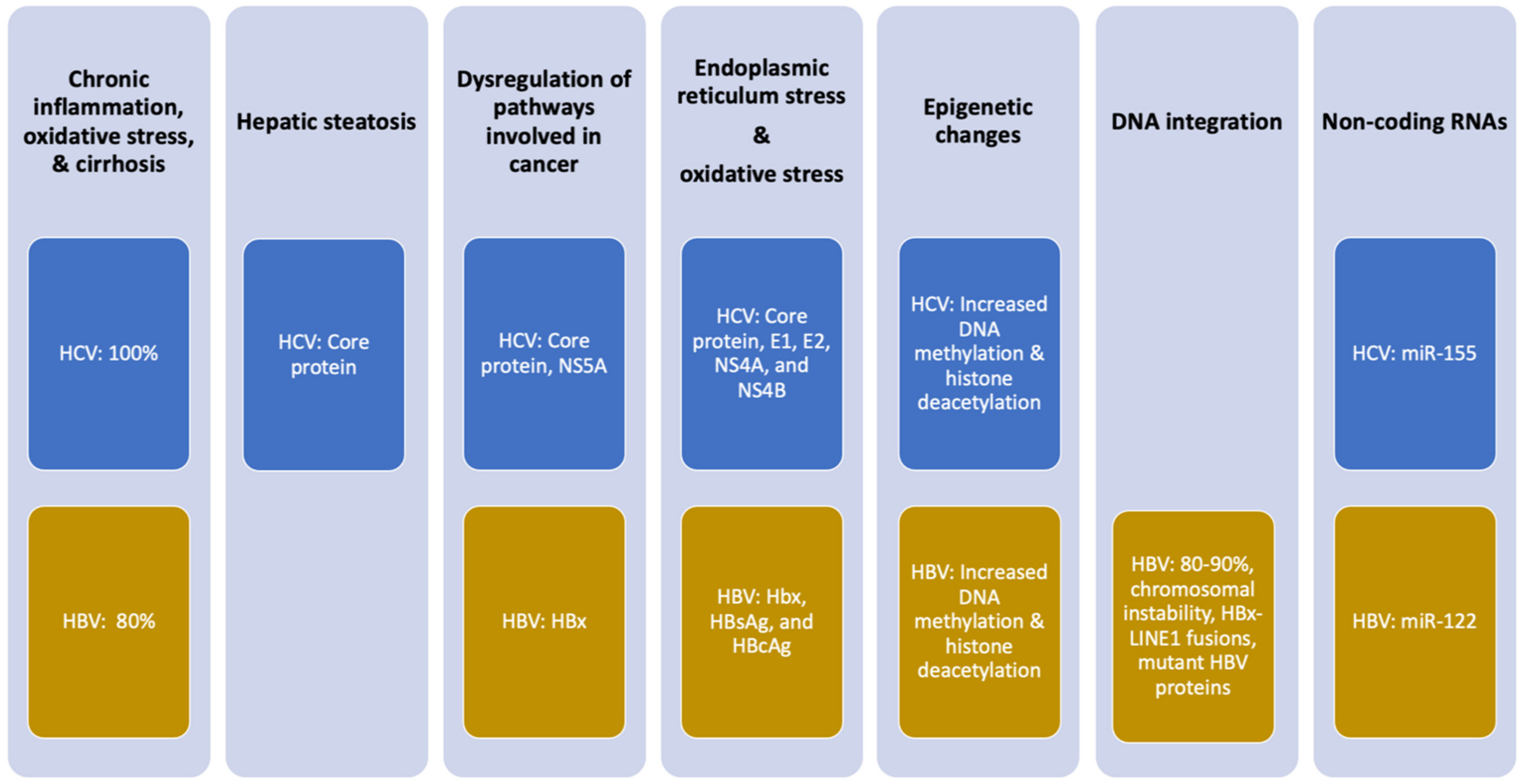
4. Molecular Mechanisms by Which HBV and HCV Induce HCC
5. Chronic Inflammation
6. Viral Proteins Deregulate Cellular Pathways and Induce Oxidative Stress
7. HCV-Induced Hepatic Steatosis
8. HBV Integration in Host DNA
9. Predictors of HCC and Screening Strategies
10. Prediction Models and Scoring Systems for HCC Development
11. Anti-HBV Therapy as Secondary HCC Prevention
12. Tenofovir or Entecavir: One Better Than the Other?
13. Can We Prevent More Cases of HCC by Treating HBV Patients Earlier?
14. Is There a Role for Statins and Aspirin?
15. Novel Treatments
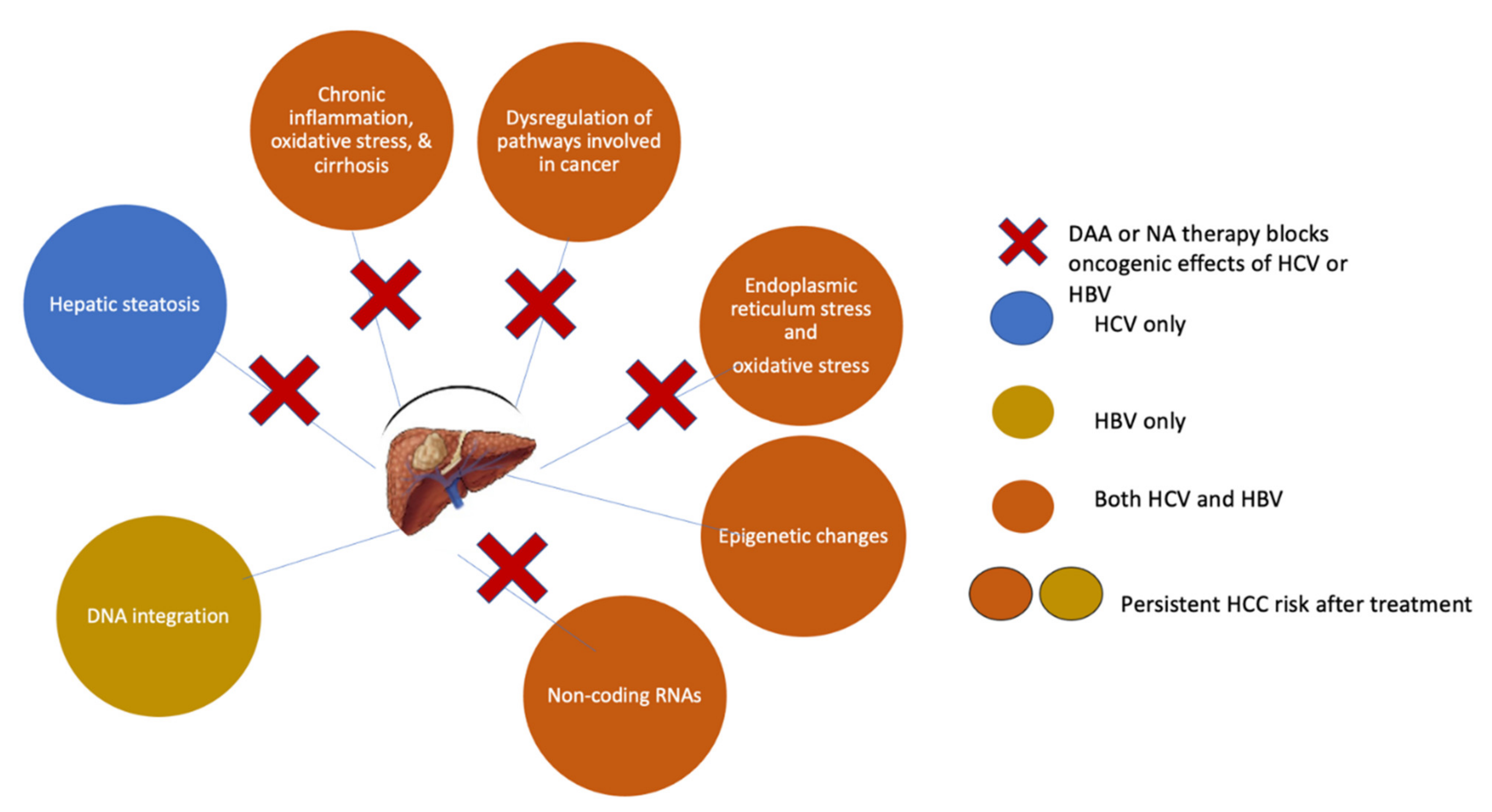
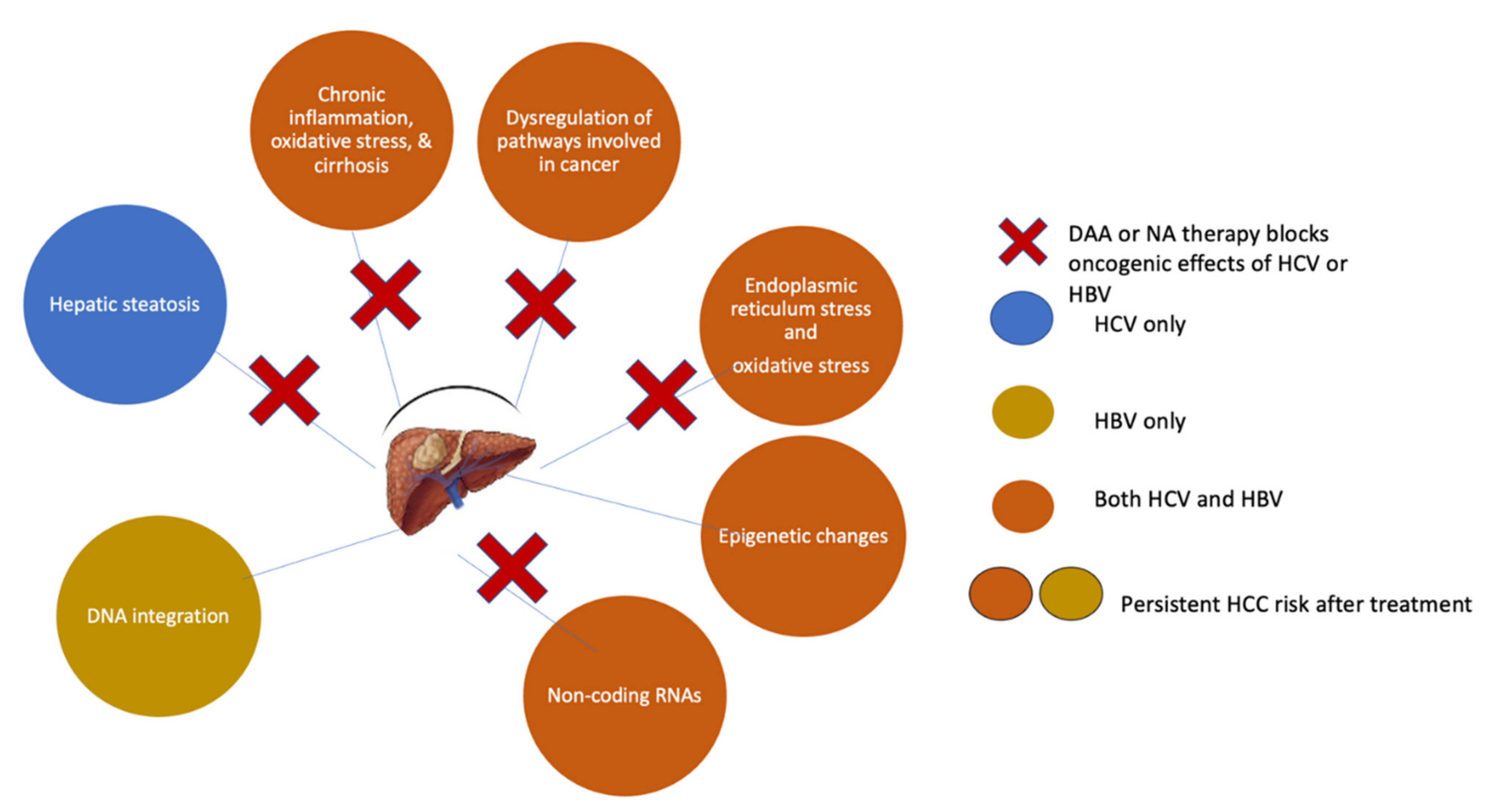
16. Anti-HCV Therapy for the Prevention of HCC
17. HCC Occurrence after DAA Therapy
18. HCC Recurrence in DAA-Treated Patients
19. Residual HCC Risk Post-SVR and the Need for Surveillance
20. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO International Agency for Research on Cancer GLOBOCAN. Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 3 December 2020).
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Valery, P.C.; Laversanne, M.; Clark, P.; Petrick, J.L.; McGlynn, K.A.; Bray, F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology 2017, 67, 600–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef]
- Raffetti, E.; Fattovich, G.; Donato, F. Incidence of hepatocellular carcinoma in untreated subjects with chronic hepatitis B: A systematic review and meta-analysis. Liver Int. 2016, 36, 1239–1251. [Google Scholar] [CrossRef]
- WHO. Global Hepatitis Report. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 7 November 2021).
- Ward, J.W.; Hinman, A.R. What Is Needed to Eliminate Hepatitis B Virus and Hepatitis C Virus as Global Health Threats. Gastroenterology 2019, 156, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridis, G.V.; Chan, H.L.-Y.; Hansen, B.E.; Janssen, H.L.; Lampertico, P. Risk of hepatocellular carcinoma in chronic hepatitis B: Assessment and modification with current antiviral therapy. J. Hepatol. 2015, 62, 956–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodoridis, G.V.; Idilman, R.; Dalekos, G.N.; Buti, M.; Chi, H.; Van Boemmel, F.; Calleja-Panero, J.L.; Sypsa, V.; Goulis, J.; Manolakopoulos, S.; et al. The risk of hepatocellular carcinoma decreases after the first 5 years of entecavir or tenofovir in Caucasians with chronic hepatitis B. Hepatology 2017, 66, 1444–1453. [Google Scholar] [CrossRef]
- Goodgame, B.; Shaheen, N.J.; Galanko, J.; El-Serag, H.B. The risk of end stage liver disease and hepatocellular carcinoma among persons infected with hepatitis C virus: Publication bias? Am. J. Gastroenterol. 2003, 98, 2535–2542. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancebo, A.; González–Diéguez, M.L.; Cadahía, V.; Varela, M.; Pérez, R.; Navascués, C.A.; Sotorríos, N.G.; Martínez, M.; Rodrigo, L.; Rodríguez, M. Annual Incidence of Hepatocellular Carcinoma Among Patients with Alcoholic Cirrhosis and Identification of Risk Groups. Clin. Gastroenterol. Hepatol. 2013, 11, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Mahale, P.; Torres, H.A.; Kramer, J.R.; Hwang, L.-Y.; Li, R.; Brown, E.L.; Engels, E.A. Hepatitis C virus infection and the risk of cancer among elderly US adults: A registry-based case-control study. Cancer 2017, 123, 1202–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160264. [Google Scholar] [CrossRef] [Green Version]
- Lemon, S.M.; McGivern, D.R. Is Hepatitis C Virus Carcinogenic? Gastroenterology 2012, 142, 1274–1278. [Google Scholar] [CrossRef] [Green Version]
- Chayanupatkul, M.; Omino, R.; Mittal, S.; Kramer, J.R.; Richardson, P.; Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Hepatocellular carcinoma in the absence of cirrhosis in patients with chronic hepatitis B virus infection. J. Hepatol. 2016, 66, 355–362. [Google Scholar] [CrossRef]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Wölfl, M.; Rutebemberwa, A.; Mosbruger, T.; Mao, Q.; Li, H.-M.; Netski, D.; Ray, S.; Pardoll, D.; Sidney, J.; Sette, A.; et al. Hepatitis C Virus Immune Escape via Exploitation of a Hole in the T Cell Repertoire. J. Immunol. 2008, 181, 6435–6446. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liao, Z.-X.; Ping, J.; Xu, D.; Wang, H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J. Gastroenterol. 2008, 43, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Hepatitis B virus X protein: A multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-Y.; Kim, C.-M.; Park, Y.-M.; Ryu, W.-S. Hepatitis B virus X protein is essential for the activation of Wnt/β-catenin signaling in hepatoma cells. Hepatology 2004, 39, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Dewantoro, O.; Gani, R.A.; Akbar, N. Hepatocarcinogenesis in viral Hepatitis B infection: The role of HBx and p53. Acta Med. Indones. 2006, 38, 154–159. [Google Scholar]
- Zhang, B.; Han, S.; Feng, B.; Chu, X.; Chen, L.; Wang, R. Hepatitis B virus X protein-mediated non-coding RNA aberrations in the development of human hepatocellular carcinoma. Exp. Mol. Med. 2017, 49, e293. [Google Scholar] [CrossRef]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawada, A.; Kanda, T.; Imazeki, F.; Yokosuka, O. Prevention of hepatitis B virus-associated liver diseases by antiviral therapy. Hepatol. Int. 2016, 10, 574–593. [Google Scholar] [CrossRef]
- Quan, H.; Zhou, F.; Nie, D.; Chen, Q.; Cai, X.; Shan, X.; Zhou, Z.; Chen, K.; Huang, A.; Li, S.; et al. Hepatitis C virus core protein epigenetically silences SFRP1 and enhances HCC aggressiveness by inducing epithelial–mesenchymal transition. Oncogene 2013, 33, 2826–2835. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yang, W.; Song, J.; Wu, Y.; Ni, B. Hepatitis B Virus X Protein-Induced Aberrant Epigenetic Modifications Contributing to Human Hepatocellular Carcinoma Pathogenesis. Mol. Cell. Biol. 2013, 33, 2810–2816. [Google Scholar] [CrossRef] [Green Version]
- Braconi, C.; Valeri, N.; Gasparini, P.; Huang, N.; Taccioli, C.; Nuovo, G.; Suzuki, T.; Croce, C.M.; Patel, T. Hepatitis C Virus Proteins Modulate MicroRNA Expression and Chemosensitivity in Malignant Hepatocytes. Clin. Cancer Res. 2010, 16, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012, 56, 1631–1640. [Google Scholar] [CrossRef]
- Pekow, J.; Bhan, A.K.; Zheng, H.; Chung, R.T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007, 109, 2490–2496. [Google Scholar] [CrossRef]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chrétien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2001, 16, 185–194. [Google Scholar] [CrossRef]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef]
- Lau, C.-C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Na Co, N.; Chan, A.W.; Li, P.-S.; Lung, R.W.; et al. Viral-Human Chimeric Transcript Predisposes Risk to Liver Cancer Development and Progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-C.; Wu, H.C.; Chen, C.-F.; Fausto, N.; Lei, H.-Y.; Su, I.-J. Different Types of Ground Glass Hepatocytes in Chronic Hepatitis B Virus Infection Contain Specific Pre-S Mutants that May Induce Endoplasmic Reticulum Stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-I.; Lu, S.-N.; Liaw, Y.-F.; You, S.-L.; Sun, C.-A.; Wang, L.-Y.; Hsiao, C.K.; Chen, P.-J.; Chen, D.-S.; Chen, C.-J. Hepatitis B e Antigen and the Risk of Hepatocellular Carcinoma. N. Engl. J. Med. 2002, 347, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-J. Risk of Hepatocellular Carcinoma Across a Biological Gradient of Serum Hepatitis B Virus DNA Level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.-S.; Chan, S.L.; Mo, F.; Chan, T.-C.; Loong, H.H.-F.; Wong, G.L.-H.; Lui, Y.Y.-N.; Chan, A.T.-C.; Sung, J.J.-Y.; Yeo, W.; et al. Clinical Scoring System to Predict Hepatocellular Carcinoma in Chronic Hepatitis B Carriers. J. Clin. Oncol. 2010, 28, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tseng, T.-C.; Yang, H.-I.; Lee, M.-H.; Batrla-Utermann, R.; Jen, C.-L.; Lu, S.-N.; Wang, L.-Y.; You, S.-L.; Chen, P.-J.; et al. Predicting Hepatitis B Virus (HBV) Surface Antigen Seroclearance in HBV e Antigen-Negative Patients With Chronic Hepatitis B: External Validation of a Scoring System. J. Infect. Dis. 2014, 211, 1566–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.-A.; Lim, Y.-S.; An, J.; Lee, D.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Chung, Y.-H.; Lee, Y.S.; Suh, D.J. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: Clinical outcomes and durability. Gut 2013, 63, 1325–1332. [Google Scholar] [CrossRef]
- Fonseca, M.A.; Ling, J.Z.J.; Al-Siyabi, O.; Co-Tanko, V.; Chan, E.; Lim, S.G.; Jie, J.L.Z. The efficacy of hepatitis B treatments in achieving HBsAg seroclearance: A systematic review and meta-analysis. J. Viral Hepat. 2020, 27, 650–662. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Iida-Ueno, A.; Enomoto, M.; Tamori, A.; Kawada, N. Hepatitis B virus infection and alcohol consumption. World J. Gastroenterol. 2017, 23, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.-C.; Lee, Y.-C.A.; Hashibe, M.; Dai, M.; Zheng, T.; Boffetta, P. Interaction between Cigarette Smoking and Hepatitis B and C Virus Infection on the Risk of Liver Cancer: A Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.L.-H.; Chan, H.L.-Y.; Wong, C.K.-Y.; Leung, C.; Chan, C.Y.; Ho, P.P.-L.; Chung, V.C.-Y.; Chan, Z.C.-Y.; Tse, Y.-K.; Chim, A.M.-L.; et al. Liver stiffness-based optimization of hepatocellular carcinoma risk score in patients with chronic hepatitis B. J. Hepatol. 2014, 60, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.-K.; Hui, R.W.; Mak, L.-Y.; Fung, J.; Cheung, K.-S.; Liu, K.S.; Wong, D.K.-H.; Lai, C.-L.; Yuen, M.-F. Association between Hepatic Steatosis, Measured by Controlled Attenuation Parameter, and Fibrosis Burden in Chronic Hepatitis B. Clin. Gastroenterol. Hepatol. 2018, 16, 575–583. [Google Scholar] [CrossRef]
- Tan, Y.; Wei, S.; Zhang, W.; Yang, J.; Yan, L. Type 2 diabetes mellitus increases the risk of hepatocellular carcinoma in subjects with chronic hepatitis B virus infection: A meta-analysis and systematic review. Cancer Manag. Res. 2019, 11, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Na Kim, M.; Han, K.-H.; Kim, S.U. Clinical application of transient elastography in patients with chronic viral hepatitis receiving antiviral treatment. Liver Int. 2014, 35, 1103–1115. [Google Scholar] [CrossRef]
- Jung, K.S.; Kim, S.U.; Ahn, S.H.; Park, Y.N.; Kim, D.Y.; Park, J.Y.; Chon, C.Y.; Choi, E.; Han, K.-H. Risk assessment of hepatitis B virus-related hepatocellular carcinoma development using liver stiffness measurement (FibroScan). Hepatology 2010, 53, 885–894. [Google Scholar] [CrossRef]
- Huang, R.; Jiang, N.; Yang, R.; Geng, X.; Lin, J.; Xu, G.; Liu, D.; Chen, J.; Zhou, G.; Wang, S.; et al. Fibroscan improves the diagnosis sensitivity of liver fibrosis in patients with chronic hepatitis B. Exp. Ther. Med. 2016, 11, 1673–1677. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.-F.; Tanaka, Y.; Fong, D.; Fung, J.; Wong, D.K.-H.; Yuen, J.C.-H.; But, D.Y.-K.; Chan, A.O.-O.; Wong, B.C.-Y.; Mizokami, M.; et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol. 2008, 50, 80–88. [Google Scholar] [CrossRef]
- Yang, H.-I.; Yuen, M.-F.; Chan, H.L.-Y.; Han, K.-H.; Chen, P.-J.; Kim, D.-Y.; Ahn, S.H.; Chen, C.-J.; Wong, V.W.-S.; Seto, W.-K. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): Development and validation of a predictive score. Lancet Oncol. 2011, 12, 568–574. [Google Scholar] [CrossRef]
- Papatheodoridis, G.; Dalekos, G.; Sypsa, V.; Yurdaydin, C.; Buti, M.; Goulis, J.; Calleja-Panero, J.L.; Chi, H.; Manolakopoulos, S.; Mangia, G.; et al. PAGE-B predicts the risk of developing hepatocellular carcinoma in Caucasians with chronic hepatitis B on 5-year antiviral therapy. J. Hepatol. 2015, 64, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.S.; Kim, S.U.; Song, K.J.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Kim, B.K.; Han, K.-H. Validation of hepatitis B virus-related hepatocellular carcinoma prediction models in the era of antiviral therapy. Hepatology 2015, 62, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Health Sector Strategy on Viral Hepatitis 2016–2021. Available online: https://apps.who.int/iris/handle/10665/246177 (accessed on 7 November 2021).
- Hsu, C.-S.; Chao, Y.-C.; Lin, H.H.; Chen, D.-S.; Kao, J.-H. Systematic Review: Impact of Interferon-based Therapy on HCV-related Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 9954. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Zeng, X.-T.; Yang, Z.; Meng, Z. Effect and Safety of Interferon for Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e61361. [Google Scholar] [CrossRef] [Green Version]
- Miyake, Y.; Kobashi, H.; Yamamoto, K. Meta-analysis: The effect of interferon on development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. J. Gastroenterol. 2009, 44, 470–475. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Zhao, W.; Zhong, Y.-D.; Xia, H.M.; Shen, L.; Zhang, N. Interferon therapy in chronic hepatitis B reduces progression to cirrhosis and hepatocellular carcinoma: A meta-analysis. J. Viral Hepat. 2009, 16, 265–271. [Google Scholar] [CrossRef]
- Cornberg, M.; Lok, A.S.-F.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.; Chang, K.-M.; et al. Guidance for design and endpoints of clinical trials in chronic hepatitis B—Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. J. Hepatol. 2019, 72, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.-J.; Yang, Y.-W.; Chen, J.-D.; You, S.-L.; Yang, H.-I.; Lee, M.-H.; Lai, M.-S.; Chen, C.-J. Significant reduction in end-stage liver diseases burden through the national viral hepatitis therapy program in Taiwan. Hepatology 2014, 61, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Newcomb, C.W.; Carbonari, D.M.; Roy, J.A.; Torgersen, J.; Althoff, K.N.; Kitahata, M.M.; Reddy, K.R.; Lim, J.K.; Silverberg, M.J.; et al. Risk of HCC With Hepatitis B Viremia Among HIV/HBV-Coinfected Persons in North America. Hepatology 2021, 74, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.-C.; Liu, C.-J.; Yang, H.-C.; Su, T.-H.; Wang, C.; Chen, C.; Kuo, S.F.; Liu, C.-H.; Chen, P.-J.; Chen, D.-S.; et al. High Levels of Hepatitis B Surface Antigen Increase Risk of Hepatocellular Carcinoma in Patients with Low HBV Load. Gastroenterology 2012, 142, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Tseng, T.-C.; Liu, C.-J.; Hsu, C.-Y.; Hong, C.-M.; Su, T.-H.; Yang, W.-T.; Chen, C.-L.; Yang, H.-C.; Huang, Y.-T.; Kuo, S.F.-T.; et al. High Level of Hepatitis B Core–Related Antigen Associated With Increased Risk of Hepatocellular Carcinoma in Patients With Chronic HBV Infection of Intermediate Viral Load. Gastroenterology 2019, 157, 1518–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between Hepatitis B Virus Mutations and the Risk of Hepatocellular Carcinoma: A Meta-Analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.L.-H.; Chan, H.L.-Y.; Yiu, K.K.-L.; Lai, J.W.-Y.; Chan, V.K.-K.; Cheung, K.K.-C.; Wong, E.W.-N.; Wong, V.W.-S. Meta-analysis: The association of hepatitis B virus genotypes and hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2013, 37, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Liaw, Y.-F.; Sung, J.J.Y.; Chow, W.C.; Farrell, G.; Lee, C.-Z.; Yuen, H.; Tanwandee, T.; Tao, Q.-M.; Shue, K.; Keene, O.; et al. Lamivudine for Patients with Chronic Hepatitis B and Advanced Liver Disease. N. Engl. J. Med. 2004, 351, 1521–1531. [Google Scholar] [CrossRef]
- Singal, A.K.; Salameh, H.; Kuo, Y.-F.; Fontana, R.J. Meta-analysis: The impact of oral anti-viral agents on the incidence of hepatocellular carcinoma in chronic hepatitis B. Aliment. Pharmacol. Ther. 2013, 38, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Terrault, N.A.; Lok, A.S.; McMahon, B.J.; Chang, K.-M.; Hwang, J.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]
- Chang, T.-T.; Liaw, Y.-F.; Wu, S.-S.; Schiff, E.; Han, K.-H.; Lai, C.-L.; Safadi, R.; Lee, S.S.; Halota, W.; Goodman, Z.; et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010, 52, 886–893. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Kim, W.R.; Loomba, R.; Berg, T.; Schall, R.E.A.; Yee, L.J.; Dinh, P.V.; Flaherty, J.F.; Martins, E.B.; Therneau, T.M.; Jacobson, I.; et al. Impact of long-term tenofovir disoproxil fumarate on incidence of hepatocellular carcinoma in patients with chronic hepatitis B. Cancer 2015, 121, 3631–3638. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; To, W.; Mak, L.; Seto, W.; Ning, Q.; Fung, J.; Lai, C.; Yuen, M. A large real-world cohort study examining the effects of long-term entecavir on hepatocellular carcinoma and HBsAg seroclearance. J. Viral Hepat. 2019, 27, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lim, J.K.; Lee, H.M.; Lok, A.S.; Nguyen, M.; Pan, C.Q.; Mannalithara, A.; Te, H.; Reddy, R.K.; Trinh, H.; et al. Lower Observed Hepatocellular Carcinoma Incidence in Chronic Hepatitis B Patients Treated With Entecavir: Results of the ENUMERATE Study. Am. J. Gastroenterol. 2016, 111, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, H.J.; Lee, J.; Cho, S.; Ko, M.J.; Lim, Y.-S. Risk of Hepatocellular Carcinoma in Patients Treated with Entecavir vs Tenofovir for Chronic Hepatitis B. JAMA Oncol. 2019, 5, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.U.; Seo, Y.S.; Lee, H.A.; Na Kim, M.; Lee, Y.R.; Lee, H.W.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Han, K.-H.; et al. A multicenter study of entecavir vs. tenofovir on prognosis of treatment-naïve chronic hepatitis B in South Korea. J. Hepatol. 2019, 71, 456–464. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Wong, G.L.-H.; Chen, C.-H.; Peng, C.-Y.; Yeh, M.-L.; Cheung, K.-S.; Toyoda, H.; Huang, C.-F.; Trinh, H.; Xie, Q.; et al. Tenofovir Versus Entecavir for Hepatocellular Carcinoma Prevention in an International Consortium of Chronic Hepatitis B. Am. J. Gastroenterol. 2019, 115, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Yoon, E.L.; Jun, D.W.; Ahn, S.B.; Lee, H.-Y.; Jeong, J.Y.; Kim, H.S.; Jeong, S.W.; Kim, S.E.; Shim, J.-J.; et al. No Difference in Incidence of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Virus Infection Treated With Entecavir vs Tenofovir. Clin. Gastroenterol. Hepatol. 2020, 18, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Yip, T.C.; Wong, V.W.; Chan, H.L.; Tse, Y.K.; Lui, G.C.; Wong, G.L. Tenofovir Is Associated with Lower Risk of Hepatocellular Carcinoma Than Entecavir in Patients With Chronic HBV Infection in China. Gastroenterology 2020, 158, 215–225. [Google Scholar] [CrossRef]
- Lee, S.W.; Kwon, J.H.; Lee, H.L.; Yoo, S.H.; Nam, H.C.; Sung, P.S.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Yoon, S.K.; et al. Comparison of tenofovir and entecavir on the risk of hepatocellular carcinoma and mortality in treatment-naïve patients with chronic hepatitis B in Korea: A large-scale, propensity score analysis. Gut 2019, 69, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Dave, S.; Park, S.; Murad, M.H.; Barnard, A.; Prokop, L.; Adams, L.A.; Singh, S.; Loomba, R. Comparative Effectiveness of Entecavir Versus Tenofovir for Preventing Hepatocellular Carcinoma in Patients with Chronic Hepatitis B: A Systematic Review and Meta-Analysis. Hepatology 2020, 73, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Hsu, Y.-C.; Chen, T.-H.; Ji, F.; Chen, I.-S.; Tsai, Y.-N.; Hai, H.; Thuy, L.T.T.; Hosaka, T.; Sezaki, H.; et al. Hepatocellular carcinoma incidence with tenofovir versus entecavir in chronic hepatitis B: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 1039–1052. [Google Scholar] [CrossRef]
- Papatheodoridi, M.; Hiriart, J.B.; Lupsor-Platon, M.; Bronte, F.; Boursier, J.; Elshaarawy, O.; Marra, F.; Thiele, M.; Markakis, G.; Payance, A.; et al. Refining the Baveno VI elastography criteria for the definition of compensated advanced chronic liver disease. J. Hepatol. 2020, 74, 1109–1116. [Google Scholar] [CrossRef]
- Yuan, J.; Peng, Y.; Hao, F.-B.; Wang, Y.-Q.; Wang, C.-R.; Zhong, G.-C. No difference in hepatocellular carcinoma risk in chronic hepatitis B patients treated with tenofovir vs entecavir: Evidence from an updated meta-analysis. Aging 2021, 13, 7147–7165. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.; Chok, K.S. The role of oral antiviral therapy in hepatitis B-related hepatocellular carcinoma. Hepatoma Res. 2017, 3, 284. [Google Scholar] [CrossRef]
- Kim, G.-A.; Lim, Y.-S.; Han, S.; Choi, J.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Lee, Y.S. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut 2017, 67, 945–952. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Kennedy, P.T.; Sandalova, E.; Jo, J.; Gill, U.; Ushiro–Lumb, I.; Tan, A.T.; Naik, S.; Foster, G.R.; Bertoletti, A. Preserved T-Cell Function in Children and Young Adults With Immune-Tolerant Chronic Hepatitis B. Gastroenterology 2012, 143, 637–645. [Google Scholar] [CrossRef]
- Kim, H.-L.; Kim, G.-A.; Park, J.-A.; Kang, H.-R.; Lee, E.-K.; Lim, Y.-S. Cost-effectiveness of antiviral treatment in adult patients with immune-tolerant phase chronic hepatitis B. Gut 2020, 70, 2172–2182. [Google Scholar] [CrossRef]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.-S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Rached, A.A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Goh, M.J.; Sinn, D.H.; Kim, S.; Woo, S.Y.; Cho, H.; Kang, W.; Gwak, G.; Paik, Y.; Choi, M.S.; Lee, J.H.; et al. Statin Use and the Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. Hepatology 2019, 71, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Statins Are Associated with a Reduced Risk of Hepatocellular Cancer: A Systematic Review and Meta-analysis. Gastroenterology 2013, 144, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S.C.; et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, M.; Heusinger-Ribeiro, J.; Goppelt-Struebe, M. Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br. J. Pharmacol. 2001, 133, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Tsan, Y.-T.; Lee, C.-H.; Wang, J.-D.; Chen, P.-C. Statins and the Risk of Hepatocellular Carcinoma in Patients with Hepatitis B Virus Infection. J. Clin. Oncol. 2012, 30, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-Y.; Hsu, Y.-C.; Tseng, H.-C.; Yu, S.-H.; Lin, J.-T.; Wu, M.-S.; Wu, C.-Y. Association of Daily Aspirin Therapy With Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis B. JAMA Intern. Med. 2019, 179, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Duberg, A.-S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Asselah, T.; Loureiro, D.; Boyer, N.; Mansouri, A. Targets and future direct-acting antiviral approaches to achieve hepatitis B virus cure. Lancet Gastroenterol. Hepatol. 2019, 4, 883–892. [Google Scholar] [CrossRef]
- Brown, J.L. Interferon therapy reduces the risk for hepatocellular carcinoma. Gut 2000, 47, 610–611. [Google Scholar] [CrossRef] [Green Version]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445. [Google Scholar] [CrossRef]
- Terrault, N.A.; Hassanein, T.I. Management of the patient with SVR. J. Hepatol. 2016, 65, S120–S129. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Boccaccio, V.; Russo, M.L.; Maisonneuve, P. Is the benefit of treating patients with cirrhosis proven? Liver Int. 2016, 36, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Reig, M.; Mariño, Z.; Perelló, C.; Iñarrairaegui, M.; Ribeiro, A.; Lens, S.; Díaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.; Vale, A.M.; Rodrigues, S.; Gonçalves, R.; Albuquerque, A.; Pereira, P.; Lopes, S.; Silva, M.; Andrade, P.; Morais, R.; et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. J. Hepatol. 2016, 65, 1070–1071. [Google Scholar] [CrossRef] [Green Version]
- Ravi, S.; Axley, P.; Jones, D.; Kodali, S.; Simpson, H.; McGuire, B.M.; Singal, A.K. Unusually High Rates of Hepatocellular Carcinoma After Treatment With Direct-Acting Antiviral Therapy for Hepatitis C Related Cirrhosis. Gastroenterology 2017, 152, 911–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, A.W.; Reddy, K.R.; Telep, L.E.; Osinusi, A.O.; Brainard, D.M.; Buti, M.; Chokkalingam, A.P. Direct-acting antiviral treatment for hepatitis C virus infection and risk of incident liver cancer: A retrospective cohort study. Aliment. Pharmacol. Ther. 2018, 47, 1278–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janjua, N.Z.; Wong, S.; Darvishian, M.; Butt, Z.A.; Yu, A.; Binka, M.; Alvarez, M.; Woods, R.; Yoshida, E.M.; Ramji, A.; et al. The impact of SVR from direct-acting antiviral- and interferon-based treatments for HCV on hepatocellular carcinoma risk. J. Viral Hepat. 2020, 27, 781–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahon, P.; Layese, R.; Bourcier, V.; Cagnot, C.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Lédinghen, V.; Ouzan, D.; et al. Incidence of Hepatocellular Carcinoma After Direct Antiviral Therapy for HCV in Patients With Cirrhosis Included in Surveillance Programs. Gastroenterology 2018, 155, 1436–1450. [Google Scholar] [CrossRef] [Green Version]
- Carrat, F.; Fontaine, H.; Dorival, C.; Simony, M.; Diallo, A.; Hezode, C.; De Ledinghen, V.; Larrey, D.; Haour, G.; Bronowicki, J.-P.; et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: A prospective cohort study. Lancet 2019, 393, 1453–1464. [Google Scholar] [CrossRef]
- Piñero, F.; Mendizabal, M.; Ridruejo, E.; Wolff, F.H.; Ameigeiras, B.; Anders, M.; Schinoni, M.I.; Reggiardo, V.; Palazzo, A.; Videla, M.; et al. Treatment with direct-acting antivirals for HCV decreases but does not eliminate the risk of hepatocellular carcinoma. Liver Int. 2019, 39, 1033–1043. [Google Scholar] [CrossRef]
- Muzica, C.M.; Stanciu, C.; Huiban, L.; Singeap, A.-M.; Sfarti, C.; Zenovia, S.; Cojocariu, C.; Trifan, A. Hepatocellular carcinoma after direct-acting antiviral hepatitis C virus therapy: A debate near the end. World J. Gastroenterol. 2020, 26, 6770–6781. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Green, P.K.; Berry, K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J. Hepatol. 2018, 68, 25–32. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Asch, S.M.; Cao, Y.; Li, L.; El-Serag, H.B. Long-Term Risk of Hepatocellular Carcinoma in HCV Patients Treated with Direct Acting Antiviral Agents. Hepatology 2019, 71, 44–55. [Google Scholar] [CrossRef]
- Tani, J.; Morishita, A.; Sakamoto, T.; Takuma, K.; Nakahara, M.; Fujita, K.; Oura, K.; Tadokoro, T.; Mimura, S.; Nomura, T.; et al. Simple scoring system for prediction of hepatocellular carcinoma occurrence after hepatitis C virus eradication by direct-acting antiviral treatment: All Kagawa Liver Disease Group Study. Oncol. Lett. 2020, 19, 2205–2212. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Tokumoto, Y.; Joko, K.; Michitaka, K.; Horiike, N.; Tanaka, Y.; Tada, F.; Kisaka, Y.; Nakanishi, S.; Yamauchi, K.; et al. Sex difference in the development of hepatocellular carcinoma after direct-acting antiviral therapy in patients with HCV infection. J. Med Virol. 2020, 92, 3507–3515. [Google Scholar] [CrossRef]
- Cheung, M.C.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettke, F.; Schlevogt, B.; Deterding, K.; Wranke, A.; Smith, A.; Port, K.; Manns, M.P.; Vogel, A.; Cornberg, M.; Wedemeyer, H. Interferon-free therapy of chronic hepatitis C with direct-acting antivirals does not change the short-term risk for de novo hepatocellular carcinoma in patients with liver cirrhosis. Aliment. Pharmacol. Ther. 2017, 47, 516–525. [Google Scholar] [CrossRef]
- Poordad, F.; Castro, R.E.; Asatryan, A.; Aguilar, H.; Cacoub, P.; Dieterich, D.; Marinho, R.T.; Carvalho, A.; Siddique, A.; Hu, Y.B.; et al. Long-term safety and efficacy results in hepatitis C virus genotype 1‒infected patients receiving ombitasvir/paritaprevir/ritonavir + dasabuvir ± ribavirin in the TOPAZ-I and TOPAZ-II trials. J. Viral Hepat. 2020, 27, 497–504. [Google Scholar] [CrossRef]
- Sangiovanni, A.; Alimenti, E.; Gattai, R.; Filomia, R.; Parente, E.; Valenti, L.; Marzi, L.; Pellegatta, G.; Borgia, G.; Gambato, M.; et al. Undefined/non-malignant hepatic nodules are associated with early occurrence of HCC in DAA-treated patients with HCV-related cirrhosis. J. Hepatol. 2020, 73, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Angeli, P.; Piovesan, S.; Noventa, F.; Anastassopoulos, G.; Chemello, L.; Cavalletto, L.; Gambato, M.; Russo, F.P.; Burra, P.; et al. Newly diagnosed hepatocellular carcinoma in patients with advanced hepatitis C treated with DAAs: A prospective population study. J. Hepatol. 2018, 69, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Boccaccio, V. HCV therapy and risk of liver cancer recurrence: Who to treat? Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-C.; Lin, Y.-S.; Chang, C.-W.; Chang, C.-W.; Wang, T.-E.; Wang, H.-Y.; Chen, M.-J. Impact of direct-acting antiviral therapy for hepatitis C–related hepatocellular carcinoma. PLoS ONE 2020, 15, e0233212. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Rich, N.E.; Mehta, N.; Branch, A.; Pillai, A.; Hoteit, M.; Volk, M.; Odewole, M.; Scaglione, S.; Guy, J.; et al. Direct-Acting Antiviral Therapy Not Associated with Recurrence of Hepatocellular Carcinoma in a Multicenter North American Cohort Study. Gastroenterology 2019, 156, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Lui, F.H.; Moosvi, Z.; Patel, A.; Hussain, S.; Duong, A.; Duong, J.; Nguyen, D.L. Decreased risk of hepatocellular carcinoma recurrence with directacting antivirals compared with no treatment for hepatitis C: A meta-analysis. Ann. Gastroenterol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Frazzoni, L.; Sikandar, U.; Metelli, F.; Sadalla, S.; Mazzella, G.; Bazzoli, F.; Fuccio, L.; Azzaroli, F. Hepatocellular Carcinoma Recurrence after Hepatitis C Virus Therapy with Direct-Acting Antivirals. A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 1694. [Google Scholar] [CrossRef]
- D’Ambrosio, R.; Aghemo, A.; Rumi, M.G.; Degasperi, E.; Sangiovanni, A.; Maggioni, M.; Fraquelli, M.; Perbellini, R.; Rosenberg, W.; Bedossa, P.; et al. Persistence of hepatocellular carcinoma risk in hepatitis C patients with a response to IFN and cirrhosis regression. Liver Int. 2018, 38, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Hamdane, N.; Jühling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Suarez, A.A.R.; et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Holmes, J.A.; Dai, C.-Y.; Huang, C.-F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef]
- Seo, Y.S.; Na Kim, M.; Kim, S.U.; Kim, S.G.; Um, S.H.; Han, K.-H.; Kim, Y.S. Risk Assessment of Hepatocellular Carcinoma Using Transient Elastography Vs. Liver Biopsy in Chronic Hepatitis B Patients Receiving Antiviral Therapy. Medicine 2016, 95, e2985. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Perrella, A.; Guarino, M.; De Luca, M.; Piai, G.; Coppola, N.; Pafundi, P.C.; Ciardiello, F.; Fasano, M.; Martinelli, E.; et al. Incidence and risk factors of early HCC occurrence in HCV patients treated with direct acting antivirals: A prospective multicentre study. J. Transl. Med. 2019, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; D’Ambrosio, R.; Iavarone, M.; Sangiovanni, A.; Aghemo, A.; Soffredini, R.; Borghi, M.; Lunghi, G.; Colombo, M.; Lampertico, P. Factors Associated With Increased Risk of De Novo or Recurrent Hepatocellular Carcinoma in Patients With Cirrhosis Treated with Direct-Acting Antivirals for HCV Infection. Clin. Gastroenterol. Hepatol. 2019, 17, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.-M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL Recommendations on Treatment of Hepatitis C 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghany, M.G.; Morgan, T.R.; Panel, A.H.C.G.; Marks, K.M.; Wyles, D.L.; Aronsohn, A.I.; Bhattacharya, D.; Broder, T.; Falade-Nwulia, O.O.; Feld, J.J.; et al. Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases–Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology 2019, 71, 686–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Beste, L.A.; Green, P.K.; Singal, A.G.; Tapper, E.B.; Waljee, A.K.; Sterling, R.K.; Feld, J.J.; Kaplan, D.E.; Taddei, T.H.; et al. Increased Risk for Hepatocellular Carcinoma Persists Up to 10 Years After HCV Eradication in Patients with Baseline Cirrhosis or High FIB-4 Scores. Gastroenterology 2019, 157, 1264–1278. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Green, P.K.; Beste, L.A.; Mun, E.J.; Kerr, K.; Berry, K. Development of models estimating the risk of hepatocellular carcinoma after antiviral treatment for hepatitis C. J. Hepatol. 2018, 69, 1088–1098. [Google Scholar] [CrossRef]
- Fan, R.; Papatheodoridis, G.; Sun, J.; Innes, H.; Toyoda, H.; Xie, Q.; Mo, S.; Sypsa, V.; Guha, I.N.; Kumada, T.; et al. aMAP risk score predicts hepatocellular carcinoma development in patients with chronic hepatitis. J. Hepatol. 2020, 73, 1368–1378. [Google Scholar] [CrossRef]
- Zangneh, H.F.; Wong, W.W.; Sander, B.; Bell, C.M.; Mumtaz, K.; Kowgier, M.; van der Meer, A.J.; Cleary, S.; Janssen, H.L.; Chan, K.K.; et al. Cost Effectiveness of Hepatocellular Carcinoma Surveillance after a Sustained Virologic Response to Therapy in Patients With Hepatitis C Virus Infection and Advanced Fibrosis. Clin. Gastroenterol. Hepatol. 2019, 17, 1840–1849. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N. HCC surveillance after SVR in patients with F3/F4 fibrosis. J. Hepatol. 2020, 74, 458–465. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Chinese University Model [40] | Guide with Age, Gender, HBV DNA, Core Promoter Mutations, and Cirrhosis (GAG Model) [53] | Risk Estimation for HCC in CHB (REACH-B Model) [54] | Modified REACH-B Model [57] | Liver Stiffness Measurement Model [47] | Score Based on Age, Gender, and Platelet Count for HCC in CHB [55] | |
|---|---|---|---|---|---|---|
| Items | Age Albumin Bilirubin Cirrhosis HBV DNA level | Age Gender BCP mutation Cirrhosis HBV DNA level | Age Gender ALT level HBeAg status HBV DNA level | Age Gender ALT level HBeAg status Liver stiffness value | Age Albumin HBV DNA level Liver stiffness value | Age Gender Platelet level |
| Negative Predictive Value | 97% at 10 years | 99% at 10 years | 98% at 10 years | 96.8% at 5 years | 99.4% at 10 years | 100% at 5 years |
| Reference | Patient Population | Follow-Up | De Novo HCC Incidence in DAA-Treated Patients |
|---|---|---|---|
| Retrospective Studies | |||
| Conti et al. [108] | n = 285; cirrhotic, DAA-treated | Mean 5.6 months | 3.26% |
| Ravi et al. [110] | n = 66; cirrhotic, DAA-treated | 6 months | 6-month rate: 9.1% |
| Cardoso et al. [109] | n = 240; cirrhotic DAA-treated | Median 12 months | 1-year rate: 7.4% |
| Singer et al. [111] | Chronic HCV, DAA-treated (n = 30,183), IFN-treated (n = 12,948), or untreated (n = 13,7502) | Mean 1.05 years | 1.18 per 100 person-years |
| Nahon et al. [113] | Compensated cirrhotic; DAA-treated (n = 336), IFN-treated with SVR (n = 495), or IFN-treated without SVR (n = 439) | Median 21.2 months | 2.6 per 100 person-years |
| Ioannou et al. [117] | DAA-treated (n = 21948), IFN-treated (n = 35871), DAA + IFN treated (n = 4535) | Mean 6.1 years | 1.32 per 100 person-years |
| Kanwal et al. [13] | n = 22,500; DAA-treated | Mean 1.02 years | 1.18 per 100 person-years |
| Kanwal et al. [118] | n = 18,076; DAA-treated, SVR | Mean 2.9 years | 3-year rate: 3% |
| Janjua et al. [112] | IFN-treated (n = 8871), DAA-treated (n = 3905), SVR | Median 1.0 year | 6.9 per 1000 person-years |
| Tani et al. [119] | DAA-treated (n = 1088) | Median 13.8 months | 3-year rate: 3.71% |
| Watanabe et al. [120] | DAA-treated (n = 1438) | Median 803 days | 3.82% |
| Prospective Studies | |||
| Cheung et al. [121] | DAA-treated (n = 406), untreated (n = 261); decompensated cirrhosis | Median 18 months | 4% |
| Mettke et al. [122] | DAA-treated (n = 158), untreated (n = 184) | Median 440 days | 2.9 per 100 person-years |
| Carrat et al. [114] | DAA-treated (n = 7344), untreated (n = 2552) | Median 33.4 months | 1.4 per 100 person-years |
| Poordad et al. [123] | DAA-treated (n = 2211) | 156 weeks from end of treatment | 1.4% |
| Piñero et al. [115] | DAA-treated (n = 1.017) | Median 16 months | Cumulative incidence 0.04 at 24 months |
| Sangiovanni et al. [124] | DAA-treated (n = 1285) | Mean 17 months | 3.1 per 100 person-years |
| Romano et al. [125] | DAA-treated (n = 3917) | Median 523 days | 0.97 per 100 person-years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, S.A.; Colombo, M. Treatment for Viral Hepatitis as Secondary Prevention for Hepatocellular Carcinoma. Cells 2021, 10, 3091. https://doi.org/10.3390/cells10113091
Alqahtani SA, Colombo M. Treatment for Viral Hepatitis as Secondary Prevention for Hepatocellular Carcinoma. Cells. 2021; 10(11):3091. https://doi.org/10.3390/cells10113091
Chicago/Turabian StyleAlqahtani, Saleh A., and Massimo Colombo. 2021. "Treatment for Viral Hepatitis as Secondary Prevention for Hepatocellular Carcinoma" Cells 10, no. 11: 3091. https://doi.org/10.3390/cells10113091
APA StyleAlqahtani, S. A., & Colombo, M. (2021). Treatment for Viral Hepatitis as Secondary Prevention for Hepatocellular Carcinoma. Cells, 10(11), 3091. https://doi.org/10.3390/cells10113091