
Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation


Abstract
1. Introduction
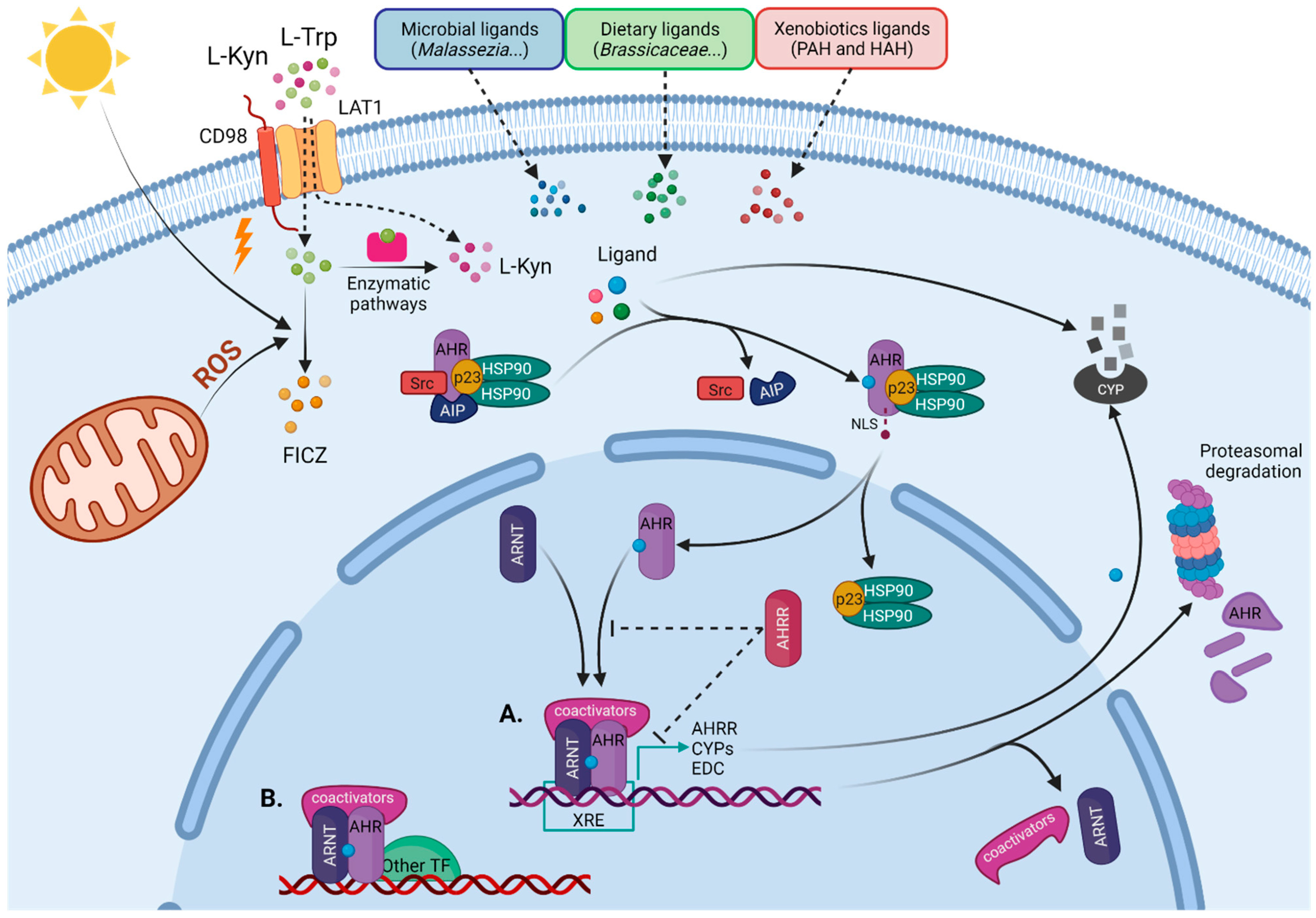
1.1. AHR as a Sensor of Environmental Cues
1.2. AHR Signaling Pathways
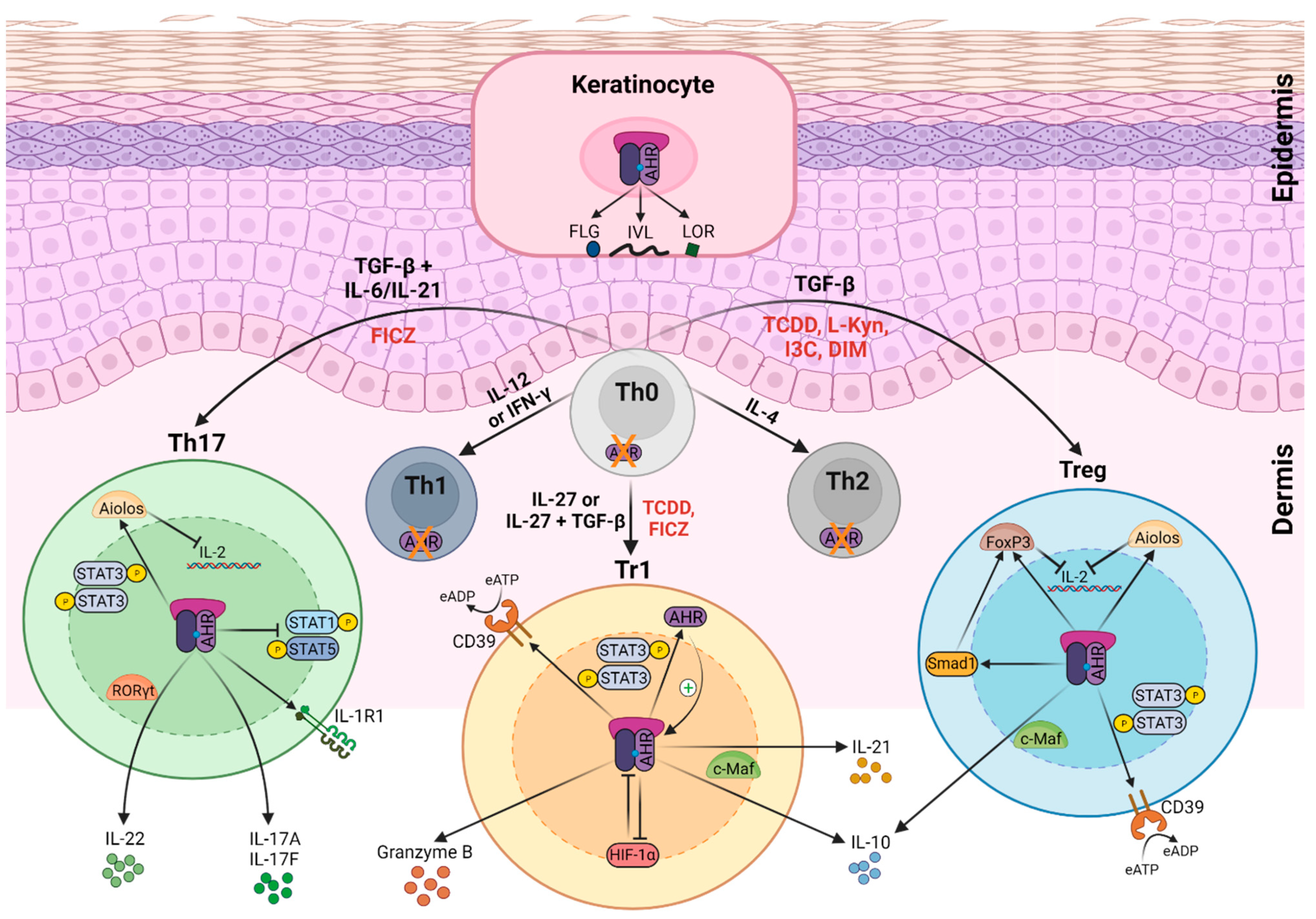
2. Role of AHR Function in Skin Immune System
AHR Function in the Epidermis
3. AHR Ligands in Skin Homeostasis and Inflammation
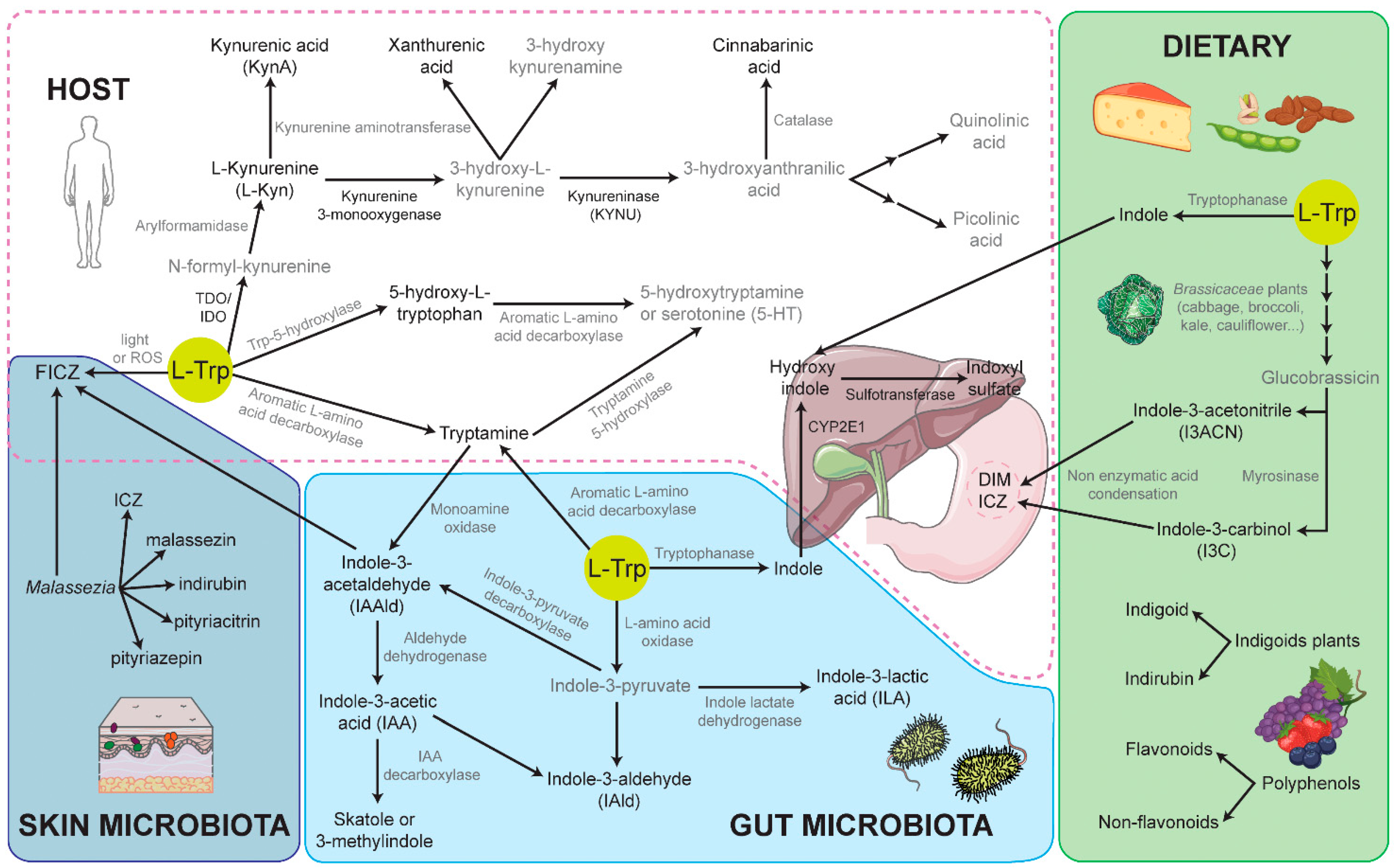
3.1. Endogenous L-Tryptophan-Derived AHR Ligands
3.2. Endogenous Ligands Derived from Photo-Oxidation of L-Trp
3.3. Exogenous AHR Ligands: Flavonoids and Indoles as a Phytochemical Dietary Source of AHR Ligands
3.4. Microbial-Derived AHR Ligands
3.5. Toxicity of AHR-Exogenous Ligand TCDD in the Skin
3.6. Tapinarof—A Novel Treatment for PS and AD

{kind=link}
{kind=link}
{kind=link}
| Origin/Source | Molecule | Effects in PS or AD |
|---|---|---|
| L-Trp-derived metabolites of L-kynurenine pathway | L-Kynurenine (L-Kyn) | Ido2-deficient mice show exacerbated IMQ-induced PS [128]. Increase Kyn/Trp ratio in PS and AD patients [129,130]. |
| Kynurenic Acid (Kyn A) | Suppresses IL-23/IL-17 in vitro secretion in DC/CD4+ T cells, respectively, after LPS stimulation [134]. | |
| Xanthurenic Acid | Not assessed in PS or AD. | |
| Cinnabarinic Acid | Not assessed in PS or AD. | |
| 3-Hydroxyanthralinic acid | Proinflammatory role suggested in PS and AD. Induces the expression of CCL20 and IL-8 in keratinocytes in vitro [130]. | |
| Quinolinic acid | Proinflammatory role suggested in PS and AD. Induces chemokines and adhesion molecules in keratinocytes and endothelial cells, respectively, in vitro [130]. | |
| 3-hydroxy-L-kynurenamine (3-HKA) | Protective role in IMQ-induced PS mouse model [133]. | |
| Serotonin pathway | 5-Hydroxytryptophan (5(OH)Trp) | Controls IMQ-induced psoriasiform dermatitis [144]. |
| Tryptamine pathway | Tryptamine | Not assessed in PS or AD. |
| Oxidative L-Trp metabolite | 6-formylindolo [3,2-b]carbazole (FICZ) | Attenuates IMQ-psoriasiform skin inflammation by increasing FLG expression and reducing IL-17 and IL-22 levels [105]. Exacerbates the DTH response by promoting Th17 cells [180]. |
| Synthetic | NPD-0614-13 NPD-0614-24 | Protective role in three-dimensional models of psoriasis [171]. |
| Dietary ligands | Indole-3-acetonitrile (I3ACN) | Not assessed in PS or AD. |
| Indole-3-carbinol (I3C) | Controls IL-17 secretion and increases Foxp3 and IL-10 expression. Controls skin inflammation in a model of DTH [180]. | |
| Indigo | Effective in the treatment of PS and AD patients [191,192,193]. Induce keratinocyte differentiation [194,195] | |
| Indirubin | Inhibits inflammatory reactions in DTH mouse model [187] | |
| 3,3′-diindolylmethane (DIM) | Decreases IL-17 secretion while increasing Treg differentiation, thus controlling skin inflammation in a model of DTH [180]. Reduces Th2 and Th17 cell proliferation, increases Treg, attenuating atopic dermatitis-related immune responses [181]. | |
| Microbiota indole ligands | Indole-3-acetylaldehyde (IAAld) | Not assessed in PS or AD. |
| Indole-3-acetic acid (IAA) | Not assessed in PS or AD. | |
| Indole-3-lactic acid (ILA) | Not assessed in PS or AD. | |
| Indole-3-aldehyde (IAld) | Metabolite significantly decreased on both lesional and non-lesional skin of patients with AD Alleviates skin inflammation in AD mouse model [225]. | |
| Malassezia | Malassezin Pityriacitrin Pityriazepin | Upregulate FLG and IVL genes in keratinocytes in vitro [229]. Induce proinflammatory mediators in keratinocytes in vitro [230,231]. Associated to exacerbated AD and seborrheic dermatitis. |
| Synthetic | 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) | Induces chloracne syndrome in humans [12]. |
| Bacterial symbionts of entomopathogenic nematodes | 3,5-dihydroxy-4-isopropylstilbene (Tapinarof, GSK2894512 or WBI-1001) | Protective role in the IMQ-induced PS model, by downregulation of inflammatory cytokines, and improvement of skin-barrier function [242]. |
4. Concluding Remarks and Future Perspectives
Funding
Data Availability Statement
Conflicts of Interest
References
- Montero-Vilchez, T.; Segura-Fernández-Nogueras, M.V.; Pérez-Rodríguez, I.; Soler-Gongora, M.; Martinez-Lopez, A.; Fernández-González, A.; Molina-Leyva, A.; Arias-Santiago, S. Skin barrier function in psoriasis and atopic dermatitis: Transepidermal water loss and temperature as useful tools to assess disease severity. J. Clin. Med. 2021, 10, 359. [Google Scholar] [CrossRef]
- Alwan, W.; Nestle, F. Pathogenesis and treatment of psoriasis: Exploiting pathophysiological pathways for precision medicine. Clin. Exp. Rheumatol. 2015, 33, S2–S6. [Google Scholar]
- Guttman-Yassk, E.; Waldman, A.; Ahluwalia, J.; Ong, P.Y.; Eichenfield, L.F. Atopic dermatitis: Pathogenesis. Semin. Cutan. Med. Surg. 2017, 36, 100–103. [Google Scholar] [CrossRef]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.-P.; et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.L.; Powers, J.L.; Matheson, R.T.; Goffe, B.S.; Zitnik, R.; Wang, A.; Gottlieb, A.B. Etanercept as monotherapy in patients with psoriasis. N. Engl. J. Med. 2003, 349, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Wharton, K.A.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Scharschmidt, T.C.; Man, M.-Q.; Hatano, Y.; Crumrine, D.; Gunathilake, R.; Sundberg, J.P.; Silva, K.A.; Mauro, T.M.; Hupe, M.; Cho, S.; et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J. Allergy Clin. Immunol. 2009, 124, 496–506. [Google Scholar] [CrossRef]
- Kawasaki, H.; Nagao, K.; Kubo, A.; Hata, T.; Shimizu, A.; Mizuno, H.; Yamada, T.; Amagai, M. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J. Allergy Clin. Immunol. 2012, 129, 1538–1546.e6. [Google Scholar] [CrossRef]
- Flohr, C.; England, K.; Radulovic, S.; McLean, W.H.I.; Campbell, L.E.; Barker, J.; Perkin, M.; Lack, G. Filaggrin loss-of-function mutations are associated with early-onset eczema, eczema severity and transepidermal water loss at 3 months of age. Br. J. Dermatol. 2010, 163, 1333–1336. [Google Scholar] [CrossRef]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef]
- Furue, M.; Iida, K.; Imaji, M.; Nakahara, T. Microbiome analysis of forehead skin in patients with atopic dermatitis and healthy subjects: Implication of Staphylococcus and Corynebacterium. J. Dermatol. 2018, 45, 876–877. [Google Scholar] [CrossRef]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the Aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Bargen, I.; Weighardt, H.; Haarmann-Stemmann, T.; Krutmann, J. Functions of the aryl hydrocarbon receptor in the skin. Semin. Immunopathol. 2013, 35, 677–691. [Google Scholar] [CrossRef]
- Sutter, C.; Yin, H.; Li, Y.; Mammen, J.; Bodreddigari, S.; Stevens, G.; Cole, J.; Sutter, T. EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 4266–4271. [Google Scholar] [CrossRef]
- Kennedy, L.H.; Sutter, C.H.; Carrion, S.L.; Tran, Q.T.; Bodreddigari, S.; Kensicki, E.; Mohney, R.P.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol. Sci. 2013, 132, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Sutter, C.H.; Bodreddigari, S.; Campion, C.; Wible, R.S.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol. Sci. 2011, 124, 128–137. [Google Scholar] [CrossRef]
- Kopf, P.; Walker, M. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol. Appl. Pharmacol. 2010, 245, 91–99. [Google Scholar] [CrossRef]
- Soshilov, A.A.; Denison, M.S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef]
- Hahn, M.E.; Karchner, S.I.; Merson, R.R. Diversity as opportunity: Insights from 600 million years of AHR evolution. Curr. Opin. Toxicol. 2017, 2, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Ohe, N.; Suzuki, M.; Mimura, J.; Sogawa, K.; Ikawa, S.; Fujii-Kuriyama, Y. Dioxin binding activities of polymorphic forms of mouse and human aryl hydrocarbon receptors. J. Biol. Chem. 1994, 269, 27337–27343. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Perdew, G.H. Transgenic humanized AHR mouse reveals differences between human and mouse AHR ligand selectivity. Mol. Cell. Biol. 2009, 1, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.; Reen, R.K.; Kusnadi, A.; Perdew, G.H. The mouse and human Ah receptor differ in recognition of LXXLL motifs. Arch. Biochem. Biophys. 2008, 471, 215–223. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Murray, I.A.; Perdew, G.H. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicol. Sci. 2010, 114, 217–225. [Google Scholar] [CrossRef]
- Forgacs, A.L.; Dere, E.; Angrish, M.M.; Zacharewski, T.R. Comparative analysis of temporal and dose-dependent TCDD-elicited gene expression in human, mouse, and rat primary hepatocytes. Toxicol. Sci. 2013, 133, 54–66. [Google Scholar] [CrossRef]
- Sogawa, K.; Fujisawa-Sehara, A.; Yamane, M.; Fujii-kuriyama, Y. Location of regulatory elements responsible for drug induction in the rat cytochrome P-450c gene. Proc. Natl. Acad. Sci. USA 1986, 83, 8044–8048. [Google Scholar] [CrossRef]
- Lusska, A.; Shen, E.; Whitlock, J.P. Protein-DNA interactions at a dioxin-responsive enhancer. Analysis of six bona fide DNA-binding sites for the liganded Ah receptor. J. Biol. Chem. 1993, 268, 6575–6580. [Google Scholar] [CrossRef]
- Durrin, L.K.; Whitlock, J.P. In situ protein-DNA interactions at a dioxin-responsive enhancer associated with the cytochrome P1-450 gene. Mol. Cell. Biol. 1987, 7, 3008–3011. [Google Scholar] [CrossRef]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P. Inducible, receptor-dependent protein-DNA interactions at a dioxin-responsive transcriptional enhancer. Proc. Natl. Acad. Sci. USA 1988, 85, 2528–2532. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef]
- MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Förster, I.; Weighardt, H.; Matthews, J. Aryl hydrocarbon receptor repressor and TiPARP (ARTD14) use similar, but also distinct mechanisms to repress aryl hydrocarbon receptor signaling. Int. J. Mol. Sci. 2014, 15, 7939–7957. [Google Scholar] [CrossRef]
- Grimaldi, G.; Rajendra, S.; Matthews, J. The aryl hydrocarbon receptor regulates the expression of TIPARP and its cis long non-coding RNA, TIPARP-AS1. Biochem. Biophys. Res. Commun. 2018, 495, 2356–2362. [Google Scholar] [CrossRef] [PubMed]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef]
- Gu, Y.-Z.; Hogenesch, J.B.; Bradfield, C.A. The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 519–561. [Google Scholar] [CrossRef] [PubMed]
- Dolwick, K.M.; Swanson, H.I.; Bradfield, C.A. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proc. Natl. Acad. Sci. USA 1993, 90, 8566–8570. [Google Scholar] [CrossRef]
- Antonsson, C.; Whitelaw, M.L.; McGuire, J.; Gustafsson, J.A.; Poellinger, L. Distinct roles of the molecular chaperone hsp90 in modulating dioxin receptor function via the basic helix-loop-helix and PAS domains. Mol. Cell. Biol. 1995, 15, 756–765. [Google Scholar] [CrossRef]
- Jain, S.; Dolwick, K.M.; Schmidt, J.V.; Bradfield, C.A. Potent transactivation domains of the Ah receptor and the Ah receptor nuclear translocator map to their carboxyl termini. J. Biol. Chem. 1994, 269, 31518–31524. [Google Scholar] [CrossRef]
- Ko, H.P.; Okino, S.T.; Ma, Q.; Whitlock, J.P. Dioxin-induced CYP1A1 transcription in vivo: The aromatic hydrocarbon receptor mediates transactivation, enhancer-promoter communication, and changes in chromatin structure. Mol. Cell. Biol. 1996, 16, 430–436. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Morales, J.L.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef]
- Enan, E.; Matsumura, F. Evidence for a second pathway in the action mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Significance of Ah-receptor mediated activation of protein kinase under cell-free conditions. Biochem. Pharmacol. 1995, 49, 249–261. [Google Scholar] [CrossRef]
- Enan, E.; Matsumura, F. Identification of c-Src as the integral component of the cytosolic Ah receptor complex, transducing the signal of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through the protein phosphorylation pathway. Biochem. Pharmacol. 1996, 52, 1599–1612. [Google Scholar] [CrossRef]
- Kudo, I.; Hosaka, M.; Haga, A.; Tsuji, N.; Nagata, Y.; Okada, H.; Fukuda, K.; Kakizaki, Y.; Okamoto, T.; Grave, E.; et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 2018, 163, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Carver, L.A.; Jackiw, V.; Bradfield, C.A. The 90-kDa heat shock protein is essential for Ah receptor signaling in a yeast expression system. J. Biol. Chem. 1994, 269, 30109–30112. [Google Scholar] [CrossRef]
- Petrulis, J.R.; Kusnadi, A.; Ramadoss, P.; Hollingshead, B.; Perdew, G.H. The hsp90 co-chaperone XAP2 alters importin β recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J. Biol. Chem. 2003, 278, 2677–2685. [Google Scholar] [CrossRef]
- Kazlauskas, A.; Sundström, S.; Poellinger, L.; Pongratz, I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol. Cell. Biol. 2001, 21, 2594–2607. [Google Scholar] [CrossRef]
- Ikuta, T.; Tachibana, T.; Watanabe, J.; Yoshida, M.; Yoneda, Y.; Kawajiri, K. Nucleocytoplasmic shuttling of the aryl hydrocarbon receptor. J. Biochem. 2000, 127, 503–509. [Google Scholar] [CrossRef]
- Reyes, H.; Reisz-Porszasz, S.; Hankinsont, O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 1992, 256, 1193–1195. [Google Scholar] [CrossRef]
- Wu, L.; Whitlock, J.P. Mechanism of dioxin action: Ah receptor-mediated increase in promoter accessibility in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4811–4815. [Google Scholar] [CrossRef]
- Morgan, J.E.; Whitlock, J.P. Transcription-dependent and transcription-independent nucleosome disruption induced by dioxin. Proc. Natl. Acad. Sci. USA 1992, 89, 11622–11626. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Wang, F.; Hsu, E.L.; Hankinson, O. Roles of coactivator proteins in dioxin induction of CYP1A1 and CYP1B1 in human breast cancer cells. Toxicol. Sci. 2009, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kurita, H.; Schnekenburger, M.; Ovesen, J.L.; Xia, Y.; Puga, A. The Ah receptor recruits IKKα to its target binding motifs to phosphorylate serine-10 in histone H3 required for transcriptional activation. Toxicol. Sci. 2014, 139, 121–132. [Google Scholar] [CrossRef]
- Ohtake, F.; Takeyama, K.I.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Vijay, K. The Aryl hydrocarbon Receptor (AhR) interacts with c-Maf to promote the differentiation of IL-27-induced regulatory type 1 (TR1) cells. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Raphaëlle; Sherr, D.H.; Sonenshein, G.E. The RelA NF-κB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Tian, Y.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-κB interactions: Mechanisms and physiological implications. Chem. Biol. Interact. 2002, 141, 97–115. [Google Scholar] [CrossRef]
- Miniero, R.; De Felip, E.; Ferri, F.; Di Domenico, A. An overview of TCDD half-life in mammals and its correlation to body weight. Chemosphere 2001, 43, 839–844. [Google Scholar] [CrossRef]
- Rannug, A.; Rannug, U. The tryptophan derivative 6-formylindolo [3,2-b]carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol. 2018, 48, 555–574. [Google Scholar] [CrossRef]
- Sakurai, S.; Shimizu, T.; Ohto, U. The crystal structure of the AhRR–ARNT heterodimer reveals the structural basis of the repression of AhR-mediated transcription. J. Biol. Chem. 2017, 292, 17609–17616. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Mimura, J.; Yamamoto, M.; Fujii-Kuriyama, Y. Molecular mechanism of transcriptional repression of AhR repressor involving ANKRA2, HDAC4, and HDAC5. Biochem. Biophys. Res. Commun. 2007, 364, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1996, 274, 28708–28715. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.J.; Whitelaw, M.L. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J. Biol. Chem. 1999, 274, 36351–36356. [Google Scholar] [CrossRef]
- N’Diaye, M.; Le Ferrec, E.; Lagadic-Gossmann, D.; Corre, S.; Gilot, D.; Lecureur, V.; Monteiro, P.; Rauch, C.; Galibert, M.D.; Fardel, O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J. Biol. Chem. 2006, 281, 19906–19915. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The aryl hydrocarbon receptor: A multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The role of the aryl hydrocarbon receptor (AHR) in immune and inflammatory diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Furue, M. Regulation of skin barrier function via competition between AHR axis versus IL-13/IL-4–JAK–STAT6/STAT3 axis: Pathogenic and therapeutic implications in atopic dermatitis. J. Clin. Med. 2020, 9, 3741. [Google Scholar] [CrossRef]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of Aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of Foxp3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell–like and Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and Th17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links Th17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef]
- Cibrian, D.; Saiz, M.L.; de la Fuente, H.; Sánchez-Díaz, R.; Moreno-Gonzalo, O.; Jorge, I.; Ferrarini, A.; Vázquez, J.; Punzón, C.; Fresno, M.; et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat. Immunol. 2016, 17, 985–996. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef]
- Castillo-González, R.; Cibrian, D.; Sánchez-Madrid, F. Dissecting the complexity of γδ T cell subsets in skin homeostasis, inflammation, and malignancy. J. Allergy Clin. Immunol. 2021, 147, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Meglio, P.D.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Cibrian, D.; Castillo-González, R.; Fernández-Gallego, N.; de la Fuente, H.; Jorge, I.; Saiz, M.L.; Punzón, C.; Ramírez-Huesca, M.; Vicente-Manzanares, M.; Fresno, M.; et al. Targeting L-type amino acid transporter 1 in innate and adaptive T cells efficiently controls skin inflammation. J. Allergy Clin. Immunol. 2020, 145, 199–214.e11. [Google Scholar] [CrossRef]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef]
- Kaper, T.; Looger, L.L.; Takanaga, H.; Platten, M.; Steinman, L.; Frommer, W.B. Nanosensor detection of an immunoregulatory tryptophan influx/kynurenine efflux cycle. PLoS Biol. 2007, 5, e257. [Google Scholar] [CrossRef]
- Kadow, S.; Jux, B.; Zahner, S.P.; Wingerath, B.; Chmill, S.; Clausen, B.E.; Hengstler, J.; Esser, C. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J. Immunol. 2011, 187, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Zaid, A.; Mackay, L.K.; Rahimpour, A.; Braun, A.; Veldhoen, M.; Carbone, F.R.; Manton, J.H.; Heath, W.R.; Mueller, S.N. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl. Acad. Sci. USA 2014, 111, 5307–5312. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. Tissue-resident memory T cells. Immunol. Rev. 2013, 255, 165–181. [Google Scholar] [CrossRef]
- Cheuk, S.; Wikén, M.; Blomqvist, L.; Nylén, S.; Talme, T.; Ståhle, M.; Eidsmo, L. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014, 192, 3111–3120. [Google Scholar] [CrossRef]
- Vu, T.T.; Koguchi-Yoshioka, H.; Watanabe, R. Skin-resident memory T cells: Pathogenesis and implication for the treatment of psoriasis. J. Clin. Med. 2021, 10, 3822. [Google Scholar] [CrossRef]
- Hong, C.-H.; Lin, S.-H.; Clausen, B.E.; Lee, C.-H. Selective AhR knockout in langerin-expressing cells abates Langerhans cells and polarizes Th2/Tr1 in epicutaneous protein sensitization. Proc. Natl. Acad. Sci. USA 2020, 117, 12980–12990. [Google Scholar] [CrossRef] [PubMed]
- Climaco-Arvizu, S.; Domínguez-Acosta, O.; Cabañas-Cortés, M.A.; Rodríguez-Sosa, M.; Gonzalez, F.J.; Vega, L.; Elizondo, G. Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci. 2016, 155, 76–84. [Google Scholar] [CrossRef]
- Kim, H.O.; Kim, J.H.; Chung, B.Y.; Choi, M.G.; Park, C.W. Increased expression of the aryl hydrocarbon receptor in patients with chronic inflammatory skin diseases. Exp. Dermatol. 2014, 23, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, H.R.; Kang, S.Y.; Jung, M.J.; Heo, N.H.; Lee, H.J.; Ryu, A.; Kim, H.O.; Park, C.W.; Chung, B.Y. Aryl hydrocarbon Receptor and autophagy-related protein microtubule-associated protein light chain 3 expression in psoriasis. Ann. Dermatol. 2021, 33, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Loertscher, J.A.; Sattler, C.A.; Allen-Hoffmann, B.L. 2,3,7,8-tetrachlorodibenzo-p-dioxin alters the differentiation pattern of human keratinocytes in organotypic culture. Toxicol. Appl. Pharmacol. 2001, 175, 121–129. [Google Scholar] [CrossRef]
- Loertscher, J.A.; Lin, T.-M.; Peterson, R.E.; Lynn Allen-Hoffmann, B. In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin. Toxicol. Pathol. 2002, 7, 465–472. [Google Scholar] [CrossRef]
- Furue, M.; Tsuji, G.; Mitoma, C.; Nakahara, T.; Chiba, T.; Morino-Koga, S.; Uchi, H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J. Dermatol. Sci. 2015, 80, 83–88. [Google Scholar] [CrossRef]
- Hashimoto-Hachiya, A.; Tsuji, G.; Murai, M.; Yan, X.; Furue, M. Upregulation of FLG, LOR, and IVL expression by rhodiola crenulata root extract via Aryl hydrocarbon receptor: Differential involvement of OVOL1. Int. J. Mol. Sci. 2018, 19, 1654. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl hydrocarbon receptor in atopic dermatitis and psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef]
- Park, J.Y.; Shigenaga, M.K.; Ames, B.N. Induction of cytochrome P4501A1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin or indolo(3,2-b)carbazole is associated with oxidative DNA damage. Proc. Natl. Acad. Sci. USA 1996, 93, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.M.; Ward, J.M.; Sundberg, J.P.; Gonzalez, F.J. Lesions of Aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997, 34, 605–614. [Google Scholar] [CrossRef]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of Aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell. Biol. 2005, 25, 9360–9368. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Mezentsev, A.; Kalachikov, S.; Raith, K.; Roop, D.R.; Panteleyev, A.A. Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J. Cell Sci. 2006, 119, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.L.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, J.; Lin, Y.; Zhang, C.; Li, W.; Qiao, H.; Fu, M.; Dang, E.; Wang, G. Aryl hydrocarbon receptor in cutaneous vascular endothelial cells restricts psoriasis development by negatively regulating neutrophil recruitment. J. Invest. Dermatol. 2020, 140, 1233–1243. [Google Scholar] [CrossRef]
- Haas, K.; Weighardt, H.; Deenen, R.; Köhrer, K.; Clausen, B.; Zahner, S.; Boukamp, P.; Bloch, W.; Krutmann, J.; Esser, C. Aryl hydrocarbon receptor in keratinocytes is essential for murine skin barrier integrity. J. Invest. Dermatol. 2016, 136, 2260–2269. [Google Scholar] [CrossRef]
- Uberoi, A.; Bartow-McKenney, C.; Zheng, Q.; Flowers, L.; Campbell, A.; Knight, S.A.B.; Chan, N.; Wei, M.; Lovins, V.; Bugayev, J.; et al. Commensal microbiota regulates skin barrier function and repair via signaling through the aryl hydrocarbon receptor. Cell Host Microbe 2021, 29, 1235–1248.e8. [Google Scholar] [CrossRef]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef]
- Wyatt, M.; Greathouse, K.L. Targeting dietary and microbial tryptophan-indole metabolism as therapeutic approaches to colon cancer. Nutrients 2021, 13, 1189. [Google Scholar] [CrossRef]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic phenotype of IFN-γ–induced IDO+ dendritic cells is maintained via an autocrine IDO–kynurenine/AhR–IDO loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Mellor, A.L.; Baban, B.; Chandler, P.; Marshall, B.; Jhaver, K.; Hansen, A.; Koni, P.A.; Iwashima, M.; Munn, D.H. Cutting edge: Induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J. Immunol. 2003, 171, 1652–1655. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor ζ-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. Tryptophan catabolism generates autoimmune-preventive regulatory T cells. Transpl. Immunol. 2006, 17, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Ho, P.P.; Youssef, S.; Fontoura, P.; Garren, H.; Hur, E.M.; Gupta, R.; Lee, L.Y.; Kidd, B.A.; Robinson, W.H.; et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 2005, 310, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Criado, G.; Šimelyte, E.; Inglis, J.J.; Essex, D.; Williams, R.O. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum. 2009, 60, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Elizei, S.S.; Pakyari, M.; Ghoreishi, M.; Kilani, R.; Mahmoudi, S.; Ghahary, A. IDO-expressing fibroblasts suppress the development of Imiquimod-induced psoriasis-like dermatitis. Cell Transplant. 2018, 27, 557–570. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Mandik-Nayak, L. IDO2: A pathogenic mediator of inflammatory autoimmunity. Clin. Med. Insights Pathol. 2016, 9, 21–28. [Google Scholar] [CrossRef]
- Fujii, K.; Yamamoto, Y.; Mizutani, Y.; Saito, K.; Seishima, M. Indoleamine 2,3-dioxygenase 2 deficiency exacerbates Imiquimod-induced psoriasis-like skin inflammation. Int. J. Mol. Sci. 2020, 21, 5515. [Google Scholar] [CrossRef]
- Llamas-Velasco, M.; Bonay, P.; José Concha-Garzón, M.; Corvo-Villén, L.; Vara, A.; Cibrián, D.; Sanguino-Pascual, A.; Sánchez-Madrid, F.; de la Fuente, H.; Daudén, E. Immune cells from patients with psoriasis are defective in inducing indoleamine 2,3-dioxygenase expression in response to inflammatory stimuli. Br. J. Dermatol. 2017, 176, 695–704. [Google Scholar] [CrossRef]
- Harden, J.L.; Lewis, S.M.; Lish, S.R.; Suárez-Fariñas, M.; Gareau, D.; Lentini, T.; Johnson-Huang, L.M.; Krueger, J.G.; Lowes, M.A. The tryptophan metabolism enzyme L-kynureninase is a novel inflammatory factor in psoriasis and other inflammatory diseases. J. Allergy Clin. Immunol. 2016, 137, 1830–1840. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Zhang, M.; Duan, Q.; Chen, C.; Sun, Q.; Liu, M.; Zheng, Y.; Shao, Y. Kynureninase contributes to the pathogenesis of psoriasis through pro-inflammatory effect. J. Cell. Physiol. 2021. epub ahead of print. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Invest. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.C.; D’Alessandro, A.; Thangaswamy, S.; Chalmers, S.; Furtado, R.; Spada, S.; Mondanelli, G.; Ianni, F.; Gehrke, S.; Gargaro, M.; et al. 3-hydroxy-L-kynurenamine is an immunomodulatory biogenic amine. Nat. Commun. 2021, 12, 4447. [Google Scholar] [CrossRef] [PubMed]
- Salimi Elizei, S.; Poormasjedi-Meibod, M.-S.; Wang, X.; Kheirandish, M.; Ghahary, A. Kynurenic acid downregulates IL-17/1L-23 axis in vitro. Mol. Cell. Biochem. 2017, 431, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Poormasjedi-Meibod, M.S.; Hartwell, R.; Kilani, R.T.; Ghahary, A. Anti-scarring properties of different tryptophan derivatives. PLoS ONE 2014, 9, e91955. [Google Scholar] [CrossRef]
- Dolivo, D.M.; Larson, S.A.; Dominko, T. Tryptophan metabolites kynurenine and serotonin regulate fibroblast activation and fibrosis. Cell. Mol. Life Sci. 2018, 75, 3663–3681. [Google Scholar] [CrossRef]
- Poormasjedi-Meibod, M.S.; Salimi Elizei, S.; Leung, V.; Baradar Jalili, R.; Ko, F.; Ghahary, A. Kynurenine modulates MMP-1 and Type-I Collagen expression via Aryl hydrocarbon receptor activation in dermal fibroblasts. J. Cell. Physiol. 2016, 231, 2749–2760. [Google Scholar] [CrossRef]
- Poormasjedi-Meibod, M.S.; Pakyari, M.; Jackson, J.K.; Salimi Elizei, S.; Ghahary, A. Development of a nanofibrous wound dressing with an antifibrogenic properties in vitro and in vivo model. J. Biomed. Mater. Res. Part A 2016, 104, 2334–2344. [Google Scholar] [CrossRef]
- Murai, M.; Tsuji, G.; Hashimoto-Hachiya, A.; Kawakami, Y.; Furue, M.; Mitoma, C. An endogenous tryptophan photo-product, FICZ, is potentially involved in photo-aging by reducing TGF-β-regulated collagen homeostasis. J. Dermatol. Sci. 2018, 89, 19–26. [Google Scholar] [CrossRef]
- Murai, M.; Yamamura, K.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M.; Mitoma, C. Tryptophan photo-product FICZ upregulates AHR/MEK/ERK-mediated MMP1 expression: Implications in anti-fibrotic phototherapy. J. Dermatol. Sci. 2018, 91, 97–103. [Google Scholar] [CrossRef]
- Nestor, M.S.; Berman, B.; Fischer, D.L.; Han, H.; Gade, A.; Arnold, D.; Lawson, A. A randomized, double-blind, active- and placebo-controlled trial evaluating a novel topical treatment for keloid scars. J. Drugs Dermatol. 2021, 20, 964–968. [Google Scholar] [CrossRef]
- Papp, A.; Hartwell, R.; Evans, M.; Ghahary, A. The safety and tolerability of topically delivered kynurenic acid in humans. A phase 1 randomized double-blind clinical trial. J. Pharm. Sci. 2018, 107, 1572–1576. [Google Scholar] [CrossRef]
- Bittinger, M.A.; Nguyen, L.P.; Bradfield, C.A. Aspartate aminotransferase generates proagonists of the aryl hydrocarbon receptor. Mol. Pharmacol. 2003, 64, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Yang, H.J.; Yang, T.H.; Su, C.C. 5-hydroxytryptophan attenuates imiquimod-induced psoriasiform dermatitis probably through inhibition of IL-17A production and keratinocyte activation. Exp. Dermatol. 2018, 27, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Cao, D.; Xiao, Y.; Kuang, Y.; Jing, D.; Li, Y.; Liu, P.; Chen, X.; Zhu, W. Serum 5-hydroxytryptamine is related to psoriasis severity in patients with comorbid anxiety or depression. Acta Derm. Venereol. 2021, 101, adv00514. [Google Scholar] [CrossRef] [PubMed]
- Matiushenko, V.; Kutasevych, Y.; Jafferany, M. Neurotransmitter imbalance in serum of psoriatic patients in exacerbation stage with comorbid psychoemotional disorders. Dermatol. Ther. 2020, 33, e13337. [Google Scholar] [CrossRef]
- Younes, S.F.; Bakry, O.A. Immunohistochemical evaluation of role of serotonin in pathogenesis of psoriasis. J. Clin. Diagn. Res. 2016, 10, EC05–EC09. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, G.; Xiang, J.; Yin, D.; Chi, R. Immunohistochemical study of serotonin in lesions of chronic eczema. Int. J. Dermatol. 2004, 43, 723–726. [Google Scholar] [CrossRef]
- Wetterberg, J.; Taher, C.; Azmitia, E.C.; El-Nour, H. Time-dependent modulation of serotonin and its receptors 1A and 2A expression in allergic contact dermatitis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1200–1205. [Google Scholar] [CrossRef]
- Rasul, A.; El-Nour, H.; Lonne-Rahm, S.B.; Fransson, O.; Johansson, C.; Johansson, B.; Zubeidi, M.; Seeberg, E.; Djurfeldt, D.R.; Azmitia, E.C.; et al. Serotonergic markers in atopic dermatitis. Acta Derm. Venereol. 2016, 96, 732–736. [Google Scholar] [CrossRef]
- Thorslund, K.; El-Nour, H.; Nordlind, K. The serotonin transporter protein is expressed in psoriasis, where it may play a role in regulating apoptosis. Arch. Dermatol. Res. 2009, 301, 449–457. [Google Scholar] [CrossRef]
- Thorslund, K.; Amatya, B.; Dufva, A.E.; Nordlind, K. The expression of serotonin transporter protein correlates with the severity of psoriasis and chronic stress. Arch. Dermatol. Res. 2013, 305, 99–104. [Google Scholar] [CrossRef]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef] [PubMed]
- Bergander, L.V.; Cai, W.; Klocke, B.; Seifert, M.; Pongratz, I. Tryptamine serves as a proligand of the AhR transcriptional pathway whose activation is dependent of monoamine oxidases. Mol. Endocrinol. 2012, 26, 1542–1551. [Google Scholar] [CrossRef]
- Rouse, M.; Singh, N.P.; Nagarkatti, P.S.; Nagarkatti, M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br. J. Pharmacol. 2013, 169, 1305–1321. [Google Scholar] [CrossRef]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Schanz, O.; Chijiiwa, R.; Cengiz, S.C.; Majlesain, Y.; Weighardt, H.; Takeyama, H.; Förster, I. Dietary AhR ligands regulate AhRR expression in intestinal immune vells and intestinal microbiota composition. Int. J. Mol. Sci. 2020, 21, 3189. [Google Scholar] [CrossRef]
- Kahalehili, H.M.; Newman, N.K.; Pennington, J.M.; Kolluri, S.K.; Kerkvliet, N.I.; Shulzhenko, N.; Morgun, A.; Ehrlich, A.K. Dietary indole-3-carbinol activates AhR in the gut, alters Th17-microbe interactions, and exacerbates insulitis in NOD mice. Front. Immunol. 2021, 11, 606441. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.N.; Swimm, A.; Sonowal, R.; Bretin, A.; Gewirtz, A.T.; Jones, R.M.; Kalman, D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc. Natl. Acad. Sci. 2020, 117, 21519–21526. [Google Scholar] [CrossRef]
- Peng, C.; Wu, C.; Xu, X.; Pan, L.; Lou, Z.; Zhao, Y.; Jiang, H.; He, Z.; Ruan, B. Indole-3-carbinol ameliorates necroptosis and inflammation of intestinal epithelial cells in mice with ulcerative colitis by activating aryl hydrocarbon receptor. Exp. Cell Res. 2021, 404, 112638. [Google Scholar] [CrossRef]
- Na, J.-I.; Kim, S.-Y.; Kim, J.-H.; Youn, S.-W.; Huh, C.-H.; Park, K.-C. Indole-3-acetic acid: A potential new photosensitizer for photodynamic therapy of acne vulgaris. Lasers Surg. Med. 2011, 43, 200–205. [Google Scholar] [CrossRef]
- Kwon, S.H.; Jeong, M.Y.; Park, K.C.; Youn, S.W.; Huh, C.H.; Na, J.I. A new therapeutic option for facial seborrhoeic dermatitis: Indole-3-acetic acid photodynamic therapy. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Grandi, V.; Baldi, I.; Cappugi, P.; Mori, M.; Pimpinelli, N. Indole 3-acetic acid-photodynamic therapy in the treatment of multiple actinic keratoses: A proof of concept pilot study. Photodiagnosis Photodyn. Ther. 2016, 16, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Helferich, W.G.; Denison, M.S. Ultraviolet photoproducts of tryptophan can act as dioxin agonists. Mol. Pharmacol. 1991, 40, 674–678. [Google Scholar] [PubMed]
- Diani-Moore, S.; Ma, Y.; Labitzke, E.; Tao, H.; David Warren, J.; Anderson, J.; Chen, Q.; Gross, S.S.; Rifkind, A.B. Discovery and biological characterization of 1-(1H-indol-3-yl)-9H-pyrido[3,4-b]indole as an aryl hydrocarbon receptor activator generated by photoactivation of tryptophan by sunlight. Chem. Biol. Interact. 2011, 193, 119–128. [Google Scholar] [CrossRef][Green Version]
- Öberg, M.; Bergander, L.; Håkansson, H.; Rannug, U.; Rannug, A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol. Sci. 2005, 85, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.D.; Helleberg, H.; Rannug, U.; Rannug, A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2- b]carbazole. Chem. Biol. Interact. 1998, 110, 39–55. [Google Scholar] [CrossRef]
- Fritsche, E.; Schäfer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hübenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar] [CrossRef] [PubMed]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef]
- Smirnova, A.; Wincent, E.; Vikström Bergander, L.; Alsberg, T.; Bergman, J.; Rannug, A.; Rannug, U. Evidence for new light-independent pathways for generation of the endogenous aryl hydrocarbon receptor agonist FICZ. Chem. Res. Toxicol. 2016, 29, 75–86. [Google Scholar] [CrossRef]
- Cardinali, G.; Flori, E.; Mastrofrancesco, A.; Mosca, S.; Ottaviani, M.; Dell’Anna, M.L.; Truglio, M.; Vento, A.; Zaccarini, M.; Zouboulis, C.C.; et al. Anti-inflammatory and pro-differentiating properties of the Aryl hydrocarbon receptor ligands NPD-0614-13 and NPD-0614-24: Potential therapeutic benefits in psoriasis. Int. J. Mol. Sci. 2021, 22, 7501. [Google Scholar] [CrossRef]
- Arfaoui, L. Dietary plant polyphenols: Effects of food processing on their content and bioavailability. Molecules 2021, 26, 2959. [Google Scholar] [CrossRef] [PubMed]
- Omar, F.; Tareq, A.M.; Alqahtani, A.M.; Dhama, K.; Sayeed, M.A.; Emran, T.B.; Simal-Gandara, J. Plant-based indole alkaloids: A comprehensive overview from a pharmacological perspective. Molecules 2021, 26, 2297. [Google Scholar] [CrossRef]
- Bjeldanes, L.F.; Kim, J.Y.; Grose, K.R.; Bartholomew, J.C.; Bradfield, C.A. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: Comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl. Acad. Sci. USA 1991, 88, 9543–9547. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Safe, S.; Bjeldanes, L. Indole-3-carbinol and diindolylmethane as aryl hydrocarbon (Ah) receptor agonists and antagonists in T47D human breast cancer cells. Biochem. Pharmacol. 1996, 51, 1069–1076. [Google Scholar] [CrossRef]
- Chen, Y.H.; Riby, J.; Srivastava, P.; Bartholomew, J.; Denison, M.; Bjeldanes, L. Regulation of CYP1A1 by indolo[3,2-b]carbazole in murine hepatoma cells. J. Biol. Chem. 1995, 270, 22548–22555. [Google Scholar] [CrossRef]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Zákostelská, Z.; Málková, J.; Klimešová, K.; Rossmann, P.; Hornová, M.; Novosádová, I.; Stehlíková, Z.; Kostovčík, M.; Hudcovic, T.; Štepánková, R.; et al. Intestinal microbiota promotes psoriasis-like skin inflammation by enhancing Th17 response. PLoS ONE 2016, 11, e0159539. [Google Scholar] [CrossRef]
- Stehlikova, Z.; Kostovcikova, K.; Kverka, M.; Rossmann, P.; Dvorak, J.; Novosadova, I.; Kostovcik, M.; Coufal, S.; Srutkova, D.; Prochazkova, P.; et al. Crucial Role of Microbiota in Experimental Psoriasis Revealed by a Gnotobiotic Mouse Model. Front. Microbiol. 2019, 10, 236. [Google Scholar] [CrossRef]
- Singh, N.P.; Singh, U.P.; Rouse, M.; Zhang, J.; Chatterjee, S.; Nagarkatti, P.S.; Nagarkatti, M. Dietary indoles suppress delayed-type hypersensitivity by inducing a switch from proinflammatory Th17 cells to anti-inflammatory regulatory T cells through regulation of microRNA. J. Immunol. 2016, 196, 1108–1122. [Google Scholar] [CrossRef]
- Wu, X.; Liu, J.; Chen, C.; Huang, Z.; Zang, Y.; Chen, J.; Dong, L.; Zhang, J.; Ding, Z. 3,3′-Diindolylmethane alleviates acute atopic dermatitis by regulating T cell differentiation in a mouse model. Mol. Immunol. 2021, 130, 104–112. [Google Scholar] [CrossRef]
- Weng, J.R.; Huang, T.H.; Lin, Z.C.; Alalaiwe, A.; Fang, J.Y. Cutaneous delivery of [1-(4-chloro-3-nitrobenzenesulfonyl)-1H-indol-3-yl]-methanol, an indole-3-carbinol derivative, mitigates psoriasiform lesion by blocking MAPK/NF-κB/AP-1 activation. Biomed. Pharmacother. 2019, 119, 109398. [Google Scholar] [CrossRef]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Miller, C.A.; Kato, T.; Saeki, K.; Matsuda, T. Indirubin and indigo are potent Aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 2001, 276, 31475–31478. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; Jha, G.G.; King, R.S. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, J.A. Indigogenic substrates for detection and localization of enzymes. Biotech. Histochem. 2007, 82, 73–103. [Google Scholar] [CrossRef]
- You, W.C.; Hsieh, C.C.; Huang, J.T. Effect of extracts from indigowood root (Isatis indigotica Fort.) on immune responses in radiation-induced mucositis. J. Altern. Complement. Med. 2009, 15, 771–778. [Google Scholar] [CrossRef]
- Kunikata, T.; Tatefuji, T.; Aga, H.; Iwaki, K.; Ikeda, M.; Kurimoto, M. Indirubin inhibits inflammatory reactions in delayed-type hypersensitivity. Eur. J. Pharmacol. 2000, 410, 93–100. [Google Scholar] [CrossRef]
- Uchiyama, K.; Takami, S.; Suzuki, H.; Umeki, K.; Mochizuki, S.; Kakinoki, N.; Iwamoto, J.; Hoshino, Y.; Omori, J.; Fujimori, S.; et al. Efficacy and safety of short-term therapy with indigo naturalis for ulcerative colitis: An investigator-initiated multicenter double-blind clinical trial. PLoS ONE 2020, 15, e0241337. [Google Scholar] [CrossRef]
- Naganuma, M.; Sugimoto, S.; Fukuda, T.; Mitsuyama, K.; Kobayashi, T.; Yoshimura, N.; Ohi, H.; Tanaka, S.; Andoh, A.; Ohmiya, N.; et al. Indigo naturalis is effective even in treatment-refractory patients with ulcerative colitis: A post hoc analysis from the INDIGO study. J. Gastroenterol. 2020, 55, 169–180. [Google Scholar] [CrossRef]
- Sugimoto, S.; Naganuma, M.; Kiyohara, H.; Arai, M.; Ono, K.; Mori, K.; Saigusa, K.; Nanki, K.; Takeshita, K.; Takeshita, T.; et al. Clinical efficacy and safety of oral Qing-Dai in patients with ulcerative colitis: A single-center open-label prospective study. Digestion 2016, 93, 193–201. [Google Scholar] [CrossRef]
- Lin, Y.K.; Wong, W.R.; Chang, Y.C.; Chang, C.J.; Tsay, P.K.; Chang, S.C.; Pang, J.H.S. The efficacy and safety of topically applied indigo naturalis ointment in patients with plaque-type psoriasis. Dermatology 2007, 214, 155–161. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chang, C.J.; Chang, Y.C.; Wong, W.R.; Chang, S.C.; Pang, J.H.S. Clinical assessment of patients with recalcitrant psoriasis in a randomized, observer-blind, vehicle-controlled trial using indigo naturalis. Arch. Dermatol. 2008, 144, 1457–1464. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chang, S.H.; Yang, C.Y.; See, L.C.; Lee, B.H.; Shih, I.H. Efficacy and safety of indigo naturalis ointment in treating atopic dermatitis: A randomized clinical trial: Indigo naturalis in treating atopic dermatitis. J. Ethnopharmacol. 2020, 250, 112477. [Google Scholar] [CrossRef]
- Lin, Y.K.; Leu, Y.L.; Yang, S.H.; Chen, H.W.; Wang, C.T.; Pang, J.H.S. Anti-psoriatic effects of indigo naturalis on the proliferation and differentiation of keratinocytes with indirubin as the active component. J. Dermatol. Sci. 2009, 54, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Chen, H.W.; Leu, Y.L.; Yang, Y.L.; Fang, Y.; Hwang, T.L. Indigo naturalis upregulates claudin-1 expression in human keratinocytes and psoriatic lesions. J. Ethnopharmacol. 2013, 145, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef]
- Jin, U.H.; Park, H.; Li, X.; Davidson, L.A.; Allred, C.; Patil, B.; Jayaprakasha, G.; Orr, A.A.; Mao, L.; Chapkin, R.S.; et al. Structure-dependent modulation of aryl hydrocarbon receptor-mediated activities by flavonoids. Toxicol. Sci. 2018, 164, 205–217. [Google Scholar] [CrossRef]
- Van der Heiden, E.; Bechoux, N.; Muller, M.; Sergent, T.; Schneider, Y.J.; Larondelle, Y.; Maghuin-Rogister, G.; Scippo, M.L. Food flavonoid aryl hydrocarbon receptor-mediated agonistic/antagonistic/synergic activities in human and rat reporter gene assays. Anal. Chim. Acta 2009, 637, 337–345. [Google Scholar] [CrossRef]
- Xue, Z.; Li, D.; Yu, W.; Zhang, Q.; Hou, X.; He, Y.; Kou, X. Mechanisms and therapeutic prospects of polyphenols as modulators of the aryl hydrocarbon receptor. Food Funct. 2017, 8, 1414–1437. [Google Scholar] [CrossRef]
- Vezza, T.; Rodríguez-Nogales, A.; Algieri, F.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Flavonoids in inflammatory bowel disease: A review. Nutrients 2016, 8, 211. [Google Scholar] [CrossRef]
- Salaritabar, A.; Darvishi, B.; HadjiakhoonDi, F.; Manayi, A.; Sureda, A.; Nabavi, S.F.; Fitzpatrick, L.R.; Nabavi, S.M.; Bishayee, A. Therapeutic potential of flavonoids in inflammatory bowel disease: A comprehensive review. World J. Gastroenterol. 2017, 23, 5097–5114. [Google Scholar] [CrossRef]
- Gálvez, J.; Coelho, G.; Crespo, M.E.; Cruz, T.; Rodríguez-Cabezas, M.E.; Concha, A.; Gonzalez, M.; Zarzuelo, A. Intestinal anti-inflammatory activity of morin on chronic experimental colitis in the rat. Aliment. Pharmacol. Ther. 2001, 15, 2027–2039. [Google Scholar] [CrossRef]
- Guazelli, C.F.S.; Fattori, V.; Colombo, B.B.; Georgetti, S.R.; Vicentini, F.T.M.C.; Casagrande, R.; Baracat, M.M.; Verri, W.A. Quercetin-loaded microcapsules ameliorate experimental colitis in mice by anti-inflammatory and antioxidant mechanisms. J. Nat. Prod. 2013, 76, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Park, M.Y.; Ji, G.E.; Sung, M.K. Dietary Kaempferol suppresses inflammation of dextran sulfate sodium-induced colitis in mice. Dig. Dis. Sci. 2012, 57, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Stivala, L.A.; Savio, M.; Carafoli, F.; Perucca, P.; Bianchi, L.; Maga, G.; Forti, L.; Pagnoni, U.M.; Albini, A.; Prosperi, E.; et al. Specific structural determinants are responsible for the antioxidant activity and the cell cycle effects of resveratrol. J. Biol. Chem. 2001, 276, 22586–22594. [Google Scholar] [CrossRef]
- Wang, H.K.; Yeh, C.H.; Iwamoto, T.; Satsu, H.; Shimizu, M.; Totsuka, M. Dietary flavonoid naringenin induces regulatory T cells via an aryl hydrocarbon receptor mediated pathway. J. Agric. Food Chem. 2012, 60, 2171–2178. [Google Scholar] [CrossRef]
- Michalski, J.; Deinzer, A.; Stich, L.; Zinser, E.; Steinkasserer, A.; Knippertz, I. Quercetin induces an immunoregulatory phenotype in maturing human dendritic cells. Immunobiology 2020, 225, 151929. [Google Scholar] [CrossRef]
- Schroeder, J.C.; DiNatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; De Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef]
- Miller, C.A. Expression of the human Aryl hydrocarbon receptor complex in yeast. J. Biol. Chem. 1997, 272, 32824–32829. [Google Scholar] [CrossRef]
- Weems, J.M.; Yost, G.S. 3-Methylindole metabolites induce lung CYP1A1 and CYP2F1 enzymes by AhR and non-AhR mechanisms, respectively. Chem. Res. Toxicol. 2010, 23, 696–704. [Google Scholar] [CrossRef]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; DeLuca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2017, 22, 598–605. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]
- Takamura, T.; Harama, D.; Fukumoto, S.; Nakamura, Y.; Shimokawa, N.; Ishimaru, K.; Ikegami, S.; Makino, S.; Kitamura, M.; Nakao, A. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol. Cell Biol. 2011, 89, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4+CD8αα+ T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Tay, A.S.L.; Li, C.; Ng, A.H.Q.; Wang, J.; Suri, B.K.; Matta, S.A.; McGovern, N.; Janela, B.; Wong, X.F.C.C.; et al. Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat. Microbiol. 2016, 1, 16106. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Luo, Y.; Zhu, Z.; Zhou, Y.; Sun, L.; Gao, J.; Sun, J.; Wang, G.; Yao, X.; Li, W. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol. 2019, 143, 2108–2119.e12. [Google Scholar] [CrossRef] [PubMed]
- Wille, G.; Mayser, P.; Thoma, W.; Monsees, T.; Baumgart, A.; Schmitz, H.J.; Schrenk, D.; Polborn, K.; Steglich, W. Malassezin—A novel agonist of the Aryl hydrocarbon receptor from the yeast Malassezia furfur. Bioorganic Med. Chem. 2001, 9, 955–960. [Google Scholar] [CrossRef]
- Gaitanis, G.; Magiatis, P.; Stathopoulou, K.; Bassukas, I.D.; Alexopoulos, E.C.; Velegraki, A.; Skaltsounis, A.-L. AhR ligands, malassezin, and indolo[3,2-b]carbazole are selectively produced by Malassezia furfur strains isolated from seborrheic dermatitis. J. Invest. Dermatol. 2008, 128, 1620–1625. [Google Scholar] [CrossRef]
- Magiatis, P.; Pappas, P.; Gaitanis, G.; Mexia, N.; Melliou, E.; Galanou, M.; Vlachos, C.; Stathopoulou, K.; Leandros, A.; Marselos, M.; et al. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. J. Invest. Dermatol. 2014, 133, 2023–2030. [Google Scholar] [CrossRef]
- Mexia, N.; George, G.; Velegraki, A.; Soshilov, A.; Denison, M.S.; Magiatis, P. Pityriazepin and other potent AhR ligands isolated from Malassezia furfur yeast. Arch. Biochem. Biophys. 2015, 571, 16–20. [Google Scholar] [CrossRef]
- Buommino, E.; Baroni, A.; Papulino, C.; Nocera, F.P.; Coretti, L.; Donnarumma, G.; De Filippis, A.; De Martino, L. Malassezia pachydermatis up-regulates AhR related CYP1A1 gene and epidermal barrier markers in human keratinocytes. Med. Mycol. 2018, 56, 987–993. [Google Scholar] [CrossRef]
- Buommino, E.; De Filippis, A.; Parisi, A.; Nizza, S.; Martano, M.; Iovane, G.; Donnarumma, G.; Tufano, M.A.; De Martino, L. Innate immune response in human keratinocytes infected by a feline isolate of Malassezia pachydermatis. Vet. Microbiol. 2013, 163, 90–96. [Google Scholar] [CrossRef]
- Buommino, E.; Nocera, F.P.; Parisi, A.; Rizzo, A.; Donnarumma, G.; Mallardo, K.; Fiorito, F.; Baroni, A.; De Martino, L. Correlation between genetic variability and virulence factors in clinical strains of Malassezia pachydermatis of animal origin. New Microbiol. 2016, 39, 216–223. [Google Scholar] [PubMed]
- Furue, M.; Tsuji, G. Chloracne and hyperpigmentation caused by exposure to hazardous aryl hydrocarbon receptor ligands. Int. J. Environ. Res. Public Health 2019, 16, 4864. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.M.; Höller, D.; Schiffer, R.; Frankenberg, S.; Neis, M.; Merk, H.F.; Jugert, F.K. Expression of multiple cytochrome P450 enzymes and multidrug resistance-associated transport proteins in human skin keratinocytes. J. Invest. Dermatol. 2001, 116, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Uchi, H.; Hashimoto-Hachiya, A.; Furue, M. Tryptophan photoproduct FICZ upregulates IL1A, IL1B, and IL6 Expression via oxidative stress in keratinocytes. Oxid. Med. Cell. Longev. 2018, 2018, 9298052. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Boyer, J.A.; Muku, G.E.; Murray, I.A.; Gowda, K.; Desai, D.; Amin, S.G.; Glick, A.B.; Perdew, G.H. Editor’s highlight: Ah receptor activation potentiates neutrophil chemoattractant (C-X-C Motif) ligand 5 expression in keratinocytes and skin. Toxicol. Sci. 2017, 160, 83–94. [Google Scholar] [CrossRef]
- Ray, S.S.; Swanson, H.I. Alteration of keratinocyte differentiation and senescence by the tumor promoter dioxin. Toxicol. Appl. Pharmacol. 2003, 192, 131–145. [Google Scholar] [CrossRef]
- Greenlee, W.F.; Dold, K.M.; Osborne, R. Actions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on human epidermal keratinocytes in culture. Vitr. Cell. Dev. Biol. 1985, 21, 509–512. [Google Scholar] [CrossRef]
- Um, J.-Y.; Kim, H.B.; Kang, S.Y.; Son, J.H.; Chung, B.Y.; Park, C.W.; Kim, H.O. 2,3,7,8-Tetrachlorodibenzo-p-dioxin regulates the expression of aryl hydrocarbon receptor-related factors and cytokines in peripheral blood mononuclear cells and CD4+ T cells from patients with atopic dermatitis and psoriasis. Ann. Dermatol. 2020, 32, 360–369. [Google Scholar] [CrossRef]
- Richardson, W.H.; Schmidt, T.M.; Nealson, K.H. Identification of an anthraquinone pigment and a hydroxystilbene antibiotic from Xenorhabdus luminescens. Appl. Environ. Microbiol. 1988, 54, 1602–1605. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, G.; Wu, H.; Webster, J.M. Identification of two pigments and a hydroxystilbene antibiotic from Photorhabdus luminescens. Appl. Environ. Microbiol. 1995, 61, 4329–4333. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J. Invest. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Vu, Y.H.; Hashimoto-Hachiya, A.; Takemura, M.; Yumine, A.; Mitamura, Y.; Nakahara, T.; Furue, M.; Tsuji, G. IL-24 negatively regulates keratinocyte differentiation induced by Tapinarof, an aryl hydrocarbon receptor modulator: Implication in the treatment of atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 9412. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Stein Gold, L.; Soung, J.; Tallman, A.M.; Rubenstein, D.S.; Gooderham, M. Efficacy and patient-reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J. Am. Acad. Dermatol. 2021, 84, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Chen, G.; Bolduc, C.; Maari, C.; Lyle, M.; Tang, L.; Webster, J.; Zhou, Y. Efficacy and safety of topical WBI-1001 in the treatment of atopic dermatitis: Results from a phase 2A, randomized, placebo-controlled clinical trial. Arch. Dermatol. 2010, 146, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Bolduc, C.; Maari, C.; Nigen, S.; Webster, J.M.; Tang, L.; Lyle, M. Efficacy and safety of topical WBI-1001 in patients with mild to moderate psoriasis: Results from a randomized double-blind placebo-controlled, phase II trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1516–1521. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for healthy skin: The emerging role of Aryl Hydrocarbon Receptors and Nuclear Factor-Erythroid 2-Related Factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Furue, M.; Izuhara, K. IL-24: A new player in the pathogenesis of pro-inflammatory and allergic skin diseases. Allergol. Int. 2020, 69, 405–411. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Gallego, N.; Sánchez-Madrid, F.; Cibrian, D. Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation. Cells 2021, 10, 3176. https://doi.org/10.3390/cells10113176
Fernández-Gallego N, Sánchez-Madrid F, Cibrian D. Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation. Cells. 2021; 10(11):3176. https://doi.org/10.3390/cells10113176
Chicago/Turabian StyleFernández-Gallego, Nieves, Francisco Sánchez-Madrid, and Danay Cibrian. 2021. "Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation" Cells 10, no. 11: 3176. https://doi.org/10.3390/cells10113176
APA StyleFernández-Gallego, N., Sánchez-Madrid, F., & Cibrian, D. (2021). Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation. Cells, 10(11), 3176. https://doi.org/10.3390/cells10113176